

Abstract
The ErbB2 tyrosine kinase receptor is an attractive target for immunotherapy, as it is overexpressed in many carcinomas. ImmunoRNases, made up of a human anti-ErbB2 scFv (single chain antibody fragment) and human RNases, have been engineered to overcome the limits of immunotoxins, made up of mouse antibodies and plant or bacterial toxins, such as immunogenicity and non-specific toxicity. Here we describe the construction and characterization of a second-generation anti-ErbB2 immunoRNase, called ERB–HP-DDADD-RNase, obtained by fusing Erbicin, a human ErbB2-directed scFv, with an inhibitor-resistant variant of human pancreatic RNase (HP-DDADD-RNase). This novel immunoRNase retains both the enzymatic activity of human pancreatic RNase and the specific binding of the parental scFv to ErbB2-positive cells, showing an affinity comparable with that of the previously reported parental immunoRNase (ERB–HP-RNase). Moreover, the novel immunoRNase is endowed with an effective and selective in vitro antiproliferative action for ErbB2-positive tumor cells, which is more potent than that of the parental immunoRNase on tumor cells expressing low levels of ErbB2, due to its resistance to the RNase inhibitor. Thus, the novel immunoRNase could represent a valuable tool for ErbB2-positive cancer therapy.
Keywords: breast cancer, ErbB2, human RNase, immunotherapy, RNase inhibitor
Introduction
Immunotherapy is of great interest today as a powerful strategy to fight cancer. One tumor-associated antigen that represents an attractive target for immunotherapy is ErbB2, a transmembrane tyrosine kinase receptor overexpressed on tumor cells of different origin, such as breast, ovary, lungs (Slamon et al., 1989; Tagliabue et al., 1991) or gastric cancer (Fukushige et al., 1986). ErbB2 is a member of the HER/ErbB transmembrane receptor tyrosine kinase family, and its overexpression leads to the activation of the signaling pathways promoting cancer growth and survival, such as mitogen-activated protein kinase and phosphatidylinositol-3-kinase (Klapper et al., 2000). On normal tissues, ErbB2 is not detectable or expressed at very low levels (Press et al., 1990). Further, ErbB2 plays a key role in the development of malignancy (Slamon et al., 1987) and is associated with more aggressive clinical behavior of breast carcinomas and poor patient prognosis (Baselga and Albanell, 2001).
The first ErbB2-targeted therapeutic immunoagent approved by the United States Food and Drug Administration was Herceptin® (trastuzumab), a humanized monoclonal antibody widely used for immunotherapy of ErbB2-positive carcinomas (Stebbing et al., 2000). Herceptin® is currently used with some success for breast cancer but it can engender cardiotoxicity, and a high fraction of breast cancer patients is resistant to Herceptin® treatment (Slamon et al., 2001; Seidman et al., 2002; Nahta et al., 2006). Moreover, carcinomas with a high expression of ErbB2, such as gastric and prostate tumors, have been found to be resistant or much less sensitive to Herceptin® (Agus et al., 1999; Gong et al., 2004).
To enhance their clinical potential, monoclonal antibodies can be equipped with radionuclides or toxins (Pastan and FitzGerald, 1991; Carter, 2001). Immunotoxins (ITs), based on toxins fused to antibodies or miniantibodies (single chain variable fragment, scFv) reactive to a certain type of tumor cells, have been designed for anticancer therapy, combining the potent toxicity of toxins with the antigen specificity of antibodies (Reiter and Pastan, 1998; Zhang et al., 2007). Nevertheless, the immunogenicity due to the mouse nature of the antibody moiety, and to the plant or bacterial origin of the toxin moieties, together with the non-specific toxicity displayed by ITs, such as vascular leak syndrome and hepatotoxicity, have greatly limited the therapeutic potential of ITs (Weiner et al., 1989; Schindler et al., 2001). A strategy to circumvent these problems is based on a specific type of immunoconjugate, an ‘immunoRNase’ (IR), in which a non-immunogenic RNase is the toxin and internalization into the target cells is mediated by the antibody moiety (Rybak and Newton, 1999; De Lorenzo and D'Alessio, 2008).
A human ErbB2-specific scFv, named Erbicin, has been obtained, which binds to cells that overexpress ErbB2, is internalized through receptor mediated endocytosis, inhibits cell proliferation (De Lorenzo et al., 2002), and recognizes an epitope different from that of Herceptin (Troise et al., 2011). Thus, Erbicin represents a valuable vehicle for delivering RNases to tumor cells. With this aim, a novel fully human IR, named ERB–HP-RNase, was previously obtained by fusing Erbicin with human pancreatic RNase (HP-RNase or RNase 1) (De Lorenzo et al., 2004), which is one of few human proteins that can manifest potent cytotoxic activity. The chimeric protein was found to retain the high binding affinity to ErbB2-positive cells of Erbicin, as well as the enzymatic activity of native human pancreatic RNase. When tested in vitro on a series of malignant cells, ERB–HP-RNase was found to discriminate between target and non-target cells, and to inhibit specifically the proliferation of ErbB2-positive cells. In particular, a positive correlation was evidenced between its cytotoxic activity and the levels of expression of ErbB2. Its antitumor potential has also been demonstrated in vivo using mice implanted with ErbB2-positive tumors that are either sensitive or resistant to Herceptin® (De Lorenzo et al., 2004; De Lorenzo and D'Alessio, 2009; Gelardi et al., 2010).
Unexpectedly, ERB–HP-RNase was found to be sensitive to the neutralizing action of the cytosolic RNase inhibitor (RI), a 50-kDa protein present in the cytosol that inhibits the ribonucleolytic activity of all monomeric members of the human pancreatic RNase family through its horseshoe-shaped binding surface (Kobe and Deisenhofer, 1995; Haigis et al., 2003; Dickson et al., 2005). The apparent inconsistency between the sensitivity to RI and the cytotoxic activity of the IR was resolved when it was shown that ERB–HP-RNase is indeed delivered to the cytosolic compartment of target cells, but in quantities that overcome the amounts of endogenous RI present in that compartment. Certainly, only the fraction of ERB–HP-RNase that evades RI would display any antitumor activity (De Lorenzo et al., 2007).
Based on the concern that the fraction of ERB–HP-RNase sequestered by the cytosolic inhibitor could not exert its antitumor activity, we designed a novel IR, named ERB–HP-DDADD-RNase, made up of Erbicin and an inhibitor-resistant variant (HP-DDADD-RNase) of human pancreatic RNase. The variant RNase was designed to suppress through Coulombic repulsion its binding to RI by the replacement of five amino acid residues crucial for both the formation and the stability of the RI·RNase complex (R39D/N67D/N88A/G89D/R91D) (Johnson et al., 2007b). For instance, when these five residues were replaced, the affinity was strongly reduced, as it was found to be 6 × 109-fold lower than that of the parental RNase (Johnson et al., 2007a,b). HP-DDADD-RNase is the most RI-evasive variant of HP-RNase reported to date.
Still, the amino acid substitutions only slightly affect the enzymatic activity of the variant RNase, which retains both conformational stability and ribonucleolytic activity comparable with that of the parental RNase (Johnson et al., 2007b). These features endow HP-DDADD-RNase with a more potent cytotoxic action, which has been exploited here for the construction of a second generation IR.
Materials and methods
Cell cultures
SKBR3 and A431 cells (both from ATCC, Rockville, MD, USA) were cultured in RPMI 1640 (Gibco BRL, Life Technologies, Paisley, UK). The KPL4 cell line from the malignant pleural effusion of a breast cancer patient with an inflammatory skin metastasis, and the JIMT-1 cell line, established from a pleural metastasis of a 62-year-old breast cancer patient resistant to Herceptin®, were grown in Dulbecco's modified Eagle's medium (Gibco BRL). Media were supplemented with 10% heat-inactivated fetal bovine serum (7.5% for JIMT-1), penicillin (100 UI ml–1), streptomycin (100 µg ml–1) and 2 mM glutamine (all from Gibco BRL) in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Antibodies
The following antibodies were used: horseradish peroxidase (HRP)-conjugated anti-His mouse monoclonal antibody (Qiagen GmnH, Hilden, Germany), anti-HER2 rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA, USA), anti-actin rabbit polyclonal antibody (Sigma-Aldrich, St Louis, MO, USA), HRP-conjugated anti-rabbit immunoglobulins from goat antiserum (Thermo Scientific, Rockford, IL, USA).
Construction of the chimeric cDNA encoding ERB–HP-DDADD-RNase
The cDNA encoding HP-DDADD-RNase was engineered by two successive PCRs. Upstream primers, named SP1 and SP2, were used to incorporate a NotI restriction site at the 5′ end and a spacer sequence, whereas a downstream primer, named AP, was used to introduce a NotI site at the 3′ end. In the first reaction, primers SP1 and AP were used. The oligonucleotide SP1 sequence (5′-GGCCCGGAAGGCGGCAGC AAAGAATCTAGAGCTAAAAA-3′) encodes the COOH-terminal half of the spacer; the oligonucleotide AP sequence (5′-ATAAGAATGCGGCCGCAGAGTCTTCAAC AGACG-3′) contains the NotI restriction site. In the second reaction, primers SP2 and AP were used. Oligonucleotide SP2 (5′-ATAAGAATGCGGCCGCAAGCGGCGGCCCGGAAGGCGG-3′) encodes the N-terminal half of the spacer preceded by the NotI site sequence.
The chimeric cDNA was cloned into the pET22b+ expression vector downstream of the sequence encoding the human anti-ErbB2 scFv, and the recombinant plasmid was fully sequenced to confirm the correct directional insertion of the RNase gene in the NotI site and the expected DNA sequence.
Expression and purification of ERB–HP-DDADD-RNase
Cultures of Escherichia coli BL21 DE3 (Novagen, Merk Millipore, Darmstadt, Germany), previously transformed with the recombinant pET22b+ expression vector, were grown at 37°C in Luria–Bertani (LB) medium containing 50 µg/ml ampicillin until the exponential phase was reached. The expression of soluble IR was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Applichem GmbH, Darmstadt, Germany) in the cell culture, which then was grown at room temperature for 4 h. The cells were harvested by centrifugation at 6000 rpm for 15 min at 4°C.
The periplasmic extract was obtained by resuspending the bacterial pellet in B-PER® buffer (Bacterial Protein Extraction Reagent; Pierce, Thermo Fisher Scientific) in the presence of EDTA-free protease inhibitors (Roche Applied Science GmbH, Mannheim, Germany). After an incubation at 25°C for 20 min by gently rotation, the supernatant containing the soluble periplasmic extract was obtained by centrifugation at 12 000 rpm for 20 min at 4°C.
The supernatant was loaded on an immobilized-metal affinity chromatography (IMAC) by incubation with cobalt-chelating resin (TALON®; Clontech, Palo Alto, CA, USA) for 2 h at 25°C by gentle rotation. After extensive washes in an appropriate buffer (phosphate-buffered saline (PBS), 0.16 M NaCl, 20 mM imidazole), the elution step was performed in the same buffer containing a higher concentration of imidazole (250 mM).
RNase activity and inhibition assays
RNase activity was tested by the acid-insoluble RNA precipitation assay as described previously (Bartholeyns et al., 1977; Borriello et al., 2011) on yeast RNA (8 mg/ml).
Briefly, yeast RNA was incubated with the enzyme under test in the reaction buffer (50 mM Tris–HCl, pH 8.0 containing 0.15 M NaCl) at 37°C for 30 min. The reaction was stopped, and undegraded RNA was precipitated by addition of 10% cold perchloric acid containing 0.25% uranyl acetate. After incubation on ice for 15 min, the samples were subjected to centrifugation at 12 000 rpm for 15 min at 4°C, and the absorbance at 260 nm of the supernatant was determined. In this assay, one unit of enzymatic activity is defined as the amount of enzyme that generates an increase of one optical density (OD) at 260 nm.
For inhibition assays, appropriate amounts of ERB–HP-RNase and ERB–HP-DDADD-RNase were pre-incubated with increasing concentrations of RI (Promega) in the presence of 2 mM dithiothreitol for 10 min at 37°C before starting the activity test described above. The SD (≤5%) was calculated on the basis of the results obtained in three independent experiments.
Enzyme-linked immunosorbant assay)
Binding assays were performed by modified enzyme-linked immunosorbent assay (ELISA), as previously described (De Lorenzo et al., 2004). Briefly, ErbB2-positive SKBR3, KPL4 or JIMT-1 cells and ErbB2-negative A431 control cells, harvested in non-enzymatic dissociation solution (Sigma-Aldrich), were washed and transferred to U-bottom microtiter plates (2 × 105 cells per well). After blocking with PBS containing 6% bovine serum albumin (BSA), the cells were incubated in the absence or presence of increasing concentrations of each IR (ERB–HP-RNase or ERB–HP-DDADD-RNase) in ELISA buffer (PBS/BSA 3%) for 90 min at 25°C by gentle shaking. After centrifugation at 1200 rpm for 7 min and the removal of supernatant, cell pellets were washed twice in 200 µl of PBS buffer, resuspended in 100 µl of ELISA buffer and incubated for 1 h with a peroxidase-conjugated anti-His antibody. After 1 h, the plates were centrifuged, washed with PBS buffer and reacted with 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich). The enzymatic reaction was stopped with 1 N HCl, and the binding values were determined from the absorbance at 450 nm and reported as the mean of three determinations. The SD (≤5%) was calculated on the basis of the results obtained in three independent experiments.
Cell lysis and western blotting
The cells were harvested in non-enzymatic dissociation solution (Sigma-Aldrich). After centrifugation at 1200 rpm for 7 min, the cell pellets were resuspended in a lysis buffer containing 10 mM Tris–HCl pH 7.4, 0.5% Nonidet-P-40, 150 mM NaCl in the presence of protease inhibitors (Roche). After incubation on ice for 20 min, the extracts were clarified by centrifugation at 12 000 rpm for 15 min at 4°C. Protein concentration was determined by the Bradford colorimetric assay (Sigma-Aldrich) and aliquots of 20 µg were analyzed by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE), followed by electroblotting onto poly(vinylidene difluoride) membranes (Merck-Millipore), as previously reported (Castellano et al., 2008).
Cytotoxicity assays
Cells were seeded in 96-well flat-bottom plates; SKBR3 cells at a density of 1.5 × 104 per well; A431, KPL-4 and JIMT-1 cells at a density of 5 × 103 per well. To test the effects of the IRs (ERB–HP-RNase or ERB–HP-DDADD-RNase) on cell growth, SKBR3 and A431 cells were incubated at 37°C for 72 h with the IRs (10–50 nM) in the culture medium.
KPL-4 and JIMT-1 cells were deprived of serum for 24 h and then treated with the IRs (10–100 nM) for 72 h in medium containing 0.1% serum. Cell counts were determined in triplicate by using the trypan blue exclusion test by an Automated Cell Counter TC10 (Bio-Rad, Richmond, CA, USA). Cell survival was expressed as a percentage of viable cells in the presence of the tested proteins, with respect to untreated control cultures. The SD (≤5%) was calculated on the basis of the results obtained in three independent experiments.
Results
Construction of ERB–HP-DDADD-RNase
For the construction of the chimeric ERB–HP-DDADD-RNase, we used a variant of human pancreatic RNase, in which one alanine and multiple aspartate residues were used to replace five residues involved intimately in the interaction with RI and thus crucial for both the formation and the stability of the RI·RNase complex. The RNase variant (R39D/N67D/N88A/G89D/R91D), named HP–DDADD-RNase, retains the ribonucleolytic activity and conformational stability of the native enzyme but has a markedly lower affinity for RI (Johnson et al., 2007b).
The chimeric construct was obtained by fusing the cDNAs encoding the inhibitor-resistant variant of human pancreatic RNase (HP-DDADD-RNase) and the anti-ErbB2 scFv Erbicin, as follows: the cDNA encoding the engineered RNase was amplified by two successive PCRs using upstream primers to incorporate the NotI restriction site at 5′ end, a spacer sequence encoding a peptide of 11 residues to separate the RNase and scFv moieties in the fusion protein, and a downstream primer to introduce a second NotI site at 3′ end. The amplification product was cloned in a T7 promoter-based E.coli expression vector (pET22b+) downstream to the sequence encoding the available human anti-ErbB2 scFv, cloned at NcoI/NotI sites. The chimeric cDNA was fully sequenced to confirm the correct directional insertion of the RNase cDNA in the NotI site and the expected DNA sequence.
The resulting construct, named ERB–HP-DDADD-RNase, has at the N-terminal end the scFv with a 15-residue linker made up of glycine and serine residues (SSGGGGSGGGGSGGS) interposed between variable domains of the heavy and light chains (VH and VL, respectively) of Erbicin, an 11-residue spacer (AAASGGPEGGS) inserted between the antibody fragment and the ribonuclease, and at the C-terminal end the RNase variant followed by a hexahistidine tag (Fig. 1).
Fig. 1.

Schematic representation of ERB–HP-DDADD-RNase. The construct was obtained by fusing the anti-ErbB2 scFv Erbicin and the engineered HP-DDADD-RNase. VH and VL, the variable domains of the heavy and light chains, respectively, of Erbicin; the linker, peptide between VH and VL domains, the spacer, peptide connecting the scFv and the RNase moieties; HP-DDADD, the variant RNase; (His)6, the 6-residue His tag.
Expression of ERB–HP-DDADD-RNase
Cultures of E.coli BL21(DE3), which had been transformed with the recombinant pET22b+ expression vector containing the cDNA of ERB–HP-DDADD-RNase, were grown in 1 l of LB medium with ampicillin to an OD600 of 0.8, induced with 1 mM of IPTG and incubated at 25°C for 4 h by shaking at a speed of 140 rpm. Cells were harvested by centrifugation at 6000 rpm for 15 min.
The periplasmic extract containing the chimeric protein was obtained by resuspending the bacterial pellet in B-PER buffer with protease inhibitors. After incubation at 25°C with gentle rotating, the supernatant, collected by centrifugation, was loaded on a cobalt-chelating resin to allow for the protein purification by IMAC (see ‘Materials and methods’).
The eluted protein was analyzed by SDS–PAGE followed by Coomassie staining. As shown in Fig. 2A, a single band of the expected molecular size was detectable, thus indicating that the heterologous protein was successfully purified. The purification yield was ∼0.5 mg/l culture. By western blotting with an anti-His antibody, the band of the expected size was visualized, thus confirming the immunoreactivity of the isolated protein (Fig. 2B).
Fig. 2.
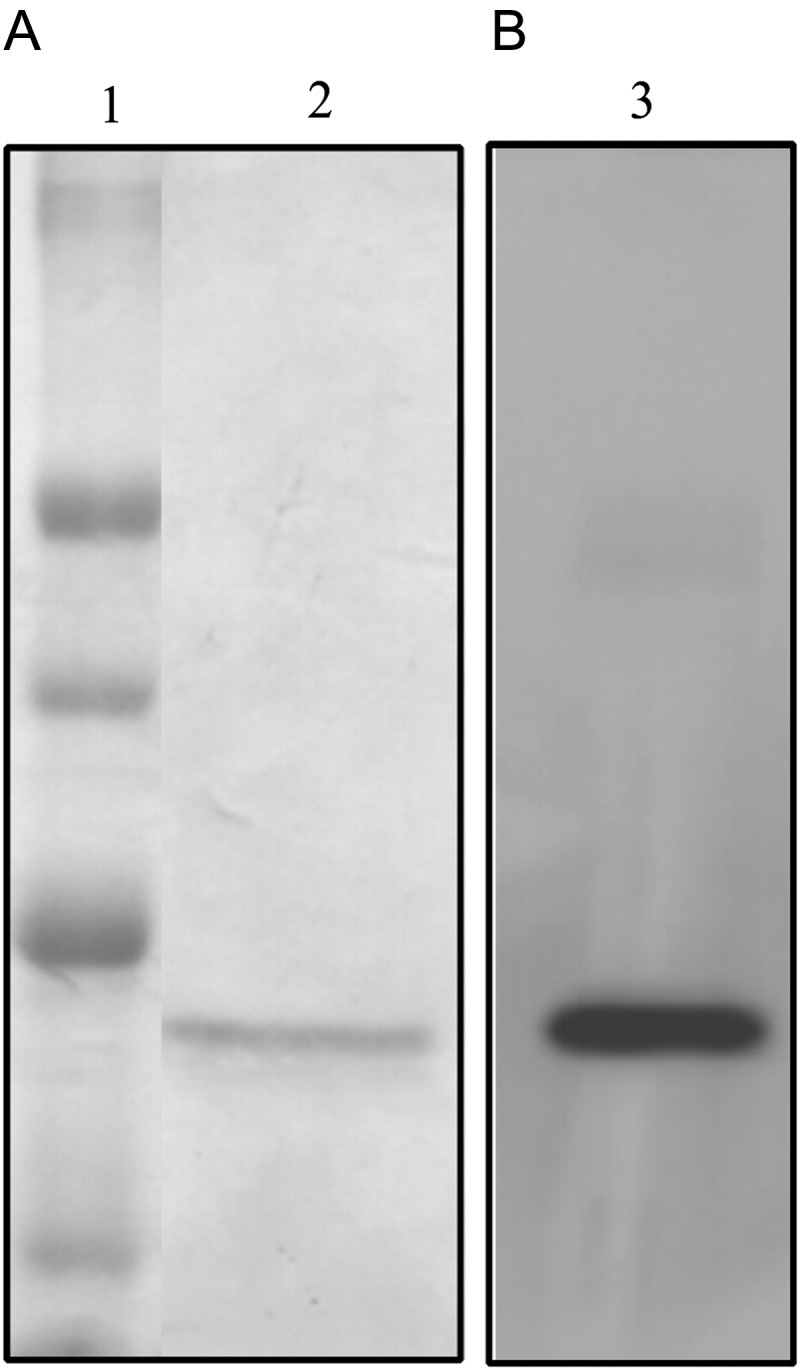
SDS–PAGE and western blotting analyses of purified ERB–HP-DDADD-RNase. (A) SDS–PAGE analysis followed by Coomassie staining of the samples eluted by IMAC. Lane 1: Molecular weight standards. Lane 2: fractions eluted from the column. (B) Western blotting analysis with an anti-His antibody. Lane 3: fractions eluted from the column.
RNase activity and inhibition of ERB–HP-DDADD-RNase
When tested by the acid-insoluble RNA precipitation assay (see Materials and methods), the novel IR was found to be active with a specific activity of 110 U/nmol. As the specific activity of the IR ERB–HP-RNase, made up of the wild-type RNase, is ∼280 U/nmol in this assay, we can conclude that the engineered IR retains about 40% of the activity of the parental IR, as expected from the data reported previously (Johnson et al., 2007b) for the variant and wild-type RNases.
The sensitivity of the engineered IR to inhibition by the human inhibitor (RI) was then tested by pre-incubating appropriate amounts of the IR with an increasing concentration of RI, prior to the activity test described above. As shown in Fig. 3, the IR ERB–HP-DDADD-RNase was found to be not susceptible to inhibition by RI, in contrast to the native IR ERB–HP-RNase, tested in parallel, which was found to be fully inhibited at an RI : IR ratio of ∼1. The percentage of inhibition was calculated as follows: % inhibition = 100% ×(1 − activity in the presence of RI/activity in the absence of RI).
Fig. 3.

Effects of the RI on the enzymatic activity of the IRs. Inhibition by RI of the catalytic activity of the parental ERB–HP-RNase (rhomboids) or the variant ERB–HP-DDADD-RNase (circles) was measured at increasing RI/IR ratios.
Binding assays of ERB–HP-DDADD-RNase to the ErbB2 receptor
The ability of the recombinant fusion protein ERB–HP-DDADD-RNase to bind to ErbB2-positive cells was analyzed by ELISA assays, performed as described in Materials and methods. ErbB2-positive SKBR3, KPL4 and JIMT-1 cells (Gelardi et al., 2010) expressing the ErbB2 receptor, and ErbB2-negative A431 control cells were used.
The results, shown in Fig. 4A, indicate that the engineered IR retains the specificity of the parental IR, as it binds with high affinity to SKBR3 cells overexpressing ErbB2, whereas no significant binding to A431 cells was detected. Furthermore, ERB–HP-DDADD-RNase shows the ability to bind also to the Herceptin®-resistant KPL4 and JIMT-1 cells with a comparable affinity to that of the parental IR.
Fig. 4.

(A) Binding curves of ERB–HP-RNase (empty symbols) or ERB–HP-DDADD-RNase (black symbols) to ErbB2-positive SKBR3 (squares), KPL4 (circles), JIMT1(triangles) or ErbB2-negative A431 (rhomboids) cell lines. (B) Quantitative analysis by western blotting of the ErbB2 receptor levels on the indicated cells. The intensity of positive bands was measured by a phosphorimager and normalized to the levels of actin, used as a standard protein.
To investigate the higher binding capacity of both IRs to SKBR3 cells compared with that observed for KPL4 and JIMT-1 cells, a quantitative analysis by western blotting was performed on lysates from the three cell types, tested for their ErbB2 levels with a commercial anti-ErbB2 mAb. The positive bands were analyzed with a phosphorimager, and the corresponding signal intensities were normalized to those obtained in the same lysates by an anti-actin antibody. The results of these experiments showed that the levels of ErbB2 in KPL4 and JIMT-1 cells are about 2- or 2.5-fold lower, respectively, than that measured in SKBR3 cells (Fig. 4B).
These results indicated that there is a positive correlation between the levels of expression of ErbB2 on a cell and the binding of the IRs to that cell.
Cytotoxic effects of ERB–HP-DDADD-RNase on tumor cells
ERB–HP-DDADD-RNase was tested for its cytotoxic effects on ErbB2-positive and ErbB2-negative cells. The cells were plated in the absence or in the presence of increasing concentrations of the IRs, ERB–HP-RNase or ERB–HP-DDADD-RNase. After 72 h of incubation at 37°C, cell survival was measured by counting trypan blue-excluding cells. As shown in Fig. 5, ERB–HP-DDADD-RNase was found to be selectively cytotoxic in a dose-dependent manner on all the antigen-positive SKBR3, KPL4 and JIMT-1 cell lines tested, whereas no effects on ErbB2-negative A431 control cells were observed.
Fig. 5.

In vitro effects of the IRs on tumor cell survival. (A) Dose–response curves of ErbB2-positive SKBR3 (squares) and ErbB2-negative A431 (rhomboids) cells, treated for 72 h with ERB–HP-RNase (empty symbols) or ERB–HP-DADD-RNase (black symbols). (B) Dose–response curves of Herceptin®-resistant KPL4 (circles) and JIMT-1 (triangles) cells, treated for 72 h with ERB–HP-RNase (empty symbols) or ERB–HP-DDADD-RNase (black symbols).
Surprisingly, despite the different sensitivity to RI of the two IRs, no different effects on cell survival were detected when ERB–HP-RNase or ERB–HP-DDADD-RNase were tested on SKBR3 cells expressing high levels of ErbB2, whereas a stronger cytotoxicity of ERB–HP-DDADD-RNase with respect to that of ERB–HP-RNase was evidenced on KPL4 and JIMT-1 cell lines, particularly when low protein concentrations were used. This finding can be explained by taking into consideration the different levels of ErbB2 receptor in the cell lines tested.
High levels of ErbB2 receptors, such as those of SKBR3 cells, allow for the internalization of large quantities of the IRs that overcome the amounts of the endogenous RI, so that only a fraction of each IR is neutralized by RI (De Lorenzo et al., 2007), whereas the remaining RI-free RNase fused to Erbicin can actively degrade cytosolic RNA and exert its cytosolic effect. Instead, on KPL4 and JIMT-1 cell lines, the lower levels of the receptor with respect to that of the SKBR3 cell line, allow for a less efficient internalization of the IRs in the cytosolic compartment, so that the low amount of intracellular ERB–HP-RNase is inhibited by RI more efficiently and its antitumor activity is strongly reduced, whereas the engineered RI-resistant ERB–HP-DDADD-RNase can still exert its full cytotoxic effect.
Discussion
Immunotherapy has been demonstrated to be a powerful strategy in anti-cancer therapy to overcome the limits of the conventional treatments. The ErbB2 receptor is an important tumor target because of its specific localization and high expression levels on many tumor cells of different origin, especially in human breast cancer.
Previously, we reported on a fully human IR, named ERB–HP-RNase, obtained by fusing the human anti-ErbB2 scFv with human pancreatic RNase (De Lorenzo et al., 2004). In the resulting fusion protein the scFv moiety allows the RNase to enter the cytosol and kill target cells, but the antitumor action of the IR is limited by its sensitivity to the RI, as a fraction of ERB–HP-RNase is blocked by RI and cannot display any antitumor activity (De Lorenzo et al., 2007).
In this study, we report on the construction, characterization and antitumor activity of a novel fully human RI-resistant IR targeting the ErbB2 receptor, called ERB–HP-DDADD-RNase. This IR was obtained by the fusion of the inhibitor-resistant variant of human pancreatic RNase (HP-DDADD-RNase) with the anti-ErbB2 scFv Erbicin. On the basis of the results of this study, we can conclude that the novel IR retains the biological action of both the antibody and the RNase variant moieties with the following advantages: it recognizes one of the most specific tumor-associated antigens, ErbB2, with an affinity comparable with that of the parental ERB–HP-RNase; it retains about 40% of the enzymatic activity of the parental ERB–HP-RNase IR but, unlike the latter, it is fully resistant to RI inhibition; it inhibits selectively and efficiently the in vitro growth of ErbB2-positive tumor cells with greater potency than that of the parental ERB–HP-RNase, likely due to its RI resistance. This differential toxicity is highlighted when the proteins are tested on cell lines with low expression of ErbB2 receptor, in which less efficient internalization of the IRs in the cytosolic compartment does not affect the cytotoxicity of the engineered RI-resistant ERB–HP-DDADD-RNase but leads to full inhibition of the parental ERB–HP-RNase.
The novel fully human ERB–HP-DDADD-RNase, on the basis of the efficacy and selectivity of its cytotoxic action on target cells, could represent a valuable tool in ErbB2-positive cancer therapy.
Funding
This work was supported by MIUR (Ministero dell' Istruzione, dell'Università e della Ricerca), Italy, and by the National Institutes of Health (R01 CA073808).
Acknowledgements
The authors wish to thank Dr Carmine Fedele for his skilled assistance and Prof G.D'Alessio for the critical reading of the manuscript.
Conflict of interest
Authors declare that they have no conflict of interest.
References
- Agus D.B., Scher H.I., Higgins B., Fox W.D., Heller G., Fazzari M., Cordon-Cardo C., Golde D.W. Cancer Res. 1999;59:4761–4764. [PubMed] [Google Scholar]
- Bartholeyns J., Wang D., Blackburn P., Wilson G., Moore S., Stein W.H. Int. J. Pept. Protein Res. 1977;10:172–175. doi: 10.1111/j.1399-3011.1977.tb02792.x. [DOI] [PubMed] [Google Scholar]
- Baselga J., Albanell J. Ann. Oncol. 2001;12(Suppl. 1):S35–41. doi: 10.1093/annonc/12.suppl_1.s35. [DOI] [PubMed] [Google Scholar]
- Borriello M., Laccetti P., Terrazzano G., D'Alessio G., De Lorenzo C. Br. J. Cancer. 2011;104:1716–1723. doi: 10.1038/bjc.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P. Nat. Rev. Cancer. 2001;1:118–129. doi: 10.1038/35101072. [DOI] [PubMed] [Google Scholar]
- Castellano I., Ruocco M.R., Cecere F., Di Maro A., Chambery A., Michniewicz A., Parlato G., Masullo M., De Vendittis E. Biochim. Biophys. Acta. 2008;1784:816–826. doi: 10.1016/j.bbapap.2008.02.003. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., Arciello A., Cozzolino R., Palmer D.B., Laccetti P., Piccoli R., D'Alessio G. Cancer Res. 2004;64:4870–4874. doi: 10.1158/0008-5472.CAN-03-3717. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., D'Alessio G. Curr. Pharm. Biotechnol. 2008;9:210–214. doi: 10.2174/138920108784567254. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., D'Alessio G. FEBS J. 2009;276:1527–1535. doi: 10.1111/j.1742-4658.2009.06896.x. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., Di Malta C., Cali G., Troise F., Nitsch L., D'Alessio G. FEBS Lett. 2007;581:296–300. doi: 10.1016/j.febslet.2006.12.034. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., Palmer D.B., Piccoli R., Ritter M.A., D'Alessio G. Clin. Cancer Res. 2002;8:1710–1719. [PubMed] [Google Scholar]
- Dickson K.A., Haigis M.C., Raines R.T. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:349–374. doi: 10.1016/S0079-6603(05)80009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige S., Matsubara K., Yoshida M., Sasaki M., Suzuki T., Semba K., Toyoshima K., Yamamoto T. Mol. Cell. Biol. 1986;6:955–958. doi: 10.1128/mcb.6.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelardi T., Damiano V., Rosa R., Bianco R., Cozzolino R., Tortora G., Laccetti P., D'Alessio G., De Lorenzo C. Br. J. Cancer. 2010;102:513–519. doi: 10.1038/sj.bjc.6605499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S.J., Jin C.J., Rha S.Y., Chung H.C. Cancer Lett. 2004;214:215–224. doi: 10.1016/j.canlet.2004.04.029. [DOI] [PubMed] [Google Scholar]
- Haigis M.C., Kurten E.L., Raines R.T. Nucleic Acids Res. 2003;31:1024–1032. doi: 10.1093/nar/gkg163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.J., Chao T.Y., Lavis L.D., Raines R.T. Biochemistry. 2007a;46:10308–10316. doi: 10.1021/bi700857u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.J., McCoy J.G., Bingman C.A., Phillips G.N., Jr, Raines R.T. J. Mol. Biol. 2007b;368:434–449. doi: 10.1016/j.jmb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper L.N., Kirschbaum M.H., Sela M., Yarden Y. Adv. Cancer Res. 2000;77:25–79. [PubMed] [Google Scholar]
- Kobe B., Deisenhofer J. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- Nahta R., Yu D., Hung M.C., Hortobagyi G.N., Esteva F.J. Nat. Clin. Pract. Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- Pastan I., FitzGerald D. Science. 1991;254:1173–1177. doi: 10.1126/science.1683495. [DOI] [PubMed] [Google Scholar]
- Press M.F., Cordon-Cardo C., Slamon D.J. Oncogene. 1990;5:953–962. [PubMed] [Google Scholar]
- Reiter Y., Pastan I. Trends Biotechnol. 1998;16:513–520. doi: 10.1016/s0167-7799(98)01226-8. [DOI] [PubMed] [Google Scholar]
- Rybak S.M., Newton D.L. Exp. Cell. Res. 1999;253:325–335. doi: 10.1006/excr.1999.4718. [DOI] [PubMed] [Google Scholar]
- Schindler J., Sausville E., Messmann R., Uhr J.W., Vitetta E.S. Clin. Cancer Res. 2001;7:255–258. [PubMed] [Google Scholar]
- Seidman A., Hudis C., Pierri M.K., Shak S., Paton V., Ashby M., Murphy M., Stewart S.J., Keefe D. J. Clin. Oncol. 2002;20:1215–1221. doi: 10.1200/JCO.2002.20.5.1215. [DOI] [PubMed] [Google Scholar]
- Slamon D.J., Clark G.M., Wong S.G., Levin W.J., Ullrich A., McGuire W.L. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon D.J., Godolphin W., Jones L.A., et al. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Slamon D.J., Leyland-Jones B., Shak S., et al. N. Engl. J. Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Stebbing J., Copson E., O'Reilly S. Cancer Treat. Rev. 2000;26:287–290. doi: 10.1053/ctrv.2000.0182. [DOI] [PubMed] [Google Scholar]
- Tagliabue E., Centis F., Campiglio M., et al. Int. J. Cancer. 1991;47:933–937. doi: 10.1002/ijc.2910470625. [DOI] [PubMed] [Google Scholar]
- Troise F., Monti M., Merlino A., Cozzolino F., Fedele C., Russo Krauss I., Sica F., Pucci P., D'Alessio G., De Lorenzo C. FEBS J. 2011;278:1156–1166. doi: 10.1111/j.1742-4658.2011.08041.x. [DOI] [PubMed] [Google Scholar]
- Weiner L.M., O'Dwyer J., Kitson J., Comis R.L., Frankel A.E., Bauer R.J., Konrad M.S., Groves E.S. Cancer Res. 1989;49:4062–4067. [PubMed] [Google Scholar]
- Zhang Q., Chen G., Liu X., Qian Q. Cell. Res. 2007;17:89–99. doi: 10.1038/sj.cr.7310143. [DOI] [PubMed] [Google Scholar]