

Abstract
Poor oral absorption is one of the limiting factors in utilizing the full potential of polar antiviral agents. The neuraminidase target site requires a polar chemical structure for high affinity binding, thus limiting oral efficacy of many high affinity ligands. The aim of this study was to overcome this poor oral absorption barrier, utilizing prodrug to target the apical brush border peptide transporter 1 (PEPT1). Guanidine oseltamivir carboxylate (GOCarb) is a highly active polar antiviral agent (prodrug) with insufficient oral bioavailability (4%) to be an effective therapeutic agent. In this report we utilize a carrier-mediated targeted prodrug approach to improve the oral absorption of GOCarb.
Acyloxy(alkyl) ester based amino acid linked prodrugs were synthesized and evaluated as potential substrates of mucosal transporters e.g. PEPT1. Prodrugs were also evaluated for their chemical and enzymatic stability. PEPT1 transport studies included [3H]Gly-Sar uptake inhibition and cellular uptake experiments using HeLa cells over-expressing PEPT1. The intestinal membrane permeability of the selected prodrugs and the parent drug were then evaluated for epithelial cell transport across Caco-2 monolayers, and in the in-situ rat intestinal jejunal perfusion model.
Prodrugs exhibited a pH dependent stability with higher stability at acidic pHs. Significant inhibition of uptake (IC50 <1mM) was observed for L-valyl and L-isoleucyl amino acid prodrugs in competition experiments with [3H]Gly-Sar, indicating a 3–6 times higher affinity for PEPT1 compared to valacyclovir; a well-known PEPT1 substrate and >30 fold increase in affinity compared to GOCarb. The L-valyl prodrug exhibited significant enhancement of uptake in PEPT1/HeLa cells, and compared favorably with the well absorbed valacyclovir. Transepithelial permeability across Caco-2 monolayers showed that these amino acid prodrugs have a 2–5 fold increase in permeability as compared to the parent drug and showed that the L-valyl prodrug (Papp = 1.7×10−6 cm/sec) has the potential to be a rapidly transported across the epithelial cell apical membrane. Significantly, only the parent drug (GOCarb) appeared in the basolateral compartment, indicating complete activation (hydrolysis) during transport. Intestinal rat jejunal permeability studies showed that L-valyl and L-isoleucyl prodrugs are highly permeable compared to the orally well absorbed metoprolol, while the parent drug had essentially zero permeability in the jejunum, consistent with its known poor low absorption.
Prodrugs were rapidly converted to parent in cell homogenates suggesting their ability to be activated endogenously in the epithelial cell, consistent with the transport studies. Additionally, L-valyl prodrug was found to be a substrate for valacyclovirase (Km=2.37 mM) suggesting a potential cell activation mechanism.
Finally we determined the oral bioavailability of our most promising candidate, GOC-L-Val, in mice to be 23% under fed conditions and 48% under fasted conditions.
In conclusion, GOC-L-Val prodrug was found to be a very promising antiviral agent for oral delivery. These findings indicate that the carrier-mediated prodrug approach is an excellent strategy for improving oral absorption of polar neuraminidase inhibitors. These promising results demonstrates that the oral peptide transporter-mediated prodrug strategy has enormous promise for improving the oral mucosal cell membrane permeability of polar, poorly absorbed antiviral agents and treating influenza via the oral route of administration.
Keywords: Prodrugs, valacyclovirase, PEPT1, influenza, neuraminidase, oral absorption, intestinal permeability, antiviral, oseltamivir, transporter, stability, caco-2 permeability, glysar, rat perfusion
Introduction
Influenza, caused by Type A and B virus strains, is one of the most frequent illnesses suffered by humankind. The infamous `Spanish flu' killed an estimated 50–100 million people worldwide and flu still poses seasonal and pandemic threats around the globe through the emergence of new strains, such as the avian flu strain H5N1. Anti-influenza agents have met with limited success due to the virus's ability to alter its surface antigen.1 Similarly, vaccines have met with only partial success due to this `antigenic drift'. The FDA has approved the M2 channel blockers amantadine (Symmetrel®) and rimantadine (Flumadine®) and the neuraminidase inhibitors zanamivir (Relenza®) and oseltamivir (Tamiflu®, Compound 2, Figure 1) as anti-influenza medications. However, the CDC recommends only use of oseltamivir, which is an oral prodrug of the active neuraminidase inhibitor, oseltamivir carboxylate (OC, compound 1, Figure 1), for prevention of flu and use of oseltamivir or zanamivir for treatment of flu due to high resistance of many current strains of influenza toward the M2 channel blockers. In the search for medications for influenza, neuraminidase, a viral membrane bound glycoside hydrolase enzyme, was determined to be a potential therapeutic target. The viral neuraminidase enzyme is essential for escape of newly synthesized virions from infected cells and spread throughout the organism.2 This enzyme has also been shown to be essential for the virus to move through the respiratory tract, the most common means of viral transmission.3
Figure 1.
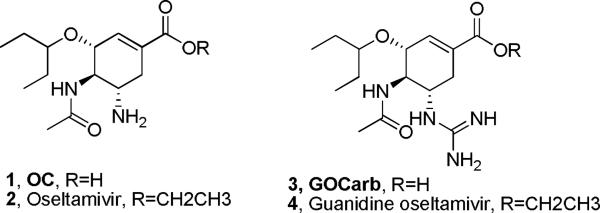
Oseltamivir and its analogues
Recent reports on oseltamivir-resistant strains of influenza virus have raised concerns about the usefulness of oseltamivir, emphasizing the urgent need for new therapies for flu treatment and prevention.4 The crystal structure of oseltamivir-resistant neuraminidase reveals an altered hydrophobic pocket in the active site of the enzyme required for oseltamivir binding that greatly diminishes the effectiveness of the oseltamivir carboxylate.5 Further, the H5N1 virus strains (His274Tyr or His294Ser substitution) have been found to be oseltamivir resistant.4c It has also been determined that the H1N1 strain (seasonal influenza A) is naturally resistant to oseltamivir with the His274Tyr mutation highly circulating in various European countries.
4-Guanidine oseltamivir carboxylate (GOCarb), a more potent guanidine analogue of OC, is a potential influenza treatment. It was found to be a potent inhibitor of both Type A and B neuraminidases and also a potent growth inhibitor (IC50 ranging from 0.64 to 7.9 nM) of a wide range of influenza A and B viruses in vitro.6 In tissue culture, GOCarb was found to be approximately 100 and 13 fold more potent than OC, against Type A and B strains, respectively and was found to be ~10 fold more potent in inhibiting human lysosomal neuraminidase as compared to OC.7 Importantly, GOCarb, is active against oseltamivir-resistant neuraminidase. However, similar to the guanidinium containing zanamivir, GOCarb shows very limited membrane permeability and, as a result, very poor oral bioavailability, limiting it utility as an influenza therapeutic.8 Significantly the ethyl ester prodrug, designed to increase logP values and hence membrane permeability, effective with OC, did not increase the oral bioavailability of GOCarb.9
In this report, we present a carrier-mediated prodrug approach, effective in increasing intestinal mucosal cell membrane permeability and oral availability, using linked amino acids as promoieties, targeting the PEPT1 membrane carrier protein. Amino acid acyloxy ester prodrugs of nucleoside analogues have been shown to be substrates of the PEPT1 peptide transporter increasing the oral absorption and bioavailability.10 Valacyclovir (Valtrex®) and valganciclovir (Valcyte®) are two well-known examples of nucleoside analogue antiviral prodrugs that utilize PEPT1 to achieve good oral bioavailability.11 Analogous carrier-mediated approach was successfully applied to zanamivir by our group that resulted in enhanced uptake of prodrugs in in-vitro and in-situ experiments.12
We describe the syntheses and characterization of L-isoleucyl, L-leucyl and L-valyl amino acid ester linked prodrugs of GOCarb. These prodrugs were evaluated for stability, activation (hydrolysis) and mucosal cell uptake and transport. Transport mechanism and membrane permeability were assessed using cell over expressing PEPT1, Caco-2 cell monolayer transport studies and single-pass intestinal jejunal perfusion (SPIP) experiments in rats. In addition, the potential activation mechanism of the prodrugs in mucosal cells was evaluated in transport experiments, cell homogenates and through the use of recombinant valacyclovirase, the enzyme that was found to hydrolyze valacyclovir in intestinal cells. Finally, the oral bioavailability of the GOC-L-Val prodrug was determined in mice.
Materials and Methods
All chemicals were reagent, analytical, HPLC or LC/MS grade. Oseltamivir phosphate was obtained from Sequoia Research Products Ltd (Pangbourne, UK). The tert-butyloxycarbonyl (Boc) protected amino acids were obtained either from Calbiochem-Novabiochem (San Deigo, CA, USA) or Alfa Aesar (Ward Hill, MA, USA). 1-bromo-1-chloro ethane was obtained from Alfa Aesar (Ward Hill, MA, USA). ACS grade solvents, trifluoroacetic acid, anhydrous dimethyl formamide (DMF), triethylamine and other reagents were procured from Aldrich Chemical Company (Milwaukee, WI, USA). HPLC and LC/MS grade water, acetonitrile, methanol, formic acid, trifluoroacetic acid were obtained from Fisher Scientific (St. Louis, MO, USA). Valacyclovir was kindly gifted by GlaxoSmithKline, Inc. (Research Triangle Park, NC, USA). Valacyclovirase was previously cloned and purified by the Amidon Lab and used as such for activation studies. Cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA) and other supplies were obtained from Corning (Corning, NY, USA) and Falcon (Lincoln Park, NJ, USA).
GOCarb Prodrug Synthesis
Compounds 11 a–c were synthesize as shown in Scheme 1.13 Briefly, oseltamivir was first converted to its guanidine analogue and the ester group was then hydrolyzed to give free carboxylic acid. The carboxyl terminal of guanidine analogue was then linked to carboxyl terminal of amino acids by the use of an acyloxy ethyl linker. The detailed synthetic procedure is given below.
Scheme 1.

Synthesis of L-valyl, L-isoleucyl and L-leucyl amino acid acyloxy ester prodrugs of GOCarb
6a–c
For the synthesis of cesium salts of boc-protected amino acids (Boc-L-Val, Boc-L-Leu, Boc-LIle), corresponding amino acids were dissolved in ethanol and the pH of the solution was adjusted to 6.5 with 1M cesium carbonate. The solvent was removed and compounds were dried under high vacuum for 24 h followed by drying over phosphorus pentaoxide (P2O5). To the stirring solutions of cesium salts of Boc-protected amino acids in DMF were added 3 equivalents of 1-bromo-1-chloro ethane and reaction kept in dark for 20 h.14 TLC using ethylacetate:hexane (7:1) showed three spots on charring with ninhydrin corresponding to the desired compound, its gem-diester and starting material. White precipitate of cesium bromide was then filtered through fine pore buchner funnel and concentrated in vacuum. Column chromatography using 1:20 to 1:15 (ethylacetate:hexane) gave pure compounds 6a–c in 20–25% yield.
8
To a mixture of oseltamivir phosphate, 7, (1g, 2.4 mmol), bis-boc thiourea (1.06g, 3.6 mmol) and triethyl amine (TEA, 1.1ml, 8 mmol) in 20 ml DMF was added Mercury (II) chloride (0.728g, 2.6 mmol) while stirring at 0 °C. Reaction was followed by thin layer chromatography (TLC) using ethyl acetate:hexane (1:3). After all starting material was reacted (approximately 1 h); the reaction mixture was diluted with ethyl acetate and filtered. It was then washed with water and brine and dried over anhydrous magnesium sulfate (MgSO4). Column chromatography using hexane; ethyl acetate (10:1) gave pure compound 8 as white powder in 89% overall yield.
9
565 mg of 8 was dissolved in 30ml tetrahydrofuran/water (2:1) at 0 °C and 10ml of 1N lithium hydroxide (LiOH) was added drop wise to the stirring solution.15 After 2–3 h completion of reaction was followed by TLC using ethyl acetate:hexane (1:1). The solution was diluted with ethyl acetate and 1N cold hydrochloric acid at 0 °C was added to the above solution drop wise till a pH ~5–6 was reached. It was then extracted 3 times with ethyl acetate, washed with brine and dried over MgSO4. Vacuum drying gave 478 mg (89% yield) of pure compound 9 as fine white powder.
10a–10c
To a stirring solution of 9 in anhydrous DMF was added 5 equivalents of TEA and, after approximately 30 minutes, 1 equivalent of acyloxy chloride esters of the corresponding amino acids (6a–c) were added dropwise to the stirring solution and reaction kept for 24 h at 40–45 °C.16 Reactions were monitored by TLC (ethyl acetate: hexane = 1:1 and chloroform:methanol = 9:1) and mass spectrometry. Some reactions required as long as 5 days for substantial yields. DMF was then vacuum dried and compounds were purified by column chromatography using ethyl acetate:hexane (1:10 to 1:1).
11a–c
For the synthesis of final compounds 11a–c; intermediates 10a–c were stirred with 40% trifloroacetic acid in dichloromethane for approximately 2 hours.15 Following completion of the reaction, compounds were vacuum dried and lyophilized as white powder. Yield was almost 100%
GOC-L-Val (11a)
1H NMR (CD3OD),, δ (ppm) 0.89–0.96 (m, 6H), 1.08–1.11 (m, 6H), 1.51–1.57 (m, 4H), 1.62–1.63 (m, 3H), 2.01 (s, 3H), 2.31–2.35 (m, 2H), 3.02 (m, 2H), 3.44–3.46 (m, 1H), 3.91–3.92 (m, 1H), 4.06 (d, J=10.0, 1H), 4.28 (s, 1H), 5.52 (s, 1H, NH), 6.88–6.90 (m, 1H), 7.04–7.07 (m, 1H), 8.00 (s, 1H, NH); ESI-MS: 470.2(M+H)+
GOC-L-Ile (11b)
1H NMR (CD3OD) δ (ppm) 0.89–0.96 (m, 9H), 1.07 (d, J=6.85 Hz, 3H), 1.48–1.56 (m, 6H), 1.62 (d, J=8.0 Hz, 3H), 2.02 (m, 3H), 2.36–2.37 (m, 2H), 3.37 (m, 2H), 3.44–3.46 (m, 1H), 3.92–3.94 (m, 1H), 4.07 (dd, J=3.0 Hz, 1H), 4.28–4.36 (m, 1H), 4.46 (m, 1H), 6.89–6.93 (m, 1H), 7.05–7.08 (m, 1H), 7.37 (s, 1H, NH), 8.00 (s, 1H, NH); ESI-MS: 484.2 (M+H)+
GOC-L-Leu (11c)
1H NMR (CD3OD) δ (ppm) 0.90–1.2 (m, 12H), 1.36–1.39 (m, 3H), 1.51–1.1.61 (m, 6H), 1.62–1.68 (m, 3H), 1.73–1.87 (m, 4H), 3.44–3.46 (m, 1H), 3.91–3.98 (m, 2H), 4.04–4.07 (m, 1H), 4.19–4.89 (m, 2H), 6.9–7.4 (m, 1H), 8.00 (s, 1H, NH); ESI-MS: 484.5 (M+H)+
Stability Studies
1. Chemical Stability
The chemical stability of GOCarb, its ethyl ester and corresponding prodrugs were studied in 100mM pH buffers (pH 1.2 HCl buffer, pH 4 acetate buffer, pH 6 & 7.4 phosphate buffer) as previously described.17 10 μL samples were taken at various time points and diluted with water containing either 5% formic acid or trifluoroacetic acid at 0°C. A final concentration of 10 μM was used for LC/MS analysis and 250 μM for HPLC analysis.
2. Metabolic Stability
2a. Hydrolysis in Cell Homogenates
Caco-2 cells (passage 32) were seeded for 22 days and washed with phosphate buffer saline (pH 7.4). The cells were scrapped and collected in 100mM phosphate buffer (pH 7.4) and spun down by centrifugation. The cells were re-suspended in phosphate buffer and lysed by sonication for a few seconds. The cell lysate was centrifuged at 10,000rpm for 15 min and the supernatant was collected. The protein amount of supernatant was determined with the Bio-Rad DC Protein Assay using bovine serum albumin as the standard. GOCarb, its ethyl ester and corresponding prodrugs were incubated with Caco-2 cell extract (500 μg/ml of protein) at 37°C for 2 hours. Samples were taken at specific time points (0, 5, 10, 15, 20 and 30 min) and mixed with acetonitrile containing 5 % formic acid. The samples were spun at 10,000 rpm at 4°C for 15 min and the supernatant was filtered using polypropylene filter (0.45μm, Whatman®) before HPLC or LC/MS analysis.
2b. Hydrolysis with Valacyclovirase
The potential activation mechanism of the prodrug was evaluated using valacyclovirase as described earlier.18 A final concentration of 250 ng/ml of valacyclovirase and 250 μM of prodrug was used for half-life measurements. Prodrugs were incubated with and without enzyme at 37 °C and samples were taken at 1, 3, 5, 10 and 30 minutes. An equal volume of acetonitrile with5% trifloro acetic acid at 0 °C was added to quench the reaction, and samples were analyzed by HPLC. To determine kinetic parameters, different concentrations (0.1 mM to 3 mM) of prodrugs were preincubated with 50 mM HEPES buffer (pH 7.4) for 3 minutes followed by addition of valacyclovirase (250 ng/ml final). The reaction mixture was quenched (after 2 minutes) using ice cold acetonitrile with 5% trifloro acetic acid. Hydrolysis due to buffers was normalized and Kcat and Vmax were calculated using GraphPad Prism v4.01 (GraphPad Software Inc., San Diego, CA).
Caco-2 Cell Monolayers Permeability
Caco-2 cells are human colonic carcinoma cell line. They have shown to express a whole range of transporters and have been used as a good model for both passive diffusion and carrier-mediated transport.19 Caco-2 permeability for the test compounds was determined as previously described.11a, 12 Briefly, Caco-2 cells were seeded on 6-well format collagen-coated transwell inserts (0.4μm pore size, area 4.7 cm2, Corning, NY) at a density of 1.0 × 105 cells/cm2 and cultured in DMEM (10% FBS, 1% nonessential amino acids, 1mM sodium pyruvate and 1% L-glutamine; Invitrogen, Carlsbad CA). Cells were grown in 5% CO2 and 90% relative humidity at 37°C. The culture medium was replaced every two days. Transepithelial electrical resistance (TEER) measurements were performed on all monolayers. Monolayers with TEER values > 250 Ω/cm2 were used for the study. On the day of the experiment, 1.5 mL of pH 6.0 MES buffer (5 mM D-glucose, 5 mM MES, 1 mM CaCl2, 1 mM MgCl2, 150 mM NaCl, 3 mM KCl and 1 mM NaH2PO4) was applied to the apical side and 2.6 mL pH 7.4 HEPES buffer (1mM CaCl2, 1mM MgCl2, 150mM NaCl, 3mM KCl and 1mM NaH2PO4) was applied to the basolateral side. The plates were equilibrated at 37°C for 15 minutes at which point the apical layer medium was aspirated and replaced with 1.5mL of drug (0.25mM) solution in pH 6.0 MES buffer and the basolateral medium was replaced with 2.6 mL of fresh HEPES buffer. Every 15 minutes, 200 μL aliquot from basolateral compartment was taken and was replaced with fresh HEPES buffer. The stability of prodrugs in the apical layer was determined by taking 10 μL aliquots of the apical solution and quenching with 90 μL of 0.1% (w/v) TFA in acetonitrile. At the end of experiment (2 hours), the integrity of the Caco-2 monolayers were checked by monitoring TEER values. Aliquots from both apical and basolateral sides were filtered (0.45 μ Whatman® 96 well filter plates) and analyzed by LC-MS.
[3H]Gly-Sar Uptake Inhibition
5.0 × 104 Caco-2 cells/well were seeded and grown for 10 days in 12-well plates. On the day of experiment, cells were washed with pH 6.0 uptake buffer (145 mM NaCl, 3 mM KCl, 1 mM NaH2PO4, 1 mM CaCl2, 0.5 mM MgCl2, 5 mM D-glucose, and 5 mM MES) and plates were incubated for 15 minutes with 1 mL fresh uptake buffer. 300 μL of solution containing 10 μM GlySar (containing ~1% 3[H] GlySar) and 0.05–5 mM compounds were added to corresponding wells and incubated at 37 °C. After 30 min, the drug solution was aspirated and cells were washed with 3 mL ice cold PBS. 0.5 mL of MeOH/Water (1:1) solution was then used to lyse the cells. The cell suspension was transferred to scintillation vials having 10 mL scintillation cocktail (ScintiVerse* LC Cocktail, Fisher Chemicals, Pittsburgh, PA) and the 3[H] label was determined using scintillation counting (Beckman LS-9000, Beckman Instruments, Fullerton, CA). Nonlinear data fitting using (Graph Pad Prism v4.0) gave IC50 values.
Uptake Studies
HeLa cells were transfected with recombinant adenovirus containing PEPT1 to overexpress PEPT1 transporter20 and direct uptake was evaluated as described earlier.11a 1.20× 105 cells/well of PEPT1/HeLa cells were seeded and grown in 6-well plates. On the day of the experiment, cells were washed with pH 6.0 uptake buffer and 0.5 ml of a 1mM solution of valacyclovir (VACV), GOCarb or the corresponding prodrugs of GOCarb were placed in each well. Plates were incubated at 37 °C for 45 minutes. At time 0, 10, 20 and 30 minutes, 20 μL aliquot of drug solution was taken and mixed with 80 μL of 0.1% TFA to determine the stability of the test drug in the cell system. Cells were then washed with ice cold PBS followed by their lysis using 0.5 ml MeOH:H2O (1:1). After one hour, cell suspension was transferred to eppendorf tubes and 20 μL of suspension was used to determine protein content (Bio-Rad DC Protein Assay kit, Bio-Rad Laboratories Inc., Hercules, CA). Protein was then precipitated from rest of the cell suspension using 5% TFA. The mixtures were then centrifuged for 10 minutes at 4 °C @ 10000 rpm and the supernatants were filtered and subjected to LC-MS analysis.
Single-Pass Intestinal Perfusion Studies (SPIP) in Rats
All SPIP animal experiments were conducted using protocols approved by the University Committee of Use and Care of Animals (UCUCA), University of Michigan, and the animals were housed and handled according to the University of Michigan Unit for Laboratory Animal Medicine guidelines. Male albino Wistar rats (Charles River, IN) weighing 250–280 g were used for all perfusion studies. Prior to each experiment, the rats were fasted overnight (12–18 hr) with free access to water. Animals were randomly assigned to the different experimental groups.
The procedure for the in situ single-pass intestinal perfusion in rats followed previously published reports.21 Briefly, rats were anesthetized with an intra-muscular injection of 1 ml/kg of ketamine-xylazine solution (9%:1%, respectively) and placed on a heated surface maintained at 37°C (Harvard Apparatus Inc., Holliston, MA). The abdomen was opened by a midline incision of 3–4 cm. A proximal jejunal segment of approximately 10 cm was carefully exposed and cannulated on two ends with flexible PVC tubing (2.29 mm i.d., inlet tube 40 cm, outlet tube 20 cm, Fisher Scientific Inc., Pittsburgh, PA). Care was taken to avoid disturbance of the circulatory system, and the exposed segment was kept moist with 37°C normal saline solution. Blank perfusion solution (10 mM MES buffer, pH 5.5, 135 mM NaCl, 5 mM KCl, and 0.1 mg/ml phenol red with an osmolarity of 290 mosm/l) was incubated in a 37°C water bath and pumped through the intestinal segment (Watson Marlow Pumps 323S, Watson-Marlow Bredel Inc, Wilmington, MA) at a flow rate of 0.5 ml/min in order to clean out any residual debris.
At the start of the study, perfusion solution containing the tested drug was perfused through the intestinal segment at a flow rate of 0.2 ml/min. Phenol red was added to the perfusion buffer as a non-absorbable marker for measuring water flux. Metoprolol was co-perfused with the test drugs as well, as a compound with known permeability that serves as a marker for the integrity of the experiment, and as a reference standard for permeability in close proximity to the low/high permeability class boundary.22 The concentrations of the drugs used in the perfusion studies were determined by dividing the highest prescribed oral dose by 250 ml, the accepted gastric volume, in order to represent the maximal drug concentration present in the intestinal segment, and were within their intrinsic solubility reported at pH 5.5. The perfusion buffer with drug was first perfused for 1 hr, in order to assure steady state conditions (as also assessed by the inlet over outlet concentration ratio of phenol red which approaches 1 at steady state). Once at steady state, samples were taken in 10 min intervals for 1 hour (10, 20, 30, 40, 50, and 60 min). All samples including perfusion samples at different time points, original drug solution, and inlet solution taken at the exit of the syringe were immediately assayed by HPLC. Following the termination of the experiment, the length of the perfused intestinal segment was accurately measured.
Oral Bioavailability in Mice
In these investigations, the oral bioavailability and pharmacokinetic characteristics of GOC-L-Val (11a) were tested in mice under fed and fasted conditions. All PK animal experiments were conducted using protocols approved by the TSRL Committee of Use and Care of Animals (NIH Assurance number A4194-01) and the animals were housed and handled according to TSRL Animal Care guidelines. Male Swiss Webster (CFW) mice (Charles River, IN) weighing approximately 25 grams were used for the PK studies. Prior to each experiment, the mice were assigned to different experimental groups (fed or fasted; GOCarb or GOC-L-Val). Food was withdrawn for fasted animals the night before dosing (12–18 hr) with free access to water. Dosing solutions containing either GOCarb or GOC-L-Val were administered by oral gavage (10 mg/kg) or intravenously (1 mg/kg) to fed and fasted mice and blood samples were taken over a 24-hour period. At each time point, five mice were sacrificed (n=5) and blood was collected by heart stick. Plasma was prepared following standard methods and samples were analyzed by LC/MS/MS for the GOCarb and its prodrugs. In all cases, only the active metabolite (GOCarb) was detected in plasma after dosing with the prodrug.
Analytical Methods
HPLC Analysis
The samples from enzymatic and chemical stability studies of GOCarb and its prodrugs were analyzed by Agilent 1100 Series HPLC system (Agilent Technologies, Inc. Santa Clara CA) equipped with two Agilent 1100 pumps, a Agilent autosampler maintained at 4 °C and a Agilent PDA detector. The system was operated by Agilent Chemstation software. A Waters Xterra® C18 reversed-phase column (5 μm, 4.6 × 250 mm) was used for sample analysis. The analytes were eluted using a gradient/isocratic method at a flow rate of 1mL/min. The mobile phases (water and acetonitrile) contained 0.1% TFA as modifier.
LC-MS Analysis
Samples from direct uptake and Caco-2 monolayer permeability studies were analyzed using a LC-MS 2010A system (Shimadzu Scientific Instruments, Columbia MD) equipped with LCMS solution Ver. 3 software. The LC-MS unit consisted of two pumps (LC-20AD), autosampler (SIL 20A HT) and QoQ Optical System. Positive electrospray ionization (ESI+) was used to produce the ions. The CDL temperature used was 250 °C and the detector voltage was maintained at 1.5kV. Samples were analyzed by Restek Allure Aqueous C18 column (5 μm, 2.1 × 50 mm, Waters Co., Milford, MA) equipped with a guard column. The compounds were eluted using water/acetonitrile gradient system (flow rate of 0.2mL/min) containing 0.1% formic acid as modifier.
LC/MS/MS Plasma Analysis
Plasma samples from the bioavailability studies were analyzed using a LC-MS-MS Quattro II system (MicroMass, Beverly, MA) equipped with MassLynx version 4 software. The unit consisted of an HP1100 (Hewlett Packard) HPLC system fitted with a Targo C18, 5 micron, 150 mm × 2.1 mm column (Higgins) linked to a Quattro II triple quad mass spectrometer. The mobile phase consisted of 15% acetonitrile and 85% formic acid (0.1%) and compounds were eluted at a flow rate of 0.2 mL/min. In the MS/MS detector, the cone voltage was maintained at 30V, collision energy at 25 eV and argon collision gas pressure was 4.0 × 10−3 mbar. Data acquisition was by multiple reaction monitoring in electrospray positive mode. Daughter ion transitions that were monitored were 327.2→179.9 for GOCarb and 470.3→327.1 for GOC-L-Val.
Data Analysis
Stability Studies
Apparent first order kinetics and rate constants were determined by using initial rates of hydrolysis. The apparent first-order degradation rate constants of prodrugs at 37°C were determined by plotting the logarithm of prodrug remaining as a function of time. The relation between the rate constant, k, and slopes of the plots are explained by the equation:
The degradation half-lives were then calculated by the equation:
Caco-2 Cell Monolayers Permeability
The apparent permeability coefficient (Papp unit: cm/sec) was calculated using the following equation:
Where dQ/dt is the steady-state flux (μmol/sec), A is the surface area (cm2) of the exposed monolayer and C0 is the initial concentration (μM) in the donor chamber. The concentrations of GOCarb and its prodrugs were analyzed by LC-MS. The stability of the prodrugs in apical layer was analyzed by taking samples in the beginning, during and at the end of the experiments and the concentration was determined by LC-MS.
Rat Perfusion
Since under steady-state perfusion conditions, some of the prodrug (Cin) will hydrolyze with time, samples were taken at every 10 minutes (up to 60 minutes) from perfusion buffer to determine actual Cin values. Similarly, after perfusion some portion of the prodrugs will be hydrolyzed or metabolized. To account for this hydrolysis and metabolism, the following equation was used:
The corrected concentration (Cout) for prodrug was then determined using the following equation:
The net water flux in the single-pass intestinal perfusion studies, resulting from water absorption and efflux in the intestinal perfused segment, was determined by measurement of phenol red, a non-absorbed, non-metabolized marker. The phenol red (0.1 mg/ml) was included in the perfusion buffer and co-perfused with the tested compound. The measured Cout/Cin ratio was corrected for water transport (to obtain the corrected ratio C'out/C'in) according to the following equation:
Where Cin phenol red is equal to the concentration of phenol red in the inlet sample, and Cout phenol red is equal to the concentration of phenol red in the outlet sample. The effective permeability (Peff) through the rat gut wall in the single-pass intestinal perfusion studies was determined assuming the “plug flow” model expressed in the following equation.23
Where Q is the perfusion buffer flow rate (0.2 ml/min), C'out/C'in is the ratio of the outlet concentration and the inlet or starting concentration of the tested compound that has been adjusted for water transport, R is the radius of the intestinal segment (set to 0.18 cm), and L is the length (typically 10–15 cm) of the intestinal segment.
PK Analysis
The pharmacokinetic parameter estimates for Cmax (ng/ml), Tmax (hrs), AUC (ng/ml*hrs) and F (%) [relative to an IV dose] were calculated using the Kinetica suite of programs (Kinetica, Ver 4.3).
Results
Prodrug Synthesis
Prodrugs of GOCarb were successfully synthesized in overall yield of approximately 20% (Scheme 1). The racemic mixture, due to the introduced chiral center at the acyloxy ethyl linkage, was used for all the studies. The cesium salts of corresponding amino acids were hygroscopic. They were lyophilized for at least 48 hours and immediately used for further reactions. Final compounds were also lyophilized to give white powders as amorphous TFA salts. Compounds 8 and 9 were boc-deprotected using 40% TFA in dichloromethane to yield control compounds GOCarb ethyl ester and GOCarb, respectively, with an overall 90% yield. All the compounds were analyzed for their purity by HPLC. All compounds had a purity of greater than 90%. The identity of all the intermediates and final compounds was confirmed by ESI-MS and [1H] NMR.
Stability Studies
1. Chemical Stability
The percentage of prodrug remaining was plotted against corresponding time points and linear regression gave a straight line from which half-lives (t1/2) were calculated and are reported in Table 1. An increase in stability for all prodrugs was noted with decreasing pH. GOCarb ethyl ester and GOCarb showed limited hydrolysis at all pHs up to 20 hours. For the prodrugs, t1/2s at pHs 1.2 and 4.0 were found to be > 8 hours. At pH 6.0, the L-isoleucyl prodrug was found to be 3–4 times more stable than L-valyl and L-leucyl prodrugs, similar to our previous findings using peptide prodrugs.24 At pH 7.4 the prodrugs were hydrolyzed more rapidly and with roughly similar t½ times.
Table 1.
Comparison of clogP values, estimated half-lives in pH 6.0 and 7.4 buffers and in Caco-2 cell homogenatesa
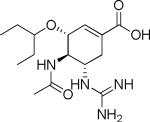
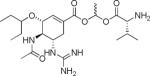
| Structure | Name | MW | cLogP | T1/2 −pH 6.0 (Min) | T1/2 pH 7.4 (Min) | T1/2 (Caco-2 cell homogenate) (Min) |
|---|---|---|---|---|---|---|

|
Valacyclovir | 324 | −1.4 | NDb 69.7 hc |
NDb >8 hc |
NDb |
|
GOCarb | 326 | −1.69 | >20h | >20h | NDb |

|
GOC ethyl ester | 354 | 1.67 | >20h | >20h | NDb |
|
GOC-L-Val | 470 | 1.47 | 212±27 | 80.5±4.9 | 14.3±7.7 |
|
GOC-L-Ile | 483 | 1.99 | 697±11 | 95±9 | 26.4±3.1 |
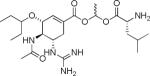
|
GOC-L-Leu | 483 | 1.99 | 174.5±23.8 | 76.4±13.7 | 23.2±6.8 |
2. Metabolic Stability
2a. Hydrolysis in Cell Homogenates
Prodrugs were incubated with Caco-2 cell homogenates and samples were analyzed by HPLC. The half-life of the prodrugs was estimated from linear regression of prodrug concentration remaining vs. time and are reported in Table 1. The half-life in buffer at pH 7.4 was used as a control. The L-isoleucyl, 11b, and L-leucyl, 11c, prodrugs showed equivalent stability and both were about 1.5 times more stable than the L-valyl, 11a, prodrug. However, all prodrugs were much more rapidly converted to parent drug than in buffer alone, with half-lives < 30 minutes. The rapid degradation in cell homogenate suggests that the parent drug may be rapidly generated in vivo by action of mucosal enzymes, such as valacyclovirase.
2b. Hydrolysis with Valacyclovirase
GOC-L-Val prodrug, 11a, was found to be a better substrate for valacyclovirase as compared to L-isoleucyl and L-leucyl prodrugs, 11b and 11c, respectively. This prodrug, when incubated with valacyclovirase, was rapidly converted to parent drug suggesting the potential activation mechanism for the prodrug. The Km was found to be 2.37 mM and Vmax was found to be 19.4 nmol/min/μg of valacyclovirase.
Buffer hydrolysis studies indicated that the prodrugs are stable enough to be absorbed intact in the duodenum and upper jejunum, where the pH is in the 5–6 range, but have the potential to release active drug immediately after absorption across the mucosal brush border membrane. Kinetics with valacyclovirase suggests that these can be potential substrates for this mucosal enzyme and valacyclovirase can catalyze the hydrolytic activation of prodrugs to give parent drug. However, other activating cellular enzymes cannot be ruled out.
Caco-2 Monolayer Permeability
Permeability was calculated from apical to basolateral (A to B) side of Caco-2 monolayer. For the prodrugs, some amount of prodrug was hydrolyzed on the apical side of the membrane by the end of the experiment (thus resulting in slightly lower apparent prodrug permeability), while only GOCarb was present on the basolateral side, suggesting all of the prodrug was hydrolyzed to active compound during its transport through the mucosal cell. The parent drug GOCarb showed negligible permeability, while its amino acid prodrugs showed 2–4 fold increase in permeability. Compared to literature values, the prodrugs showed moderate permeability; which compared favorably with valacyclovir, a drug with up to 80% bioavailability in humans. These results are summarized in Figure 2.
Figure 2.
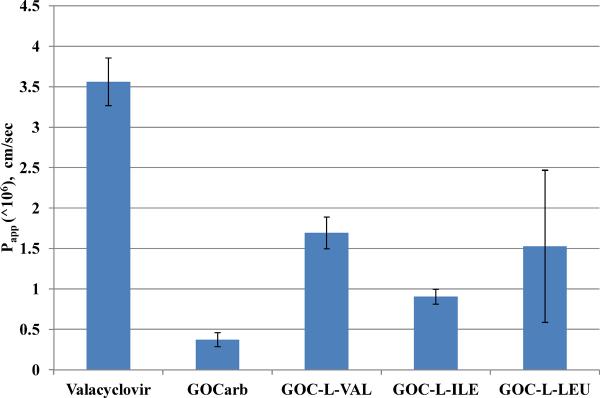
Apical to basolateral (A to B) apparent permeability (Papp in cm/sec) of valacyclovir, GOCarb and its amino acid prodrugs (0.25 mM) across Caco-2 monolayers as measured by LC/MS. Data presented as mean ± SEM, n = 3.
[3H]Gly-Sar Uptake Inhibition
The increased permeability of the prodrugs in the Caco-2 system was a result of either increased passive permeability or transporter-mediated permeability. If the permeability was carrier-mediated, we postulated that it is likely that the transporter is PEPT1 due to the amino acid promoiety on the prodrug. To examine this possibility, the affinities of GOCarb and its amino acid ester prodrugs (11a–c) were evaluated using the well-known, non metabolized PEPT1 substrate Gly-Sar in uptake inhibition assays in Caco-2 cells. Valacyclovir, a known substrate of PEPT1, was used as positive control. Parent compound GOCarb showed little competition in the presence of [3H]Gly-Sar, reflecting its poor affinity for the transporter in Caco-2 cells. However, all the prodrugs showed significant competition with [3H]Gly-Sar, indicating that the prodrugs have an affinity for the transporter. In fact, the GOC-L-Val and GOC-L-Ile prodrugs showed better inhibition of Gly-Sar uptake than valacyclovir with IC50 (mM) values of 0.19±0.01 and 0.45±0.18, respectively. Compared to the parent drug, these prodrugs showed 15–30 fold more affinity for the PEPT1 transporter. These results are summarized in Figure 3. Significantly, the much higher affinities of the valyl and isoleucyl prodrugs for PEPT1 are consistent with previous observations with amino acid ester prodrugs of floxuridine and gemcitabine.25
Figure 3.

[3H]Gly-Sar Caco-2 competitive inhibition by GOCarb and its amino acid prodrugs. Uptake inhibition in Caco-2 Cells denoted by IC50 values in mM. Data presented as Mean ± SEM, n = 3.
Uptake Studies
The potent uptake inhibition of Gly-Sar uptake in the Caco-2 cells strongly suggests that the prodrugs are substrates for PEPT1. To further investigate these findings, the direct uptake of the compounds was determined in HeLa cells that overexpressed PEPT1. In these studies, HeLa cells were transfected with a recombinant adenovirus that carried the PEPT1 gene, and then were incubated with the parent or prodrugs. The extent of influx of the compounds into the cells was measured using LC-MS analysis and was compared against valacylovir as a positive control and against non- transfected HeLa cells as a negative control (Figure 4). As expected, GOCarb showed virtually no uptake into transfected or non- transfected cells. However, the GOC-L-Val showed >6 fold increase in uptake in the PEPT1 overexpressing cells versus normal cells which was comparable to the positive control, valacyclovir. The GOC-L-Ile prodrug showed a less dramatic increase in uptake in the PEPT1 expressing cells with > 2 increase over control cells and the GOC-L-Leu showed no increase over control. As shown in Figure 3, GOC-L-Leu showed significant [3H] GlySar competitive inhibition but does not appear to show significant uptake by transfected HeLa cells (Figure 4). This suggests that GOC-L-Leu has good affinity for PEPT1 but not efficiently transported by PEPT1. It should be noted that even in the normal HeLa cells, all of the prodrugs showed a significant increase in uptake compared to the parent GOCarb, likely due to their increase lipophicility brought about by addition of the promoiety to the carboxyl group of the GOCarb. Further, primarily parent drug was observed to be present in the cells, indicating rapid cellular hydrolysis (data not shown). As summarized in Figure 5, the GOC-L-Val shows a significant advantage for PEPT1-mediated uptake, followed by GOC-L-Ile (moderate advantage) and GOC-L-Leu (little to no advantage).
Figure 4.

Direct uptake (mmol/mg of protein) for GOCarb and its amino acid prodrugs in wild type HeLa cells (control) and in PEPT1 over-expressing HeLa Cells. Data presented as Mean ± SEM, n = 3.
Figure 5.

Ratio of GlySar uptake inhibition and PEPT1 uptake for amino acid GOCarb prodrugs to their parent showing increased affinity and increased uptake for the prodrugs.
Single-pass Jejunal Perfusions in Rats
The cell culture studies show the potential for increased absorption the prodrugs, primarily mediated by PEPT1. To investigate absorption in a more rigorous model, we tested the permeability of GOC-L-Val and GOC-L-Ile, the two prodrugs showing the best PEPT1 affinity and uptake, against the parent compound in the in situ perfusion system.21b For comparative purposes, the effective permeabilities of acyclovir and its amino acid prodrug valacylovir were also examined. The effective permeability was measured as a function of disappearance of compound(s) after corrections for water flux, hydrolysis and possible metabolism. As shown in Figure 6, GOCarb has very low permeability in the jejunum, consistent with the known low absorption in rats shown by Li et al.9 However, GOC-L-Val showed a high permeability (Peff for GOC-L-Val was found to be 3.89e−5 ± 9.06e−6), higher than that of metoprolol, the high permeability internal standard routinely used in these types of experiments. GOC-L-Ile prodrug also showed significant permeability in rat perfusion experiments (Peff for GOC-L-Ile = 1.29e−5 ± 5.11e−6), though not to the extent of the L-Val prodrug, which showed an approximate 13 fold increase in permeability over its parent compound. These results are comparable to the results for valacyclovir, a well-known substrate for the PEPT1 transporter,11a which showed a similar increase in permeability from parent to prodrug.
Figure 6.

The permeability coefficient (Peff, cm/sec) obtained in in-situ rat perfusion study for metoprolol (reference standard), GOCarb and its amino acid prodrugs at 0.1mM. Acyclovir and valacyclovir Peff values (0.01mM) were reported previously.11a Permeability was observed as disappearance of prodrug in perfusate. Data presented as Mean ± SEM, n = 4.
Oral Bioavailability in Mice
To extend the enhanced permeability findings, the oral bioavailability of GOC-L-Val compared to GOCarb was tested in mice following oral gavage. The pharmacokinetic parameter estimates from the study are summarized in Table 2. GOCarb shows a bioavailability of approximately 5% in the fasted and fed state, as was previously reported by Li et al.9 In contrast, as shown in Figure 7, the bioavailability of GOC-L-Val after oral administration compounds was significantly greater than the GOCarb, with a bioavailability of 23% and 48% under fed and fasted conditions, respectively, representing a 5 to 10 fold increase in absorption over the parent compound. It should be noted that only GOCarb was detected in plasma after oral dosing of the GOC-L-Val prodrug, indicating that, once absorbed, the prodrug was rapidly cleaved to its parent compound. This resulted in plasma levels of GOCarb that were approximately 10 times higher than levels found after oral administration of the GOCarb parent compound (Figure 8). Significantly, the concentration of GOCarb after oral administration of the GOC-L-Val for the first 4 hours after dosing is 40 to 100 times greater than the IC50 of GOCarb (0.9 nM) for the H1N1 NWS/33 strain of influenza.9
Table 2.
Pharmacokinetic parameter estimates of GOCarb (GOC) and GOC-L-Valine following oral dosing in fasted and fed mice.
| Drug | Dose (mg/kg) | Route | Tmax hrs | Cmax ng/ml | AUC24hr (ng/mL· hr) |
|---|---|---|---|---|---|
| GOC | 1 | IV | - | - | 487 ± 206 |
| GOC | 10 | po-fasted | 1.3 ± 0.6 | 50.9 ± 35.8 | 278 ± 52 |
| GOC | 10 | po-fed | 3.0 ± 1.2 | 65.4 ± 26.8 | 291 ± 87 |
| GOC-L-Val | 1 | IV | - | - | 641 ± 52 |
| GOC-L-Val | 10 | po-fasted | 2.2 ± 1.1 | 541.0 ± 226.9 | 3096 ± 558 |
| GOC-L-Val | 10 | po-fed | 2.4 ± 1.3 | 405.1 ± 297.3 | 1442 ± 338 |
Figure 7.

Oral bioavailability (%) comparison of GOCarb (GOC) and GOC-L-Val after oral administration of 10 mg /kg of GOCarb or GOC-L-Val.
Figure 8.

A comparison of the GOCarb (GOC) plasma levels after oral administration of 10 mg eq GOC/kg of GOC-L-Val (□), GOC (Δ) or IV administration of 1 mg (eq. GOC)/kg GOC (◆) to fasted animals (n=5). The dashed line at 5 ng/ml is the approximate EC50 for GOC versus the A/NWS/33 virus.
Discussion
Obtaining good oral absorption for polar therapeutic agents presents a challenge to the medicinal chemist, who must deal with conflicting molecular and physical requirements. In order to achieve good mucosal cell penetration and bioavailability a drug must generally be relatively lipophilic. However, polar molecules such as nutrients, e.g. zwitterionic amino acids, and a few polar drugs do exhibit good epithelial cell apical membrane permeability by utilizing nutrient carriers. Examples of drugs utilizing epithelial cell apical membrane carrier-mediated transport are β-lactam antibiotics, ACE inhibitors and anti-viral compounds such as valacyclovir and valganciclovir.18 Thus, for certain polar drugs, the solution to the absorption challenge lies in a prodrug strategy that modifies the polar drug sufficiently to be recognized by a given nutrient carrier.
This prodrug strategy comes with its own risks and challenges; the prodrug must have the structural characteristics recognized by the dipeptide transporter with a promoiety that is stable enough to withstand the gastrointestinal environment and will break down rapidly and completely once absorbed. We have succeeded in making a prodrug of the potent neuraminidase inhibitor GOCarb with these qualities.
The properties of GOCarb are typical of a polar molecule. It has a negative log P (−1.69) and poor intestinal permeability (<5 % in rats). Structurally, it is a zwitterion with a guanidinium group and a free carboxylic acid. Significantly, a simple ester prodrug of GOCarb, as was used with the only oral neuraminidase product on the market, oseltamivir, did not improve its poor bioavailability. With this in mind, we modified the compound using a promoiety that contained a series of amino acids with their carboxyl group linked to the GOCarb carboxyl group through an acyloxy ethyl linker. These prodrugs were shown to have sufficient stability at physiological pH and were shown to rapidly breakdown in intestinal cell homogenates. This hydrolysis was attributed, at least in part, to valacyclovirase, an enzyme known to hydrolyze a variety of amino acid prodrugs, particularly its name sake, valacyclovir. Exact mechanism of prodrug hydrolysis is not detailed in this study. However, similar to (acyloxy) alkyl esters prodrugs,26 cleavage of prodrug moiety will likely be initiated at the carbonyl carbon of the valine. The presence of carboxylic acid metabolite (GOCarb), in the receiver compartment in the transport experiments suggests that the overall cleavage process is very efficient in the cells and likely efficient in vivo. Secondly, examination of the transport mechanism of these prodrugs indicated that the L-valyl acycloxy ethyl prodrug, GOC-L-Val was a good substrate for the dipeptide transporter, giving a comparable increase of uptake of the prodrug versus the parent to that of valacyclovir. Further examination of the potential for oral absorption of this prodrug, using in situ single pass perfusion system, showed that the compound had a comparable permeability to metoprolol, which is commonly used as an internal, high permeability standard that is known to be exclusively absorbed by passive route. Finally, we showed that GOC-L-Val was well absorbed in mice, showing 48% bioavailability in fasted mice and 23% bioavailability in fed mice.
In summary, the results presented in these investigations demonstrate that we have successfully used a prodrug strategy that takes advantage of the peptide transporter-cellular activation mechanism to significantly improve the oral absorption and systemic availability of a poorly-absorbed, polar antiviral agent.
Acknowledgement
This project was supported by NIH Grant RO1 GM 037188 and R43AI081396
References
- 1.(a) Couch RB. Advances in influenza virus vaccine research. Ann N Y Acad Sci. 1993;685:803–12. doi: 10.1111/j.1749-6632.1993.tb35946.x. [DOI] [PubMed] [Google Scholar]; (b) Belshe RB, Burk B, Newman F, Cerruti RL, Sim IS. Resistance of influenza A virus to amantadine and rimantadine: results of one decade of surveillance. J Infect Dis. 1989;159(3):430–5. doi: 10.1093/infdis/159.3.430. [DOI] [PubMed] [Google Scholar]; (c) Hayden FG, Hay AJ. Emergence and transmission of influenza A viruses resistant to amantadine and rimantadine. Curr Top Microbiol Immunol. 1992;176:119–30. doi: 10.1007/978-3-642-77011-1_8. [DOI] [PubMed] [Google Scholar]
- 2.Palese P, Tobita K, Ueda M, Compans RW. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61(2):397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 3.(a) Burnet FM. Mucins and mucoids in relation to influenza virus action. IV. Inhibition by purified mucoid of infection and hemagglutination with the virus strain WSE. Aust. J. exp. Biol. med. Sci. 1948;26:381–387. doi: 10.1038/icb.1948.39. [DOI] [PubMed] [Google Scholar]; (b) Klenk H-D, R. R. The molecular biology of influenza virus pathogenicity. Adv. Virus Res. 1988;34:247–280. doi: 10.1016/S0065-3527(08)60520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors--a review. Antiviral Res. 2000;47(1):1–17. doi: 10.1016/s0166-3542(00)00103-0. [DOI] [PubMed] [Google Scholar]; (b) McKimm-Breschkin JL, Selleck PW, Usman TB, Johnson MA. Reduced sensitivity of influenza A (H5N1) to oseltamivir. Emerg Infect Dis. 2007;13(9):1354–7. doi: 10.3201/eid1309.07-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Marie-Anne Rameix-Welti S. v. d. W. N. N. Sensitivity of H5N1 influenza viruses to oseltamivir: an update. Future Virology. 2008;3(2):157–165. [Google Scholar]; (d) de Jong MD, Thanh TT, Khanh TH, Hien VM, Smith GJD, Chau NV, Cam BV, Qui PT, Ha DQ, Guan Y, Peiris JSM, Hien TT, Farrar J. Oseltamivir Resistance during Treatment of Influenza A (H5N1) Infection. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]; (e) Moscona A. Oseltamivir Resistance -- Disabling Our Influenza Defenses. 2005;353:2633–2636. doi: 10.1056/NEJMp058291. [DOI] [PubMed] [Google Scholar]; (f) Gubareva LV, Webster RG, Hayden FG. Comparison of the activities of zanamivir, oseltamivir, and RWJ-270201 against clinical isolates of influenza virus and neuraminidase inhibitor-resistant variants. Antimicrob Agents Chemother. 2001;45(12):3403–8. doi: 10.1128/AAC.45.12.3403-3408.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins PJ, Haire LF, Lin YP, Liu J, Russell RJ, Walker PA, Skehel JJ, Martin SR, Hay AJ, Gamblin SJ. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature. 2008;453(7199):1258–61. doi: 10.1038/nature06956. [DOI] [PubMed] [Google Scholar]
- 6.Woods JM, Bethell RC, Coates JA, Healy N, Hiscox SA, Pearson BA, Ryan DM, Ticehurst J, Tilling J, Walcott SM. 4-Guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid is a highly effective inhibitor both of the sialidase (neuraminidase) and of growth of a wide range of influenza A and B viruses in vitro. 1993;37:1473–1479. doi: 10.1128/aac.37.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Itzstein M, Wu W-Y, Kok GB, Pegg MS, Dyason JC, Jin B, Phan TV, Smythe ML, White HF, Oliver SW, Colman PM, Varghese JN, Ryan DM, Woods JM, Bethell RC, Hotham VJ, Cameron JM, Penn CR. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363(6428):418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ryan DM, Ticehurst J, Dempsey MH, Penn CR. Inhibition of influenza virus replication in mice by GG167 (4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid) is consistent with extracellular activity of viral neuraminidase (sialidase) 1994;38:2270–2275. doi: 10.1128/aac.38.10.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hayden FG, Treanor JJ, Betts RF, Lobo M, Esinhart JD, Hussey EK. Safety and efficacy of the neuraminidase inhibitor GG167 in experimental human influenza. Jama. 1996;275(4):295–9. [PubMed] [Google Scholar]
- 9.Li W, Escarpe PA, Eisenberg EJ, Cundy KC, Sweet C, Jakeman KJ, Merson J, Lew W, Williams M, Zhang L, Kim CU, Bischofberger N, Chen MS, Mendel DB. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob Agents Chemother. 1998;42(3):647–53. doi: 10.1128/aac.42.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97(3):1109–34. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- 11.(a) Han H, de Vrueh RL, Rhie JK, Covitz KM, Smith PL, Lee CP, Oh DM, Sadee W, Amidon GL. 5'-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm Res. 1998;15(8):1154–9. doi: 10.1023/a:1011919319810. [DOI] [PubMed] [Google Scholar]; (b) Sugawara M, Huang W, Fei YJ, Leibach FH, Ganapathy V, Ganapathy ME. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci. 2000;89(6):781–9. doi: 10.1002/(SICI)1520-6017(200006)89:6<781::AID-JPS10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Varghese Gupta S, Gupta D, Sun J, Dahan A, Tsume Y, Hilfinger J, Lee K-D, Amidon GL. Enhancing the Intestinal Membrane Permeability of Zanamivir: A Carrier Mediated Prodrug Approach. Molecular Pharmaceutics. 2011;8(6):2358–2367. doi: 10.1021/mp200291x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shitara E, Nishimura Y, Nerome K, Hiramoto Y, Takeuchi T. Synthesis of 6-acetamido-5-amino- and -5-guanidino-3, 4-dehydro-N-(2-ethylbutyryl)- 3-piperidinecarboxylic acids related to zanamivir and oseltamivir, inhibitors of influenza virus neuraminidases. Org Lett. 2000;2(24):3837–40. doi: 10.1021/ol000261d. [DOI] [PubMed] [Google Scholar]
- 14.Gomes P, Santos MI, Trigo MJ. Castanheiro, R.; Moreira, R., Improved Synthesis of Amino Acid and Dipeptide Chloromethyl Esters Using Bromochloromethane. Synthetic Communications. 2003;33(10):1683–1693. [Google Scholar]
- 15.Masuda T, Yoshida S, Arai M, Kaneko S, Yamashita M, Honda T. Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives. Chem Pharm Bull (Tokyo) 2003;51(12):1386–98. doi: 10.1248/cpb.51.1386. [DOI] [PubMed] [Google Scholar]
- 16.Nudelman A, Gnizi E, Katz Y, Azulai R, Cohen-Ohana M, Zhuk R, Sampson SR, Langzam L, Fibach E, Prus E, Pugach V, Rephaeli A. Prodrugs of butyric acid. Novel derivatives possessing increased aqueous solubility and potential for treating cancer and blood diseases. Eur J Med Chem. 2001;36(1):63–74. doi: 10.1016/s0223-5234(00)01199-5. [DOI] [PubMed] [Google Scholar]
- 17.Gupta D, Gupta SV, Lee K-D, Amidon GL. Chemical and Enzymatic Stability of Amino Acid Prodrugs Containing Methoxy, Ethoxy and Propylene Glycol Linkers. Molecular Pharmaceutics. 2009;6(5):1604–1611. doi: 10.1021/mp900084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim I, Chu X.-y., Kim S, Provoda CJ, Lee K-D, Amidon GL. Identification of a Human Valacyclovirase. Biphenyl Hydrolase-like Protein as Valacyclovir Hydrolase. Journal of Biological Chemistry. 2003;278(28):25348–25356. doi: 10.1074/jbc.M302055200. [DOI] [PubMed] [Google Scholar]
- 19.(a) Hidalgo IJ, Li J. Carrier-mediated transport and efflux mechanisms in Caco-2 cells. Advanced Drug Delivery Reviews. 1996;22(1–2):53–66. [Google Scholar]; (b) Liang R, Fei YJ, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, Hediger MA, Ganapathy V, Leibach FH. Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J Biol Chem. 1995;270(12):6456–63. doi: 10.1074/jbc.270.12.6456. [DOI] [PubMed] [Google Scholar]; (c) Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175(3):880–5. doi: 10.1016/0006-291x(91)91647-u. [DOI] [PubMed] [Google Scholar]
- 20.(a) Croyle MA, Roessler BJ, Hsu C-P, Sun R, Amidon GL. Beta Cyclodextrins Enhance Adenoviral-Mediated Gene Delivery to the Intestine. Pharmaceutical Research. 1998;15(9):1348–1355. doi: 10.1023/a:1011985101580. [DOI] [PubMed] [Google Scholar]; (b) Hsu CP, Hilfinger JM, Walter E, Merkle HP, Roessler BJ, Amidon GL. Overexpression of human intestinal oligopeptide transporter in mammalian cells via adenoviral transduction. Pharm Res. 1998;15(9):1376–81. doi: 10.1023/a:1011993303397. [DOI] [PubMed] [Google Scholar]
- 21.(a) Amidon GL, Sinko PJ, Fleisher D. Estimating human oral fraction dose absorbed: a correlation using rat intestinal membrane permeability for passive and carrier-mediated compounds. Pharm Res. 1988;5(10):651–4. doi: 10.1023/a:1015927004752. [DOI] [PubMed] [Google Scholar]; (b) Kim JS, Mitchell S, Kijek P, Tsume Y, Hilfinger J, Amidon GL. The suitability of an in situ perfusion model for permeability determinations: utility for BCS class I biowaiver requests. Mol Pharm. 2006;3(6):686–94. doi: 10.1021/mp060042f. [DOI] [PubMed] [Google Scholar]
- 22.Dahan A, West BT, Amidon GL. Segmental-dependent membrane permeability along the intestine following oral drug administration: Evaluation of a triple single-pass intestinal perfusion (TSPIP) approach in the rat. Eur J Pharm Sci. 2009;36(2–3):320–9. doi: 10.1016/j.ejps.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Fagerholm U, Johansson M, Lennernas H. Comparison between permeability coefficients in rat and human jejunum. Pharm Res. 1996;13(9):1336–42. doi: 10.1023/a:1016065715308. [DOI] [PubMed] [Google Scholar]
- 24.Tsume Y, Hilfinger JM, Amidon GL. Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: increased transporter affinity and metabolic stability. Mol Pharm. 2008;5(5):717–27. doi: 10.1021/mp800008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Landowski CP, Lorenzi PL, Song X, Amidon GL. Nucleoside ester prodrug substrate specificity of liver carboxylesterase. J Pharmacol Exp Ther. 2006;316(2):572–80. doi: 10.1124/jpet.105.092726. [DOI] [PubMed] [Google Scholar]; (b) Song X, Lorenzi PL, Landowski CP, Vig BS, Hilfinger JM, Amidon GL. Amino acid ester prodrugs of the anticancer agent gemcitabine: synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol Pharm. 2005;2(2):157–67. doi: 10.1021/mp049888e. [DOI] [PubMed] [Google Scholar]
- 26.(a) Perry CM, Brogden RN. Cefuroxime axetil. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1996;52(1):125–58. doi: 10.2165/00003495-199652010-00009. [DOI] [PubMed] [Google Scholar]; (b) Frampton JE, Brogden RN, Langtry HD, Buckley MM. Cefpodoxime proxetil. A review of its antibacterial activity, pharmacokinetic properties and therapeutic potential. Drugs. 1992;44(5):889–917. doi: 10.2165/00003495-199244050-00011. [DOI] [PubMed] [Google Scholar]
- 27.Bender DM, Peterson JA, McCarthy JR, Gunaydin H, Takano Y, Houk KN. Cyclopropanecarboxylic acid esters as potential prodrugs with enhanced hydrolytic stability. Org Lett. 2008;10(3):509–11. doi: 10.1021/ol702892e. [DOI] [PubMed] [Google Scholar]