

Abstract
Tumor growth is often accompanied by the accumulation of myeloid cells in the tumors and lymphoid organs. These cells can suppress T cell immunity, thereby posing an obstacle to T cell-targeted cancer immunotherapy. In this study, we tested the possibility of activating tumor-associated myeloid cells to mediate antitumor effects. Using the peritoneal model of B16 melanoma, we show that peritoneal cells (PEC) in tumor-bearing mice (TBM) had reduced ability to secrete nitric oxide (NO) following in vitro stimulation with interferon gamma and lipopolysaccharide, as compared to PEC from control mice. This reduced function of PEC was accompanied by the influx of CD11b+ Gr-1+ myeloid cells to the peritoneal cavity. Nonadherent PEC were responsible for most of the NO production in TBM, whereas in naïve mice NO was mainly secreted by adherent CD11b+ F4/80+ macrophages. Sorted CD11b+ Gr-1− monocytic and CD11b+ Gr-1+ granulocytic PEC from TBM had a reduced ability to secrete NO following in vitro stimulation (compared to naïve PEC), but effectively suppressed proliferation of tumor cells in vitro. In vivo, treatment of mice bearing established peritoneal B16 tumors with anti-CD40 and CpG resulted in activation of tumor-associated PEC, reduction in local tumor burden and prolongation of mouse survival. Inhibition of NO did not abrogate the antitumor effects of stimulated myeloid cells. Taken together, the results indicate that in tumor-bearing hosts, tumor-associated myeloid cells can be activated to mediate antitumor effects.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1236-2) contains supplementary material, which is available to authorized users.
Keywords: Myeloid cells, Anti-CD40, CpG, Immunotherapy
Introduction
Advanced cancer can induce immunosuppression (reviewed in [1]). This immunosuppression, especially of T cells, is considered to be one of the mechanisms by which tumors evade immune-mediated destruction [2]. Several types of immune cells, including T cells, dendritic cells and macrophages (Mϕ), can be functionally suppressed by tumors. In particular, tumor-associated Mϕ (TAM) have been categorized as alternatively activated M2 Mϕ due to the influence of tumor-derived factors [3, 4]. Monocytes and Mϕ from tumor-bearing animals can suppress T cell function [5], and conversely, CD4+CD25+ T regulatory cells can exert direct suppressive effects on monocytes and Mϕ [6]. While Mϕ outside of the tumor compartment may remain unsuppressed [7], TAM are functionally inhibited, mediate immunosuppression and promote tumor growth [3, 8].
In addition to immunosuppressive TAM, immature myeloid cells accumulating in tumors and associated lymphoid organs in tumor-bearing hosts can also mediate suppression of T cell functions [9–11]. In mice, these myeloid-derived suppressor cells (MDSC) represent a heterogeneous population of myeloid cells that express both CD11b and Gr-1[11]. In addition, murine MDSC can express IL-4Rα and varying levels of F4/80, depending on the tumor model [8, 12, 13]. Immunosuppressive activities of MDSC are attributed, in part, to their production of nitric oxide (NO) or arginase in response to tumor-produced PGE2 [14], which depletes arginine necessary for T cell functions [15]. In addition to suppressing T cell responses, MDSC have been found to inhibit Mϕ functions in TBM [16].
Although TAM have been reported to promote tumor growth, and the histological detection of abundant TAM has been associated with poor prognosis for patients with certain cancers [17, 18], Mϕ in TBM can also become antitumor effector cells following proper activation. Thus, disruption of the immunosuppressive IL-10 pathway in combination with the Mϕ-activating agents CpG and LEC/CCL16 influenced TAM to become potent antitumor effectors, presumably causing M2 inhibitory Mϕ to convert to M1 effector cells [19]. However, a potential role of TAM and other tumor-associated myeloid cells as antitumor effector cells has not been well characterized. We have previously shown that a combination of two distinct immunomodulators, anti-CD40 mAb (anti-CD40) and class B oligodeoxynucleotides containing unmethylated CpG motifs (CpG), induced a strong synergistic activation of Mϕ resulting in antitumor effects in mice [20–22]. These studies, for the most part, involved subcutaneous tumors, whereas functional and phenotypic analysis was performed on peritoneal Mϕ. In this study, we hypothesized that tumor-associated myeloid cells could induce antitumor effects following activation in vitro and in vivo. Specifically, using a peritoneal B16 model, we sought to determine whether anti-CD40 and CpG treatment can activate tumor-associated myeloid cells in TBM to mediate antitumor effects. The results presented here show that myeloid cells at, or near, sites of tumor growth are indeed capable of being activated in vitro and in vivo to mediate antitumor effects.
Materials and methods
Mice and cell lines
Female C57BL/6 mice, 8–12 weeks old, were obtained from Taconic, Germantown, NY. C57BL/6-Tg(CAG-EGFP)1Osb/J transgenic mice with an “enhanced” green fluorescent protein (EGFP) cDNA were obtained from Jackson Labs (Bar Harbor, ME) and bred at the UW-Madison animal facility. Mice were housed at the UW-Madison animal facility, and animal experiments were performed under protocols approved by the Animal Care and Use Committees of UW-Madison. The murine B16 melanoma tumor cell line was grown in RPMI 1640 complete medium supplemented with 10% FCS (Sigma Chemicals, St. Louis, MO), 2 mM l-glutamine, and 100 U/ml of penicillin/streptomycin (all from Life Technologies, Inc., Grand Island, NY) at 37°C in a humidified 5% CO2 atmosphere.
Antibodies and reagents
Anti-CD40 was prepared from the FGK 45.5 hybridoma cell line as described previously [20]. Toll-like receptor 9 (TLR9) agonist CpG1826 was purchased from Coley Pharmaceuticals Group, Wellesley, MA. Bacterial LPS from Salmonella enteritidis was purchased from Sigma Chemical, St. Louis, MO. Mouse recombinant IFN-γ was purchased from eBioscience, San Diego, CA.
In vivo tumor models and therapy
C57BL/6 mice were injected subcutaneously (s.c.) or intraperitoneally (i.p.) with 1 × 105 B16 melanoma cells in 0.1 or 0.5 ml PBS, respectively (day 0). For tumor therapy, the mice with i.p. tumors were injected i.p. with 0.5-mg anti-CD40 on days 4, 11 and 18 after tumor implantation and 50-μg CpG on days 7 and 14. Antitumor effects were evaluated by extended survival of the mice and by the decrease in the number of CD45− B16 cells in PEC as detected by flow cytometry.
Activation of peritoneal cells (PEC)
PEC were obtained via a peritoneal cavity lavage with 5 ml of cold RPMI 1640 complete medium, supplemented with 1 IU/ml of heparin (American Pharmaceutical Partners, Inc., Schaumburg, IL) when collected from TBM. Collected PEC were placed into 96-well flat-bottomed cell culture plates (Corning Inc, Corning, NY) at a concentration of 2–2.5 × 106 cells/ml (or 1 × 106 cells/ml for sorted cell populations), 0.1 ml/well. The peritoneal Mϕ population was enriched by allowing PEC to adhere to plastic for 1.5–2 h, followed by removal of nonadherent cells. For in vitro activation, total PEC, nonadherent cells, or adherent Mϕ were stimulated with 10 U/ml of IFN-γ and 1 ng/ml of LPS, unless stated otherwise, for 48 h. For in vivo activation, mice were injected i.p. with 0.5 mg of anti-CD40 in 0.5 ml PBS. On day 3, PEC were harvested, enriched as described above and incubated for 48 h either in medium alone or in the presence of LPS (10 ng/ml).
Mϕ: mediated tumoristasis in vitro
Tumoristatic activity of Mϕ was determined by the inhibition of DNA synthesis in tumor cells. Briefly, adherent Mϕ were stimulated in vitro as described above and simultaneously co-cultured with B16 tumor cells (1 × 104/well) for 48 h. To estimate DNA synthesis, cells were pulsed with 3H-TdR (1 μCi/well) during the last 6 h of incubation. 3H-TdR-incorporation was determined by β-scintillation of total cells harvested from the cell culture clusters onto glass fiber filters (Packard, Meriden, CT), using the Packard Matrix 9600 Direct β-counter (Packard, Meriden, CT). Results are presented as counts per 5 min for triplicate wells ± SE.
Nitric oxide production
Peritoneal Mϕ were prepared and co-cultured with B16 cells for 48 h, as described above in the Mϕ cytostatic assay. Supernatants were collected and nitrite accumulation was determined using Griess reagent (Sigma, St. Louis, MO). Equal volumes of supernatants and Griess reagent were mixed for 10 min, and the A570 was measured by a microplate reader and compared to a standard nitrite curve ranging from 0–125 μM.
Inhibition of cytotoxic factors
Inducible NO synthase inhibitor L-NAME (Sigma St. Louis, MO) was added to the cultures at a dose of 10 mM to neutralize the effect of NO. To neutralize arginase, Nω-hydroxy-nor-l-arginine, diacetate salt (nor-NOHA, EMD Biosciences Inc., San Diego, CA) was used at a dose of 100 μM. To block TNFα, anti-TNFα mAb, clone MP6-XT3 (BD Biosciences, San Jose, CA) was used at a final concentration of 10 μg/ml. To neutralize NO in vivo, L-NAME was either injected i.p. at a dose of 50 mg/kg twice a day or given in the drinking water at a dose of 0.5 g/l for 4 days during anti-CD40/CpG treatment.
Flow cytometric analysis and sorting
PEC from treated and control C57BL/6 mice were harvested and stained with anti-F4/80 APC mAb, anti-F4/80 FITC mAb, anti-Gr-1 PE mAb, anti-CD11b APC mAb, anti-CD45 FITC mAb (all from eBioscience, San Diego, CA), anti-CD40 PE mAb, anti-CD80 PE mAb, or anti-CD86 PE mAb (all from BDPharmingen, San Diego, CA) for 40 min at 4°C. Isotype-matched irrelevant rat IgG FITC, IgG APC and IgG PE, purchased from eBioscience or BDPharmingen, were used as background controls. After washing the cells in ice-cold PBS supplemented with 0.5–2% FCS (flow buffer), the cell pellet was resuspended in 0.3-ml flow buffer and analyzed by flow cytometry using a FACSCalibur flow cytometer and FlowJo software (Ashland, OR). Data were collected for 10,000 live events per sample.
Immunohistochemistry
PEC (104–105 cells in 100 μl media with FCS) were centrifuged at 800 rpm for 3 min using a Shandon Cytospin 2. After air-drying for 5 min, slides were fixed in 100% methanol for 2 min, allowed to dry and stained horizontally with Wright-Giemsa Stain (Sigma) for 45 s. An equal volume of glass filtered water was immediately added to the Wright-Giemsa stain solution, and after staining in the dilute solution for 10 min, the slides were washed off with glass filtered water and destained horizontally with glass filtered water for 5 min. Slides were allowed to dry and mounted with glass coverslips using Cytoseal 60. Pictures of cells were taken at 40× magnification with an Optronics camera using the attached computer software (Magnafire 2.1).
In vivo depletion of Mϕ
Peritoneal Mϕ were depleted in vivo with clodronate liposomes as described [23]. Clodronate was a gift of Roche Diagnostics GmbH, Mannheim, Germany. Clodronate liposomes were prepared as described [23] and injected i.p. in TBM on day 9 post tumor cell implantation (0.3 ml) and on day 13 (0.2 ml). Depletion of Mϕ with clodronate liposomes was confirmed in naïve mice by elimination of more than 95% of PEC positively stained with both anti-CD11b APC and anti-F4/80 FITC.
Adoptive transfer of EGFP PEC
PEC were removed from EGFP mice and counted, and 4.8 × 106 cells were injected i.p. in wild-type C57BL/6 mice (day 0). To facilitate survival of adoptively transferred cells, the recipient mice were depleted of T cells by a mixture of anti-CD4 and anti-CD8 given i.p. on days −1 and 4. On day 1, half of the mice were injected i.p. with 1 × 105 B16 cells. PEC were removed on day 8, stained with anti-CD11b APC mAb and anti-Gr-1 PE mAb, and analyzed by flow cytometry.
Statistical analysis
A two-tailed Student’s t test was used to determine significance of differences between experimental and relevant control values. For survival studies, the Gehan–Breslow–Wilcoxon test was used.
Results
Functional and phenotypic changes in PEC from TBM
Our previous studies demonstrated that anti-CD40 and CpG induced antitumor effects which involved Mϕ [20]. In this study, we sought to determine how anti-CD40 and CpG treatment affects TAM and other myeloid cells in tumor-bearing mice. We hypothesized that CD40/TLR9 ligation of TAM in tumor-bearing mice can change them from being functionally suppressed to becoming antitumor effector cells.
We asked first whether B16 tumor growth in mice resulted in suppressed Mϕ function (as determined by decreased NO production in response to in vitro stimulation with IFN-γ and LPS) and altered Mϕ phenotype compared to Mϕ from naïve mice. We wanted to evaluate these Mϕ features for TAM and for Mϕ obtained from sites other than the tumor (non-TAM) in TBM. We used two tumor models to address these questions. Peritoneal cells (PEC) were obtained either from syngeneic C57BL/6 mice bearing advanced s.c. B16 tumors (a source of non-TAM) or from mice bearing advanced i.p. B16 tumors (a source of TAM). PEC from mice bearing i.p. B16 tumors, rather than the cells from s.c. tumors, were chosen as a source of TAM because of the better accessibility, viability and yield of these PEC for functional and phenotypic characterization, compared to TAM that can be recovered from s.c. B16 tumors. The results show that adherent PEC from mice with advanced s.c. B16 tumors, killed on day 18 post tumor cell injection (Mean tumor volume 514 ± 31 mm3, n = 4), secreted a similar amount of NO (Fig. 1a) and displayed a similar ability to inhibit tumor cell proliferation in vitro (data not shown) compared to adherent PEC from naïve mice. There was a small increase in the percentage of CD11b+ Gr-1+ cells in the spleens, but not in the peritoneal cavities, of s.c. TBM (Fig. 1c). In contrast to the s.c. model, adherent PEC from mice bearing i.p. B16 tumors had reduced NO production (Fig. 1b) and displayed a different surface phenotype. Specifically, expression of the F4/80 marker was downregulated on PEC from TBM, and a new population of CD11b+ Gr-1+ cells emerged comprising 55% of PEC (Fig. 1d). CD11b+ PEC from TBM expressed IL-4Rα, but did not express mannose receptor, CD206 (data not shown). These results suggest that PEC from TBM are suppressed and phenotypically altered when the mice bear peritoneal tumor, but when the tumor is s.c. at a distant site.
Fig. 1.

Effect of s.c. and i.p. B16 tumor on function and phenotype of PEC. C57BL/6 mice were injected s.c. (a, c) or i.p. (b, d) with 105 B16 cells. Eighteen (a, c) or 14 (b, d) days later, PEC from TBM and naïve mice were removed and analyzed for NO production following stimulation with LPS or IFNγ and LPS (a, b) and phenotype using flow cytometry (c, d). In mice bearing s.c. tumors, spleen cells were analyzed for expression of CD11b and Gr-1 as well (c). NO activity is expressed as nitrite levels for 4 mice per group (mean ± SEM). Phenotypes of PEC and spleen cells are shown on representative dot plots. The experiments were repeated 2–3 times with similar results
Next we looked at the kinetics of both functional and phenotypic changes in PEC obtained from mice injected i.p. 14–15 days earlier with 105 B16 cells. The results show that as tumor burden increases with time, adherent PEC from TBM gradually lose their ability to secrete NO upon stimulation with IFNγ and LPS (Fig. 2a). A similar, but less complete loss of NO secretory ability was also seen with the total PEC population from TBM (Fig. 2a). These changes in PEC were not observed on days 3 or 7, but were observed after day 11 post tumor cell injection and were more pronounced by day 14. The percentage of total PEC that co-expressed CD11b+ and Gr-1+ increased starting 1 week post tumor cell injection (Fig. 2b). The decreased ability of PEC from TBM to secrete NO (Fig. 2a) correlated with an increased number of CD11b+ Gr-1+ cells in the peritoneal cavities (Fig. 2b) of these mice (Pearson correlation coefficient −0.87, p < 0.001). In contrast to B16 tumor cell injection, daily i.p. injections of B16 supernatants for 14 days failed to induce both CD11b+ Gr-1+ PEC accumulation and NO production inhibition (data not shown). When mice were injected i.p. with 1.6 × 107 B16 tumor cells (rather than 105 cells, as in Fig. 2a) and adherent PEC were tested for NO production 4 days later, there was no suppression of NO production compared with control PEC (Supplemental Figure, A). This observation that a large number of tumor cells in the peritoneum for 4 days does not inhibit NO production suggests that the in vivo tumor-induced suppression of Mϕ function is more dependent on the duration of exposure to the peritoneal tumor than on the total number of peritoneal tumor cells.
Fig. 2.

Kinetics of functional and phenotypic changes in PEC during tumor growth. C57BL/6 mice were injected i.p. with 105 B16 cells (day 0). On days 3, 7, 11 and 14, mice were killed and PEC were removed. a PEC were placed in 96-well plate, and total (unseparated) or plastic-adherent cells were stimulated with IFN-γ (10 U/ml) and LPS (1 ng/ml). The supernatants were collected 48 h later, and NO activity was determined by nitrite levels. The results are presented as the mean ± SEM nitrite concentration from 4 mice per group. Dash signifies value below detection limit. b PEC were also analyzed by flow cytometry for CD11b, F4/80 and Gr-1 expression. The numbers indicate percentages of cells in each quadrant
Using flow cytometric analysis of PEC with anti-CD45 mAb to distinguish between CD45+ host hematopoietic-derived cells and CD45− nonhematopoietic cells (primarily tumor cells), we found tumor cells in PEC from TBM. To determine whether these contaminating tumor cells may account for the suppressed activity of tumor-associated PEC, we tested the in vitro effect of different doses of B16 cells on NO production by adherent PEC from naïve mice. We found that B16 cells had a dose-dependent immunosuppressive effect on NO production, starting at a tumor cell: Mϕ ratio of 1:1 (Supplemental Figure, B). In contrast, in some of our experiments adherent PEC from day 14 TBM had markedly reduced NO production when the percentage of B16 cells in PEC was only 10%, suggesting that more than just the presence of tumor cells in PEC is responsible for the observed inhibition of NO production.
Ability of PEC from TBM to produce NO resides in nonadherent cells
Adherence to plastic is one of the main features of Mϕ that allows for their enrichment for functional studies. In our previous studies, we have found that the majority of adherent PEC from naïve mice were CD11b+ F4/80+ cells that produced NO upon in vitro stimulation [24]. To test whether nonadherent PEC rather than, or in addition to, adherent PEC in TBM are capable of producing NO upon in vitro stimulation, PEC from TBM were separated into nonadherent cells (removed from the plastic by gentle pipetting) and adherent cells (removed from the plastic with scraping in EDTA solution after removal of nonadherent cells). Nonadherent PEC in TBM were comprised mostly of F4/80dim/− Gr-1+ (Ly6G+ Ly6C+) cells (Fig. 3a) and were the main source of NO production (Fig. 3b). In both naïve and TBM, cells producing NO were phagocytes, as shown by the reduction in NO levels in mice treated with clodronate liposomes (Fig. 3b). These results suggest that a majority of NO-producing phagocytic PEC from TBM, in contrast to PEC from naïve mice, do not adhere to plastic. Consistent with this suggestion, an MTT adherence test showed that adherence of PEC to plastic was reduced in TBM on day 14 (data not shown).
Fig. 3.
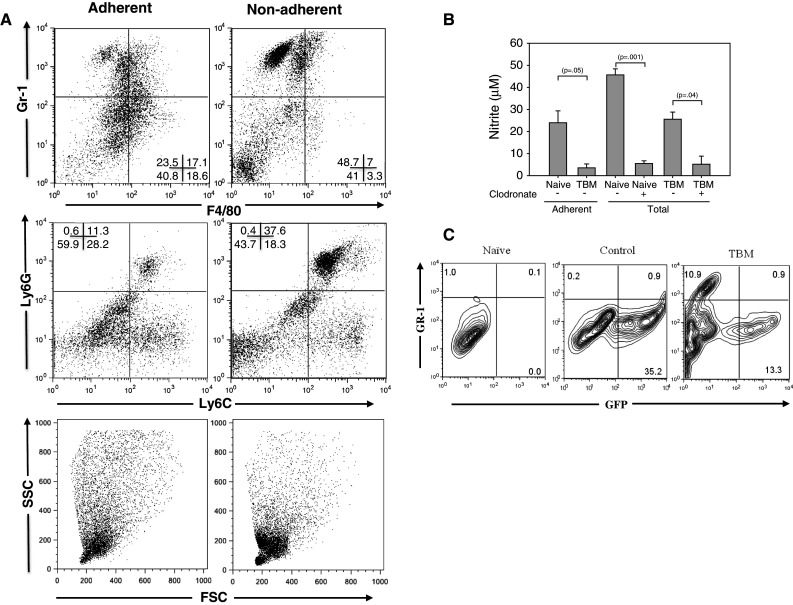
Functional and phenotypic profile of PEC in tumor-bearing mice. C57BL/6 mice were injected i.p. with 105 B16 cells. Fourteen days later, PEC were collected from these mice and from naïve mice and allowed to adhere to plastic for 1.5–2 h. Nonadherent cells were removed by gentle pipetting, and adherent cells were detached from plastic by scraping after N/A cells were washed off. a Representative dot plots of PEC from naïve and tumor-bearing mice showing expression of Gr-1, F4/80, Ly6G and Ly6C. b NO production by adherent and total (unseparated) PEC from naïve and tumor-bearing mice. Phagocytic cells were depleted from the peritoneal cavity by injecting the mice i.p. with clodronate liposomes 2 days before collecting PEC on day 13 post tumor cell inoculation. Mean ± SEM of triplicate average values of PEC obtained individually from 2–3 mice per group. c To determine whether resident Mϕ acquire the Gr-1+ phenotype during tumor growth, C57BL/6 mice were given a mixture of anti-CD4 and anti-CD8 (day 0, 5) and injected i.p. with 4.8 × 106 PEC from naïve EGFP transgenic mice (day 1).One day later, half of the recipient mice were injected i.p. with 105 B16 cells (TBM), while the other half did not receive B16 cells (control). Seven days later, PEC were removed and analyzed for expression of GFP and Gr-1 by flow cytometry after gating on CD11b+ cells, and compared to naïve mice that had not received either B16 or EGFP+ PEC. The results of one out of two representative experiments are shown. The numbers indicate percentages of cells in each quadrant
Resident PEC do not acquire the Gr-1+ phenotype during tumor growth
To determine whether resident peritoneal Mϕ acquire the Gr-1+ phenotype during tumor growth, we performed adoptive transfer of PEC from naïve GFP transgenic mice before injecting the recipient mice with tumor cells. The results in Fig. 3c show that both control (i.e., non-TBM) and TBM contained CD11b+ Gr1− donor cells (GFP+), and that the TBM also contained a population of Gr-1+ PEC that were not seen in the control mice. Importantly, almost all of the CD11b+ Gr-1+ PEC in TBM were GFP negative and thus of recipient origin. In addition, host PEC (i.e., EGFP− cells) in TBM contained a population of F4/80dim cells (both Gr-1− and Gr-1+), which was not observed in adoptively transferred EGFP PEC from TBM or in PEC from naïve mice (data not shown). Furthermore, a three-day culture of B16 cells with naïve peritoneal Mϕ or the Mϕ cell line RAW 264.7 did not result in the appearance of Gr-1+ cells (Supplemental Figure, C). Together these results indicate that resident PEC do not change to the F4/80dimGr-1+ phenotype when exposed to B16 (either in vitro or in the peritonea of mice bearing i.p. tumors). These results suggest that bone marrow-derived CD11b+ F4/80dim Gr-1+ immature cells are attracted to the peritoneal cavity in response to the presence of peritoneal tumor cells.
Characterization of sorted PEC from TBM
The change in adherence and presence of B16 tumor cells in PEC from TBM interfered with the accuracy of our culture assay systems to quantitatively compare NO production by identical numbers of PEC subpopulations from naïve and TBM, and interfered with our assay to evaluate the effect of PEC on tumor cell proliferation in vitro. To circumvent these technical problems, we used flow cytometry to sort PEC from tumor-bearing and naïve mice, and were then able to evaluate and directly compare the contribution of specific cell subsets in these functional assays.
In TBM, CD11b+Gr-1+ cells, independent of the level of Gr-1 expression (Fig. 4a), secreted less NO than CD11b+ Mϕ from naïve mice (populations 2 and 3, vs. population 1 in Fig. 4c), but still suppressed B16 cell proliferation similarly to CD11b+ cells from naïve mice (Fig. 4d). When mixed at the 1:1 ratio, CD11b+Gr-1high cells did not suppress NO production or antitumor effect by naïve Mϕ in response to IFNγ and LPS (data not shown). Among CD11b+Gr-1+ cells from TBM, F4/80+ and F4/80− PEC (Fig. 4e) had a similar ability to both secrete NO and suppress tumor cell proliferation (Fig. 4g, h). Histological evaluation showed that CD11b+Gr-1high cells (population 3 in Fig. 4b) and CD11b+Gr-1highF4/80− cells (population 5 in Fig. 4f) consisted mostly of granulocytes with a smaller percentage of Mϕ/monocytic cells, while a reverse relationship (predominant Mϕ/monocytes with few granulocytes) was observed in CD11b+Gr-1int/− cells (population 2 in Fig. 4b) and CD11b+Gr-1highF4/80+ cells (population 6 in Fig. 4f). In an attempt to obtain more pure cell populations, CD11b+ PEC were sorted according to their Gr-1 expression and side/forward scatter characteristics (using the gates for granulocytes and monocytes) as shown in Fig. 4i. As a result, relatively pure populations of granulocytes (population 7) and Mϕ/monocytes (populations 8 and 9), respectively, were obtained (Fig. 4j). When each of these populations was stimulated with IFNγ and LPS, each secreted no or minimal NO (Fig. 4k), but still suppressed, to a similar extent, B16 cell proliferation (Fig. 4l). An additional group of sorted cells, CD11b+Gr-1high PEC gated on large cells based on forward scatter and comprising Mϕ according to histological examination, showed similar responses (data not shown). Taken together, these sorting experiments show that following in vitro stimulation with IFNγ and LPS, granulocytic and monocytic CD11b+ PEC from TBM produce less NO than PEC from naïve mice, confirming the results with unsorted PEC in Fig. 2a, but still effectively suppress tumor cell proliferation in vitro.
Fig. 4.
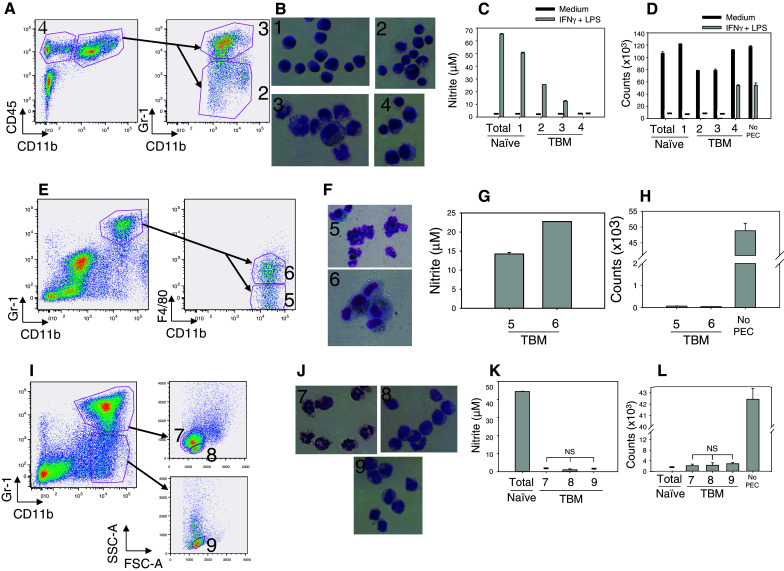
Functional activity of sorted PEC from tumor-bearing mice. PEC were obtained 14–15 days after injecting C57BL/6 mice i.p. with 1 × 105 B16 cells. PEC were sorted based on their phenotype (a, e, i) and also forward-side scatter characteristics (i). The results of three such experiments are presented (a–d, e–h and i–l). The sorting strategy of PEC from TBM is depicted in a, e and i. The morphology of sorted cell fractions from naïve and TBM is presented in b, f and j. The functional data show nitrite levels produced by IFNγ + LPS-stimulated sorted cell fractions (c, g, k) and the ability of these activated sorted cells to inhibit the proliferation of B16 cells in vitro (d, h, l). The numbers designate the sorted cell fraction for that experiment; namely in a, the designations 2, 3 and 4 correspond to the populations shown in b, c, d, where “1” depicts sorted CD11b+ PEC from naïve mice. Similarly, in e, the designations 5 and 6 correspond to those same designated populations in f, g and h. In i, the designations 7, 8 and 9 correspond to those designations in j, k and l. “Total” refers to unsorted PEC from naïve mice. Dashes signify values below nitrite detection limit (c, k) or below 500 cpm (d, l)
Anti-CD40/CpG therapy enhances the antitumor efficacy of tumor-associated myeloid cells
The experiments with sorted PEC showed that tumor-associated myeloid cells can be activated in vitro with IFN-γ and LPS to suppress proliferation of tumor cells. We hypothesized that PEC in TBM can be similarly activated in vivo to mediate antitumor effects against peritoneal tumors. In our previous studies, we had shown that anti-CD40 activates Mϕ via an IFNγ-dependent mechanism [24], and CpG provides a triggering signal to anti-CD40-primed Mϕ in a manner similar to that of LPS, resulting in synergistic antitumor effects against subcutaneous tumors [20]. To determine whether anti-CD40 and CpG given i.p. induce antitumor effects against peritoneal B16 tumors, B16 tumor cells were injected i.p. (day 0) and mice were treated with anti-CD40 on days 4, 11 and 18, and with CpG on days 7 and 14. The combined results of two experiments (Fig. 5a) show that i.p. treatment of TBM with anti-CD40 and CpG resulted in the extended survival of mice compared with control-treated mice (21 vs. 15 median survival days, respectively, p = 0.0111 using Gehan–Breslow–Wilcoxon test). This extended survival corresponded to the grossly visible reduction in tumor load in the peritoneal cavity as shown on day 14 (Fig. 5b). Next, we characterized the phenotype and function of PEC following anti-CD40 and CpG therapy. Control-treated and anti-CD40/CpG-treated TBM had similarly increased levels of CD11+Gr-1+ PEC compared to non-TBM (Fig. 5c). However, anti-CD40/CpG-treated TBM had an increased percentage of Ly6C+ cells (a marker of monocytic lineage [10, 11]), whereas the percentage of Ly6G+ cells (a marker of granulocytic lineage [10, 11]) remained unchanged (Fig. 5c), suggesting that anti-CD40/CpG treatment leads to the prevalence of monocytic cells in the peritoneal cavity. Histological examination of PEC from anti-CD40/CpG-treated mice revealed tumor cells surrounded by monocytes and PMNs (data not shown). When anti-CD40 treatment was started at an early time point, day 4 post tumor cell implantation, tumor-associated PEC regained their ability to secrete NO in response to IFNγ and LPS in vitro (Fig. 5d). When anti-CD40 treatment was started at a later time point, day 11 post tumor cell implantation, NO production increased only marginally (Fig. 5e). Yet, a substantial antitumor effect in the peritoneal cavities of such treated mice was observed as revealed by the reduced number of CD45− PEC in anti-CD40/CpG-treated mice (Fig. 5f). Histological examination of sorted CD45− PEC from B16 tumor-bearing mice, gated to exclude red blood cells, showed that 94% of them were tumor cells (data not shown). Together, these results suggest that, in addition to their activation in vitro, tumor-associated myeloid PEC can be activated in vivo to mediate antitumor effects.
Fig. 5.

Antitumor effects of anti-CD40/CpG therapy in vivo are accompanied by the activation of tumor-associated PEC. C57BL/6 mice were injected i.p. with 105 B16 cells. Anti-CD40/CpG treatments were administered on day 4/7, 11/14, and an additional injection of anti-CD40 was given in some experiments on day 18. Control mice received rat IgG and PBS, respectively. a The mice were followed for survival. The results of two combined experiments are shown (n = 16 mice per group). b Photographs of representative mice showing peritoneal tumor loads were taken on day 14 post B16 tumor cell injections; a naïve mouse is shown for comparison. The peritoneal tumor appears hemorrhagic. c Percentage of CD11b+Gr-1+ cells, as well as Ly6G+ and Ly6C+ cells, are shown (mean ± SEM, 3 mice per group). d–f Tumor-bearing mice were treated with anti-CD40/CpG on days 4/7 and 10 (d) or on days 11/14 (e, f) post tumor cell implantation. PEC were isolated on day 14 (d) or 15 (e, f), and adherent cells were tested for NO production (d, e). Dashes signify values below detection limit. The antitumor effect was determined by the percentage of B16 tumor cells (CD45− PEC) using flow cytometry (f). Mean ± SEM of 4 mice per group. * p < 0.001
Role of NO, arginase and TNFα in the antitumor effects of stimulated myeloid cells
Our experiments have shown that NO production by PEC from naïve mice correlates with their antitumor effects (Fig. 3b, c); however, this correlation was not observed with sorted PEC from TBM (Fig. 4) or with the antitumor effect of anti-CD40/CpG in vivo (Fig. 5e, f). We therefore investigated the role of NO in antitumor responses in vitro and in vivo. In vitro, the iNOS inhibitor L-NAME substantially reduced nitrite levels (Fig. 6a, c), but did not reduce the antitumor activity of sorted polymorphonuclear neutrophils (PMN) or monocytes from TBM (Fig. 6b). Interestingly, when CD11b+ Gr-1+ myeloid cells were kept as a single population and not further separated into granulocyte and monocyte fractions, L-NAME significantly reduced, but did not abrogate, the in vitro antitumor effect (Fig. 6d). Inhibition of other cytotoxic molecules, such as arginase and TNF, did not reduce the in vitro antitumor activity of CD11b+ Gr-1+ cells stimulated with IFN-γ and LPS (Fig. 6d). Inhibition of both iNOS and TNFα in naïve PEC reduced the in vitro antitumor effect to a greater degree than iNOS inhibition alone, but did not have an additive effect in tumor-bearing PEC (Fig. 6d). In vivo, treatment of TBM with iNOS inhibitor L-NAME and anti-CD40/CpG did not reduce, but rather augmented the antitumor effect of anti-CD40/CpG in the mice that survived (Fig. 6e). Together, these results argue against NO playing a major role in the antitumor effects mediated by tumor-associated myeloid cells stimulated in vitro or in vivo.
Fig. 6.

Role of NO, arginase and TNFα in the antitumor effects mediated by myeloid cells. PEC were obtained 15 (a, b) or 14 (c, d) days following injecting C57BL/6 mice i.p. with 105 B16 cells. PEC were sorted based on their CD11b+Gr-1+ phenotype (a–d) and also forward-side scatter characteristics, resulting in populations enriched for polymorphonuclear cells (PMN) and monocytes (Mon) (a, b) as also was verified by histology (not shown). PEC from naïve mice were used as the control. The inhibitors of NO, arginase and TNFα were added as described in “Materials and methods.” The functional data show mean ± SEM of nitrite levels produced by IFN-γ + LPS-stimulated sorted cell fractions (a, c) and the ability of the sorted cells to inhibit proliferation of B16 cells in vitro (b, d). Dashes signify values below detection limit (a). e Tumor-bearing mice were treated with anti-CD40 on day 11 and CpG on day 14 post tumor cell implantation. To neutralize NO in vivo, L-NAME was injected either i.p. at the dose of 50 mg/kg twice a day or given in the drinking water at the dose of 0.5 g/l on days 11–14. PEC were isolated on day 15, and the percentage of B16 tumor cells (CD45− PEC) was determined by flow cytometry. The combined data of two experiments are presented. Mean ± SEM of 10 mice per group or 7 mice for the last group (3 of 5 mice treated with anti-CD40/CpG/L-NAME died from toxicity following i.p. injections)
Discussion
Tumor-induced suppression is one of the obstacles for successful immunotherapy. Most experimental attention has been devoted to investigating the mechanisms of T cell immunosuppression in tumor-bearing hosts, as T cells are considered to be the best effector candidate for immunotherapy due to their greater specificity. T cell suppression in TBM has been attributed to the action of T regulatory cells [25] and MDSC [9–11]. Among other mechanisms, MDSC can suppress T cells by producing NO [26–28]. Although NO inhibits T cell function, it also can kill tumor cells [29], and NO secretion by TAM can be an important mechanism of antitumor effects of cytotoxic T cells [30]. Therefore, activation of cells, such as Mϕ and MDSC, may be an alternative immunotherapy strategy, which may involve NO and does not require T cells. In support of this hypothesis, we have previously shown that treatment of TBM with anti-CD40 caused T cell-independent tumor growth suppression which involved NO production by Mϕ [31]. The synergistic antitumor effects were achieved by the combination of anti-CD40 and CpG [20], even in mice whose T cell and NK functions were inhibited by chemotherapy [22, 32]. In this study, we show that both granulocytic and monocytic myeloid cells at the site of tumor growth are capable of being activated to mediate antitumor effects.
Our results show that peritoneal Mϕ in mice bearing advanced s.c. B16 tumors are not immunologically inert. In fact, they produced high levels of NO in response to IFNγ and LPS, similar to those produced by Mϕ in naïve mice. These findings are similar to those obtained with the D1-DMBA-3 mammary tumor model [33] and are consistent with the results of Danna et al. [7], which show that the systemic function of Mϕ is not inhibited in mice bearing advanced s.c. 4T1 tumors. The reported inhibited ability of peritoneal Mϕ from mice bearing s.c. B16 melanoma to produce NO [34] may be due to the fact that these Mϕ were elicited, rather than resident, as in our experiments. Therefore, our results in the s.c. B16 melanoma model indicate that tumor growth does not induce systemic suppression of Mϕ functions.
To analyze the phenotype and function of tumor-associated cells, we chose the peritoneal model as it offers the possibility of locally determining the number and phenotype of both host and tumor cells while avoiding the loss of cell populations caused by enzymatic digestion of s.c. tumors. Our results using NO production as the readout confirm the studies showing reduced NO production by immunosuppressed TAM [33]. The ability of B16 tumor cells to suppress NO production by adherent PEC 2 weeks after i.p. injection of 105 B16 cells, but not 4 days after i.p. injection of 1.6 × 107 B16 cells, suggests that prolonged interaction of tumor cells and Mϕ is required for Mϕ suppression. These functional findings suggest that TAM from peritoneal TBM may express the M2 phenotype, similar to data we reported for the s.c. B16 model in our previous study, where TAM expressed high surface levels of B7-H1 and intracellular levels of IL-10 and IL-4, but negligible levels of IFN-γ, TNF-α and IL-12 [32].
It has been shown that tumor growth leads to expansion of CD11b+ Gr-1+ cells in mice [9–11, 14]. In this study, we confirmed this observation in the peritoneal B16 model and found an inverse correlation of the percentage of CD11b+ Gr-1+ PEC and the NO levels they produced. The phenotypic change of Mϕ from Gr-1− to Gr-1+ does not appear to be tumor-specific, as it was described in response to certain agents, such as dextrans [35], treatments with anti-CD40/CpG [22] or chemotherapy [32], or infections, such as toxoplasmosis [36]. We found that CD11b+ F4/80bright Gr-1− Mϕ present in naïve mice disappeared from the peritoneal cavity of TBM and were replaced with CD11b+ F4/80dim/− Gr-1+ cells. The results of in vivo experiments using adoptive transfer of PEC from EGFP transgenic mice (Fig. 3c) and short-term in vitro experiments show that resident PEC do not acquire the Gr-1+ phenotype in response to the tumor’s presence, suggesting that bone marrow-derived (potentially immature) cells with the CD11b+ F4/80dim/− Gr-1+ phenotype enter the peritoneal cavity in response to the tumor. In agreement with this suggestion, it has been recently shown that circulating monocytes in TBM reduce expression of F4/80 and acquire expression of Gr-1[37]. The experiments with sorted PEC from TBM (Fig. 4) revealed that these CD11b+ Gr-1+ myeloid cells can be activated to secrete NO and mediate antitumor effects. This finding is in agreement with recent results showing that CD11b+ Gr-1+ MDSC can be activated in vivo with IFN-γ and LPS to secrete NO and suppress T cells [38]. Similarly, it was shown that cyclophosphamide induces the expansion of early myeloid cells, inhibiting tumor cell growth by a mechanism that involves NO [39]. Although there are anecdotal reports on the antitumor effects of MDSC, we believe our data show, for the first time, that tumor-induced myeloid cells can be specifically targeted by immunotherapy to mediate antitumor effects in vitro and in vivo.
It is possible that sorted CD11b+ PEC from TBM displayed antitumor activity in vitro because they were separated from B16 cells and therefore released from tumor-related suppression. However, the antitumor activity of the sorted CD11b+ Gr-1+ PEC from TBM was not inhibited by adding B16 cell supernatant to the assay (data not shown). Moreover, our results show that anti-CD40 and CpG treatment of mice with an advanced peritoneal B16 tumor (tumor cells injected i.p. 10 days earlier) resulted in significant reduction in local tumor load (Fig. 5). This in vivo result suggests that, even in the presence of growing tumor, myeloid PEC can be activated to mediate antitumor effects in vivo (Fig. 5).
TAM have been reported to facilitate tumor growth and serve as a negative prognostic factor for patients with certain cancers [17, 18]. The studies presented here show, in addition, that tumor-associated myeloid cells including TAM can be activated in vitro and in vivo to mediate antitumor effects. One of the mechanisms by which anti-CD40 and CpG affect tumor-associated cells can involve switching the Mϕ phenotype from M2 to M1. This assertion is supported by previous data in which anti-CD40 plus CpG immunotherapy in a s.c. B16 model downregulated the expression of B7-H1, IL-10 and IL-4 in TAM, and upregulated the expression of CD40, CD80, CD86, MHC class II, IFN-γ, TNF-α and IL-12 (32). These results are in agreement with the findings by Guiducci et al. [19] that demonstrated that CpG plus anti–interleukin-10 receptor antibody switched TAM from M2 to M1, resulting in antitumor activity. Our past [21, 24, 32] and present data are consistent with recent preclinical and clinical data showing that, in mice and in patients with pancreatic cancer, antitumor effects induced by agonistic anti-CD40 appear to involve M1 macrophages [40].
The nature of antitumor effector cells in the peritoneal cavity of TBM is not entirely clear. When flow cytometry was used to sort PEC based on their phenotype and size/granularity, tumor-associated PEC that become antitumor effector cells in vitro in response to IFN-γ and LPS consisted of two major groups of cells: CD11b+Gr-1high F4/80− granulocytes and CD11b+Gr-1+/− F4/80dim monocytes/Mϕ. These findings are in agreement with reports showing that MDSC consist of granulocytes and monocytes/Mϕ in mice [27, 41] and in people with cancer [41]. Each cell type was equally effective in suppressing B16 cell proliferation in vitro. We found that in TBM, NO-producing PEC capable of phagocytosing clodronate liposomes (as evidenced by the reduction in NO production) were mainly nonadherent to plastic. Together with the downregulation of the Mϕ marker F4/80, these results suggest that either Mϕ change their phenotype and properties in TBM, or that functional PEC are not mature Mϕ at all, but are rather MDSC or monocytes. In support of the second hypothesis, the results in Fig. 3a show that most nonadherent PEC in TBM have high expression of Ly6C, a marker that is highly expressed on precursor cells and downregulated when monocytes mature in the blood [42].
The mechanism of the antitumor effect mediated by activated myeloid cells is not completely understood. The main factors implicated in the antitumor mechanisms of activated Mϕ are NO [29–31, 43, 44], TNFα [31, 44, 45] or arginase [46]. Our in vitro results indicate that NO does not play a major role in tumor cell suppression by sorted PEC from TBM. Our in vivo data using iNOS inhibitor L-NAME (Fig. 6e) suggest that NO can even suppress the antitumor effect of anti-C40 + CpG immunotherapy. This result (Fig. 6e) may be analogous to a recently published study in which L-NAME treatment in vivo augmented the antitumor effect of IL-12 [47]. The mechanism of the enhanced antitumor effect by iNOS inhibition is not clear. It is known that NO may have protumor and antitumor effects depending on the level and persistence of NO in the tissue, microenvironment and tumor cell sensitivity [48]. It has been shown that iNOS inhibitors reduced the growth of human gastric tumors [49] or melanomas [50] in immunodeficient mice, an effect that was associated with reduced angiogenesis. Whether inhibition of angiogenesis or another mechanism (e.g., increased lymphatic contraction suppressed by CD11b+ Gr-1+ cells via NO production [51]) is responsible for the increased antitumor effect of iNOS inhibitor L-NAME in our model warrants further investigation.
In addition to a limited role of NO, our in vitro results suggest no role of TNFα and arginase in tumor cell suppression (Fig. 6). It is possible that the mechanism of tumor killing by activated myeloid cells is similar to that of Mϕ activated by cyclophosphamide and IL-12 [52]. This mechanism was found to be NO-independent and contact-dependent. It cannot be excluded, however, that NO may play a role in suppressing tumor metastasis, while being ineffective against primary tumors, as was recently shown for IL-2/anti-CD40 immunotherapy [53]. It appears that anti-CD40/CpG immunotherapy can induce antitumor effects of myeloid cells at least by two mechanisms: by directly activating them to mediate antitumor effects, as found in this study, and indirectly by reducing their suppression of antitumor T cells, as was recently reported for CpG immunotherapy [54]. Given the expansion of myeloid cells in cancer patients [55], strategies for activating these cells against the tumor, as shown in this study, might be a novel approach for cancer immunotherapy that warrants further investigation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental figure. Effect of B16 cells in vivo and in vitro on PEC function and phenotype. A. C57BL/6 mice were injected i.p. with 1.6x107 B16 cells or left untreated. PEC were obtained 4 days later, placed in 96-well plate, and plastic-adherent cells were stimulated with IFN-γ (10 U/ml) and LPS (1 ng/ml). The supernatants were collected 48 hr later and NO activity was determined by nitrite levels. The results are presented as the Mean ± SEM nitrite concentration from 4 mice per group. B. PEC were obtained from naïve C57BL/6 mice, and 2x105 PEC were allowed to adhere to plastic for 2 hr followed by removal of nonadherent cells before adding B16 cells at indicated doses to adherent PEC in the presence of IFN-γ and LPS. NO activity in supernatants was determined 48 hr later. The results are presented as the Mean ± SEM nitrite concentration from triplicates. C. PEC obtained from naïve C57BL/6 mice (5x106) and RAW 247.7 macrophage cell line cells (2.5x106) were placed in the wells of the 6-well plates. PEC were allowed to adhere to plastic for 2 hr followed by removal of nonadherent cells. The cells were incubated in medium with or without gamma-irradiated (200 Gy) B16 cells (2.5x106) for 3 days, after which the cells were analyzed for co-expression of F4/80 and Gr-1 by flow cytometry. The representative dot plots are shown. (DOC 163 kb)
Acknowledgments.
We thank Songwon Seo for performing correlative statistical analysis and Jennifer Levenson for breeding EGFP transgenic mice. This work was supported by NIH-NCI grants CA87025 and CA32685, a grant from the Midwest Athletes Against Childhood Cancer (MACC) Fund and support from The Crawdaddy Foundation.
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- IFN-γ
Interferon gamma
- Mϕ
Macrophages
- PEC
Peritoneal cells
- TLR
Toll-like receptor
- TAM
Tumor-associated macrophages
- TNFα
Tumor necrosis factor alpha
- TBM
Tumor-bearing mice
References
- 1.Souza AP, Bonorino C. Tumor immunosuppressive environment: effects on tumor-specific and nontumor antigen immune responses. Expert Rev Anticancer Ther. 2009;9:1317–1332. doi: 10.1586/era.09.88. [DOI] [PubMed] [Google Scholar]
- 2.Ostrand-Rosenberg S, Sinha P, Danna EA, Miller S, Davis C, Dissanayake SK. Antagonists of tumor-specific immunity: tumor-induced immune suppression and host genes that co-opt the anti-tumor immune response. Breast Dis. 2004;20:127–135. doi: 10.3233/bd-2004-20113. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarized phagocyte population: role in tumour progression. Eur J Cancer. 2004;40:1660–1667. doi: 10.1016/j.ejca.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Mullins DW, Martins RS, Elgert KD. Tumor-derived cytokines dysregulate macrophage interferon-gamma responsiveness and interferon regulatory factor-8 expression. Exp Biol Med (Maywood) 2003;228:270–277. doi: 10.1177/153537020322800305. [DOI] [PubMed] [Google Scholar]
- 5.Jaffe ML, Arai H, Nabel GJ. Mechanisms of tumor-induced immunosuppression: evidence for contact-dependent T cell suppression by monocytes. Mol Med. 1996;2:692–701. [PMC free article] [PubMed] [Google Scholar]
- 6.Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol. 2005;66:222–230. doi: 10.1016/j.humimm.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danna EA, Sinha P, Gilbert M, Clements VK, Pulaski BA, Ostrand-Rosenberg S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004;64:2205–2211. doi: 10.1158/0008-5472.CAN-03-2646. [DOI] [PubMed] [Google Scholar]
- 8.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 9.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 10.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 12.Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, Young HA, Zanovello P. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol. 2000;165:6723–6730. doi: 10.4049/jimmunol.165.12.6723. [DOI] [PubMed] [Google Scholar]
- 13.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 14.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–4513. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 15.Popovic PJ, Zeh HJ, Ochoa JB. Arginine and immunity. J Nutr. 2007;137:1681S–1686S. doi: 10.1093/jn/137.6.1681S. [DOI] [PubMed] [Google Scholar]
- 16.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 17.Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. 2002;7:177–189. doi: 10.1023/A:1020304003704. [DOI] [PubMed] [Google Scholar]
- 18.Knowles H, Leek R, Harris AL. Macrophage infiltration and angiogenesis in human malignancy. Novartis Found Symp. 2004;256:189–200. doi: 10.1002/0470856734.ch14. [DOI] [PubMed] [Google Scholar]
- 19.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 20.Buhtoiarov IN, Lum H, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J Immunol. 2006;176:309–318. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 21.Wu Q, Buhtoiarov IN, Sondel PM, Rakhmilevich AL, Ranheim EA. Tumoricidal effects of activated macrophages in a mouse model of chronic lymphocytic leukemia. J Immunol. 2009;182:6771–6778. doi: 10.4049/jimmunol.0801847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson EE, Buhtoiarov IN, Baldeshwiler MJ, Felder MAR, Van Rooijen N, Sondel PM, Rakhmilevich AL. Enhanced T cell-independent antitumor effect of cyclophosphamide combined with anti-CD40 mAb and CpG in mice. J Immunother. 2011;34:76–84. doi: 10.1097/CJI.0b013e318200b28a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 24.Buhtoiarov IN, Lum H, Berke G, Paulnock D, Sondel PM, Rakhmilevich AL. CD40 ligation induces antitumor reactivity of murine macrophages via an IFN gamma-dependent mechanism. J Immunol. 2005;174:6013–6022. doi: 10.4049/jimmunol.174.10.6013. [DOI] [PubMed] [Google Scholar]
- 25.Elkord E, Alcantar-Orozco EM, Dovedi SJ, Tran DQ, Hawkins RE, Gilham DE. T regulatory cells in cancer: recent advances and therapeutic potential. Expert Opin Biol Ther. 2010;10:1573–1586. doi: 10.1517/14712598.2010.529126. [DOI] [PubMed] [Google Scholar]
- 26.Hegardt P, Widegren B, Sjögren HO. Nitric-oxide-dependent systemic immunosuppression in animals with progressively growing malignant gliomas. Cell Immunol. 2000;200:116–127. doi: 10.1006/cimm.2000.1625. [DOI] [PubMed] [Google Scholar]
- 27.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia W, Jackson-Cook C, Graf MR. Tumor-infiltrating, myeloid-derived suppressor cells inhibit T cell activity by nitric oxide production in an intracranial rat glioma + vaccination model. J Neuroimmunol. 2010;223:20–30. doi: 10.1016/j.jneuroim.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortega A, Carretero J, Obrador E, Estrela JM. Tumoricidal activity of endothelium-derived NO and the survival of metastatic cells with high GSH and Bcl-2 levels. Nitric Oxide. 2008;19:107–114. doi: 10.1016/j.niox.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. 2010;185:6706–6718. doi: 10.4049/jimmunol.0903411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumor-necrosis factor-α. Immunology. 2006;118:261–270. doi: 10.1111/j.1365-2567.2006.02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, Rakhmilevich AL. Antitumor synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumor associated macrophages. Immunology. 2011;132:226–239. doi: 10.1111/j.1365-2567.2010.03357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. 1996;183:1323–1329. doi: 10.1084/jem.183.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naama HA, McCarter MD, Mack VE, Evoy DA, Hill AD, Shou J, Daly JM. Suppression of macrophage nitric oxide production by melanoma: mediation by a melanoma-derived product. Melanoma Res. 2001;11:229–238. doi: 10.1097/00008390-200106000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Atochina O, Daly-Engel T, Piskorska D, McGuire E, Harn DA. A schistosome-expressed immunomodulatory glycoconjugate expands peritoneal Gr1(+) macrophages that suppress naive CD4(+) T cell proliferation via an IFN-gamma and nitric oxide-dependent mechanism. J Immunol. 2001;167:4293–4302. doi: 10.4049/jimmunol.167.8.4293. [DOI] [PubMed] [Google Scholar]
- 36.Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caso R, Silvera R, Carrio R, Iragavarapu-Charyulu V, Gonzalez-Perez RR, Torroella-Kouri M. Blood monocytes from mammary tumor-bearing mice: early targets of tumor-induced immune suppression? Int J Oncol. 2010;37:891–900. doi: 10.3892/ijo_00000740. [DOI] [PubMed] [Google Scholar]
- 38.Greifenberg V, Ribechini E, Rössner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009;39:2865–2876. doi: 10.1002/eji.200939486. [DOI] [PubMed] [Google Scholar]
- 39.Pelaez B, Campillo JA, Lopez-Asenjo JA, Subiza JL. Cyclophosphamide induces the development of early myeloid cells suppressing tumor cell growth by a nitric oxide-dependent mechanism. J Immunol. 2001;166:6608–6615. doi: 10.4049/jimmunol.166.11.6608. [DOI] [PubMed] [Google Scholar]
- 40.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 42.Sunderkötter C, Nikolic T, Dillon MJ, van Rooijen N, Stehling M, Drevets DA, Leenen PJM. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 43.Cui SJ, Reichner JS, Mateo RB, Albina JE. Activated murine macrophages induce apoptosis in tumor-cells through nitric-oxide-dependent or nitric-oxide-independent mechanisms. Cancer Res. 1994;54:2462–2467. [PubMed] [Google Scholar]
- 44.Klostergaard J, Leroux ME, Hung MC. Cellular-models of macrophage tumoricidal effector mechanisms in vitro: characterization of cytolytic responses to tumor-necrosis-factor and nitric-oxide pathways in vitro. J Immunol. 1991;147:2802–2808. [PubMed] [Google Scholar]
- 45.Decker T, Lohmannmatthes ML, Gifford GE. Cell-associated tumor-necrosis-factor (TNF) as a killing mechanism of activated cytotoxic macrophages. J Immunol. 1987;138:957–962. [PubMed] [Google Scholar]
- 46.Ellyard JI, Quah BJ, Simson L, Parish CR. Alternatively activated macrophage possess antitumor cytotoxicity that is induced by IL-4 and mediated by arginase-1. J Immunother. 2010;33:443–452. doi: 10.1097/CJI.0b013e3181cd8746. [DOI] [PubMed] [Google Scholar]
- 47.Egilmez NK, Harden JL, Virtuoso LP, Schwendener RA, Kilinc MO. Nitric oxide short-circuits interleukin-12-mediated tumor regression. Cancer Immunol Immunother. 2011;60:839–845. doi: 10.1007/s00262-011-0998-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 49.Wang GY, Ji B, Wang X, Gu JH. Anti-cancer effect of iNOS inhibitor and its correlation with angiogenesis in gastric cancer. World J Gastroenterol. 2005;1:3830–3833. doi: 10.3748/wjg.v11.i25.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, Hailemichael Y, Jayaraman P, Myers JN, Grimm EA, Overwijk WW. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res. 2010;16:1834–1844. doi: 10.1158/1078-0432.CCR-09-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, Ruddle NH, Jain RK, Fukumura D, Padera TP. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci USA. 2011;108:18784–18789. doi: 10.1073/pnas.1116152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002;62:5069–5075. [PubMed] [Google Scholar]
- 53.Weiss JM, Ridnour LA, Back T, Hussain SP, He P, Maciag AE, Keefer LK, Murphy WJ, Harris CC, Wink DA, Wiltrout RH. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. J Exp Med. 2010;207:2455–2467. doi: 10.1084/jem.20100670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zoglmeier C, Bauer H, Noerenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immune suppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17:1765–1775. doi: 10.1158/1078-0432.CCR-10-2672. [DOI] [PubMed] [Google Scholar]
- 55.Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, Zanon A, Rossi CR, Nitti D, Bronte V, Zanovello P. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182:6562–6568. doi: 10.4049/jimmunol.0803831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure. Effect of B16 cells in vivo and in vitro on PEC function and phenotype. A. C57BL/6 mice were injected i.p. with 1.6x107 B16 cells or left untreated. PEC were obtained 4 days later, placed in 96-well plate, and plastic-adherent cells were stimulated with IFN-γ (10 U/ml) and LPS (1 ng/ml). The supernatants were collected 48 hr later and NO activity was determined by nitrite levels. The results are presented as the Mean ± SEM nitrite concentration from 4 mice per group. B. PEC were obtained from naïve C57BL/6 mice, and 2x105 PEC were allowed to adhere to plastic for 2 hr followed by removal of nonadherent cells before adding B16 cells at indicated doses to adherent PEC in the presence of IFN-γ and LPS. NO activity in supernatants was determined 48 hr later. The results are presented as the Mean ± SEM nitrite concentration from triplicates. C. PEC obtained from naïve C57BL/6 mice (5x106) and RAW 247.7 macrophage cell line cells (2.5x106) were placed in the wells of the 6-well plates. PEC were allowed to adhere to plastic for 2 hr followed by removal of nonadherent cells. The cells were incubated in medium with or without gamma-irradiated (200 Gy) B16 cells (2.5x106) for 3 days, after which the cells were analyzed for co-expression of F4/80 and Gr-1 by flow cytometry. The representative dot plots are shown. (DOC 163 kb)