

Abstract
Tremendous progress has been made in the past two decades in molecular genetics of heritable skin diseases, and pathogenic mutations have been identified in as many as 500 distinct human genes. This progress has resulted in improved diagnosis with prognostic implications, refined genetic counseling, and has formed the basis for prenatal and presymptomatic testing as well as preimplantation genetic diagnosis. However, there has been relatively little progress in developing effective and specific treatments for these often devastating diseases. Very recently, however, a number of novel molecular strategies, including gene therapy, cell-based approaches, and protein replacement therapy have been explored for treatment of these conditions. This overview will focus on the prototypic heritable blistering disorders, epidermolysis bullosa and related keratinopathies, in which significant progress has been recently made towards treatment, and illustrate how some of the translational research therapies have already entered the clinical arena.
THE CLINICAL SPECTRUM OF GENODERMATOSES
Heritable skin disorders represent a broad group of conditions in which at one end of the spectrum the cutaneous findings can be relatively minor, primarily of cosmetic concern and limited to the skin, hair and/or nails. In contrast, at the other end of the spectrum, the cutaneous manifestations, often as part of multi-system pathology, can cause significant morbidity and untimely demise of the affected individuals. Heritable skin diseases can present a diagnostic challenge to practicing dermatologist for several reasons (Pulkkinen et al., 2002). First, many of these conditions are relatively rare, and the physician may not be aware of the nuances and salient diagnostic features of the clinical presentations. This difficulty has been compounded by historically complex classification schemes, often riddled with eponyms. Secondly, in many of these conditions, there is considerable intra- and interfamilial heterogeneity, which, when combined with incomplete penetrance and/or late-onset manifestations, can obscure the timely diagnosis. Thirdly, in many of these diseases, the diagnosis is primarily made on the basis of clinical presentation, and histopathologic and ultrastructural findings are often non-specific or not diagnostic. However, over the past two decades, with the advent of molecular genetics in general and completion of the human genome project in particular, molecular diagnostics have become a standard for confirmation of the diagnosis, with potential for earlier detection. In fact, as many as 500 different genes are now known to harbor mutations in a manner that the genetic lesions explain the cutaneous manifestations in these conditions (Feramisco et al., 2009).
Examination of the mutation databases in different heritable skin diseases reveals both obvious candidate genes as well as a number of surprises. An example of clearly identifiable candidate genes is epidermolysis bullosa (EB), a group of heritable blistering disorders manifesting with fragility of skin and mucous membranes (Fine et al., 2008). In fact, just about two decades ago, when very little was known of the molecular pathology of this disorder, we made a prediction that mutations in the genes encoding the protein components of the structural attachment complexes at the epidermal-dermal basement membrane zone could explain the fragility of skin in different forms of EB (Uitto and Christiano, 1992). This prediction has since been proven correct by demonstration of a number of mutations in as many as 15 distinct genes encoding proteins necessary for the physiologic integrity of the skin (Uitto et al., 2010).
In contrast the obvious candidate genes, many of the recently disclosed genes with mutations in heritable skin disorders have turned out to be surprisingly unpredictable, and their role in skin biology in many cases was not recognized before the identification of specific mutations in these genes. An example of such a condition is pseudoxanthoma elasticum (PXE), an ectopic mineralization disorder, which has been shown to result from mutations in the ABCC6 gene that is expressed primarily in the liver and kidneys, and at very low levels, if at all, in affected tissues, including the skin. The ABCC6 gene encodes a transmembrane efflux transporter protein, and it has been postulated that as a result of loss-of-function mutations there is an absence of physiologically circulating anti-mineralization factors, a situation which allows ectopic calcification of the peripheral connective tissues to ensue (Uitto et al., 2011).
Apart from the tremendous progress in understanding the molecular basis of different genodermatoses, until very recently there has been relatively little progress in developing effective and specific treatment strategies for these, mostly intractable, disorders. However, identification of specific mutations in the candidate genes and elucidation of the consequences of such mutations at the mRNA and protein levels, have provided a basis for the development of novel therapeutic approaches, taking advantage of progress in molecular and cell biology in general. This overview will highlight a select number of genodermatoses, with a focus on EB and related blistering disorders, in which significant progress has been made towards treatment, and illustrate how some of these interventional modalities have recently entered the realm of clinical trials (Uitto et al., 2010).
PRECLINICAL MODEL SYSTEMS
A number of model systems have been developed to examine the pathomechanistic consequences of mutations in heritable skin diseases, and many of these systems are also being utilized for development of molecular therapies. Particularly valuable towards understanding of diseasemechanisms has been the development of transgenic animal models which recapitulate the clinical features noted in patients; these genetically modified animals have played a major role in advancing our understanding of the disease mechanisms in different forms of EB (Bruckner-Tuderman et al., 2010; Natsuga et al, 2010). Besides providing direct evidence for the structural role of many of the basement membrane zone adhesion molecules, the development of transgenic mice with EB phenotypes has provided novel information on the complex secondary effects mediated by signaling pathways and other systems that modify the EB phenotypes. In addition to transgenic animals, EB phenotypes have been observed in a number of animal species, both domestic and wild, as a result of naturally occurring mutations (Jiang and Uitto, 2005; Bruckner-Tuderman et al., 2010). In many cases, the suitability of these animal models of human disease for preclinical testing of gene-, protein-, and cell-based molecular therapies has been documented.
While traditionally, mice have provided the preferred platform to serve as a model for human diseases, often with remarkable similarity to the human phenotype both at the genetic, gross morphological, histopathological, and ultrastructural levels, mouse systems can also have considerable limitations. These include the relatively long developmental lifespan, and the time it takes to develop a knockout mouse. Moreover, in some cases, development of the knockout mouse as a model of the corresponding human disease has turned out not to be feasible due to the absence of the corresponding gene in the mouse genome, or because the mutations in the mouse gene result in embryonic lethality (Li et al., 2007; Sercu et al., 2007). Also, quite frequently, ablation of the mouse gene fails to result in a detectable phenotype. Such limitations, together with cost containment issues, have prompted the search for alternative model systems to study heritable skin diseases.
One of the alternate model systems utilizes zebrafish (Danio rerio), a small freshwater fish, which can be easily maintained in the laboratory setting (Lieschke and Currie, 2007). Zebrafish embryos develop and mature very rapidly, so that the development of various organs, including skin, is largely completed by days 5–6 post fertilization (dpf) (Li et al., 2011). Specifically, at 6 dpf, the epidermis is composed of two cell layers clearly separated from the underlying connective tissue stroma by a basement membrane that depicts the presence of hemidesmosomal structures (Sonawane et al., 2005). Examination of the developing zebrafish skin by scanning electron microscopy reveals well demarcated keratinocytes with the surface contour containing microridges which are well organized by 6 dpf. Also, the zebrafish epidermis and the dermal-epidermal basement membrane have characteristic landmark features, with demonstration of the corresponding gene expression. The zebrafish has, therefore, been suggested to serve as a suitable model system to study heritable skin diseases (Li et al., 2011).
In addition to in vivo systems, cell cultures in vitro, including epidermal keratinocytes and dermal fibroblasts, provide experimental systems to study the consequences of gene mutations in heritable disorders. These cells can be examined for phenotypic consequences, such as migration and proliferation anomalies, as well as for changes in their gene expression profiles. The mutant cells can also be incorporated into artificial skin equivalent systems ex vivo with perturbations in the morphology and functionality of skin layers, such as the stratum corneum and its barrier function. Finally, treatment of cultured cells by corrective pharmacologic means, gene replacement approaches, or manipulation of the degree of differentiation has advanced our understanding of the diseases and provided a platform to develop novel treatments. Particularly intriguing are recent observations that cells, such as dermal fibroblasts, can be converted to induced pluripotent stem cells (iPSC) with subsequent differentiation to essentially any cell type in the body. This technology has the potential to develop unprecedented approaches for patient-specific regenerative medicine for heritable skin disorders.
MOLECULAR THERAPIES FOR EPIDERMOLYSIS BULLOSA
All forms of dystrophic epidermolysis bullosa (DEB) result from mutations in the COL7A1 gene that encodes the anchoring fibril protein, type VII collagen, which is normally synthesized and secreted by both keratinocytes and fibroblasts (Stanley et al., 1985; Chen et al., 1994). In recessive forms of DEB (RDEB) the pathogenic mutations typically lead to reduced expression of type VII collagen (Christiano and Uitto, 1996; Aumailley et al., 2006; Chung and Uitto, 2011). The therapeutic goal in RDEB, therefore, is to increase type VII collagen expression at the dermal-epidermal junction (DEJ) to enable the skin to withstand trauma-induced blistering. To achieve this goal, various strategies are being pursued using gene, cell, protein and drug based approaches (Uitto et al., 2010). Reported clinical trials in patients with RDEB, however, have thus far been limited to cell therapy, either involving intradermal injections of allogeneic fibroblasts, derived from unrelated adult or neonatal donors, or bone marrow derived stem cell therapy.
Allogeneic Fibroblast Cell Therapy for RDEB
In 2008, we reported a first-in-man clinical trial, in which single intradermal injections of allogeneic fibroblasts were assessed in five subjects with RDEB (Wong et al., 2008). In skin biopsies from some individuals, fibroblast injection led to an increase in type VII collagen expression that was sustained for several months. Injection of male fibroblasts into a female subject, which allowed tracking of the cells with a Y-chromosome probe, indicated that the donor fibroblasts were undetectable 2 weeks after injection, yet there was no clinical evidence of inflammation, and the clinicopathologic benefits were sustained for several months (Wong et al., 2008). Given the prolonged increase in type VII collagen following allogeneic fibroblast injection, therefore, a key question has been whether the new collagen at the DEJ is derived directly from the donor cells (“direct release”) or indirectly through enhanced synthesis of mutant type VII collagen by the recipient’s own keratinocytes and/or fibroblasts (“indirect release”).
In support of the “direct release” theory, injection of COL7A1 gene corrected human RDEB fibroblasts or normal control human fibroblasts (at sufficient cell density) into immune deficient mice was shown previously to lead to deposition of new human type VII collagen at the dermal-epidermal junction (Woodley et al., 2003). Moreover, injection of normal human fibroblasts into Col7a1 hypomorphic mice generated new human type VII collagen at the mouse DEJ (Fritsch et al., 2008; Kern et al., 2009). Favoring the direct release of type VII collagen from fibroblasts were also the observations that (1) mutant fibroblasts did not exert any paracrine effect (in murine studies), (2) following fibroblast injection there was no upregulation of cytokines or growth factors known to increase type VII collagen expression, and (3) species-specific antibodies to type VII collagen showed that human type VII collagen was present at the mouse DEJ following injections of human fibroblasts (Kern et al., 2009). In contrast, data consistent with “indirect release” stemmed from observations that allogeneic fibroblast injection was associated with (1) increased type VII collagen expression within basal keratinocytes (in humans), (2) an absence of mature anchoring fibrils following treatment, (3) increased expression of recipient patient-specific mutant COL7A1 alleles at mRNA level, (4) a greater increase in type VII collagen at the DEJ in patients who expressed more type VII collagen at baseline and who therefore had an enhanced capacity to increase synthesis of their own mutant type VII collagen, and (5) some increased expression of type VII collagen following the irritant stimulus of saline injection (Wong et al., 2008; Nagy et al., 2011). Moreover, recent investigations in one patient with recessive DEB identified heparin-binding epidermal growth factor-like growth factor (HB-EGF) as a novel putative growth factor induced by fibroblast injections with the capacity to increase type VII collagen expression (predominately from the patient’s keratinocytes and therefore from mutant alleles) (Nagy et al., 2011; Uitto, 2011a) (Figure 1). HB-EGF has not previously been known to influence type VII collagen expression, but exposure of patient and control keratinocytes and fibroblasts led to increased COL7A1 gene expression with evidence of increased AP1 transcription factor probably accounting for the enhanced type VII collagen expression (Nagy et al., 2011).
Figure 1.
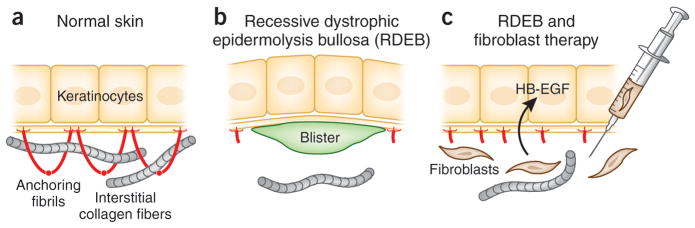
Postulated mechanism by which fibroblast therapy may ameliorate the blistering tendency in RDEB. (a) In normal skin, keratinocytes synthesize type VII collagen molecules (red), which assemble into anchoring fibrils. These fibrils entrap the interstitial collagen fibers in the dermis, securing the stable association at the dermal-epidermal junction. (b) In some patients with RDEB, there are only a few rudimentary anchoring fibrils, allowing formation of blisters below the lamina densa as a result of minor trauma. (c) Allogeneic fibroblasts injected directly into dermis elicit a subclinical immune reaction that leads to synthesis of heparin binding-EGF-like growth factor (HB-EGF), which upregulates the synthesis and assembly of patient’s own mutated type VII collagen. The increase in the rudimentary anchoring fibrils, which are partially functional, stabilizes the association of epidermis to the underlying dermis and ameliorates the blistering tendency. (Adapted from Uitto, 2011a).
With regards to wound healing in RDEB, however, one randomized, double-blind study has shown no statistical differences in the extent or rate of re-epithelialization of chronic erosions following injections of fibroblasts or saline, although other studies assessing the impact of allogeneic fibroblasts in wound healing are ongoing (Venugopal et al., 2010; Yan and Murrell, 2010; John A. McGrath, unpublished data). One interesting observation has been that in the pre-injection monitoring of wounds in recessive DEB, many of the wounds, perhaps somewhat surprisingly, often show spontaneous healing as well as breakdown with change in the shape and size of eroded areas. Notably, within a particular area, the healing of some older wounds is often compounded by the appearance of adjacent newer wounds (John A. McGrath and Gabriela Petrof, unpublished observations). It is evident that careful selection and monitoring of wounds during clinical trials of fibroblast cell therapy in RDEB is paramount in evaluating whether the approach has clinical utility or whether alternative cell therapy or other therapeutic modalities might represent a better therapeutic intervention. For now, the current data and cumulative clinical experience indicate that for a subgroup of individuals with RDEB, notably those with mild-moderate severity of the disease and some baseline expression of type VII collagen at the DEJ, allogeneic fibroblast cell therapy might be clinically useful. From a practical perspective, injection of cell volumes of >0.25 ml per cm2 to any one site can be painful and adequate analgesia (topical or systemic) may be necessary if multiple injections are planned. For patients with more generalized disease, associated with extensive scarring and contractures as well as with absence of type VII collagen protein at the DEJ, alternative cell therapy or other strategic approaches may be more effective.
Bone Marrow Stem Cell Therapy for RDEB
Cell-based therapy for heritable skin diseases has recently been extended to include bone marrow derived adult stem cells (BMDCs). These cells are known to play a crucial role in skin homeostasis, however, it has become clear that the plasticity of BMDCs also enables their differentiation into cell types responsible for skin regeneration after injury. Chronic wounding, such as in EB, has been shown to stimulate the engraftment of BMDCs to the skin and their incorporation and differentiation into non-hematopoietic skin structures (Badiavas and Falanga, 2003; Badiavas et al., 2003; Tamai et al., 2011).
A number of mouse studies have been conducted to evaluate the potential of BMDCs for the treatment of RDEB. For example, bone marrow transfer into the fetal circulation of Col7a1−/− mice resulted in deposition of type VII collagen around developing hair follicles, as well as reduction in severity of blistering in neonatal animals and extension of the overall lifespan of mutant mice (Chino et al., 2008). In another study, hematopoietic and non-hematopoietic populations were infused into unconditioned Col7a1−/− mice at birth or soon thereafter (Tolar et al., 2009). Strikingly, three of 13 (23%) recipient mice of adult bone marrow survived for several months, with evidence of skin engraftment of donor cells, production of type VII collagen in the skin, and formation of Col7a1-positive anchoring fibrils (Tolar et al., 2009). Additionally, a model of milder junctional EB, the Col17a1−/− mouse, was used to test the skin differentiation capacity of various bone marrow-derived subpopulations of cells (Fujita et al., 2010). These studies demonstrated that both purified hematopoietic stem cells (HSCs) and cultured mesenchymal stem cells (MSC) provided amelioration of clinical symptoms and an improved survival rate (Fujita et al., 2010; Tamai et al., 2011; Tolar et al., 2009). These findings established that adoptive transfer of type VII collagen producing bone marrow cell populations is sufficient for the partial correction of the basement membrane zone defect in Col7a1−/− murine recipients. Collectively, these preclinical studies provide evidence for the potential of BMDCs as a source for regeneration of damaged skin in genetic skin diseases.
The first clinical trial of allogeneic whole bone marrow was recently reported in seven children with RDEB (Wagner et al., 2010). New type VII collagen was noted at the dermal-epidermal junction and clinical improvement was sustained for at least 1 year after bone marrow transplantation (BMT). Although the results are promising, two of the seven children died of complications of the procedure which utilized traditional chemoablative pre-conditioning of the recipient. A second clinical BMT trial has been initiated, using reduced intensity chemotherapy prior to transplantation (Kiuru et al., 2010). In addition to bone marrow transplantation, a pilot study on two patients with severe RDEB suggested that intradermal injection of allogeneic MSCs into chronic ulcerated sites can accelerate re-epithelization of these wounds (Conget et al., 2010). The improved wound healing, which lasted for four months, was attributed to replenishment of type VII collagen which was undetectable in these patients with RDEB prior to the procedure. Other studies are now examining whether intradermal or intravenous injection of BM-derived MSCs from the same donor as the transplanted cells in BMT improves clinical outcome. These early observations support the usefulness of BM stem cell populations in correction of the basement membrane defect in heritable skin diseases, such as RDEB (Petrova et al., 2010).
INDUCED PLURIPOTENT STEM CELL THERAPY
The generation of induced pluripotent stem cells (iPSCs) from human fibroblasts was first reported in 2007 (Takahashi et al., 2007; Yu et al., 2007), in studies demonstrating that exogenous expression of a limited number of transcription factors was sufficient to reprogram somatic cells into an embryonic stem cell (ESC)-like state. iPSCs express ESC markers, have unlimited proliferative capacity, and can differentiate into all three germ layers, thus harnessing the full therapeutic spectrum attributed to ESC. Previous stem cell-based therapies were of limited success due to the difficulty in obtaining sufficient numbers of undifferentiated cells (adult stem cells, including BMDCs) and the problem of immune rejection (ESC). With the derivation of iPSCs, regenerative medicine was provided with a new method to circumvent these obstacles and obtain a renewable source of immunocompatible, patient-specific cells. In recent years, patient-specific iPSCs (PS-iPSCs) have been generated from several human diseases to investigate disease mechanisms, test potential drugs, and especially develop cell-based therapies. A proof of concept for iPSC-based therapy was previously provided in a study in which a sickle cell anemia mouse model was successfully treated with hematopoietic stem cells differentiated from gene-corrected iPSCs, derived from autologous mouse skin (Hanna et al., 2007).
In the field of heritable skin disorders, PS-iPSCs have recently been generated from patients with dyskeratosis congenita (Agarwal et al., 2010) and RDEB (Tolar et al., 2010; Itoh et al., 2011; Uitto, 2011b) (Figure 2). However, in order to move iPSCs-based therapy to the clinic, several technical challenges must be overcome. To begin with, integration factor-free iPSCs must be generated, because integration of viral transgenes carrying oncogenes, such as MYC and KLF4, into the genomic DNA of iPSCs may lead to their reactivation in vivo and subsequently to tumor formation (Okita et al., 2007). Rapid development in this field, however, has recently produced several alternative methods, including plasmid-based derivation (Okita et al., 2008), recombinant proteins (Kim et al., 2009), integration-free viral vectors (Yu et al., 2009; Zhou and Freed, 2009), and mRNA (Warren et al., 2010), to generate integration-free iPSCs. Secondly, an efficient way to correct gene defects in PS-iPSCs must be employed. Several methods of correcting gene mutations have been developed, with the most promising approach using a type of homologous recombination (HR) based gene targeting. iPSCs, in contrast to other somatic cells, are suitable for HR-based gene correction due their unlimited, karyotypically stable proliferation. The first report of gene correction in PS-iPSCs from a patient with gyrate atrophy was recently published (Howden et al., 2011), providing a proof-of-concept for this technique. However, the low frequency of HR in mammalian cells still prevents gene targeting from being applied in a broader therapeutic context. Recent advances in generating customized zinc finger nucleases (ZFNs), however, have paved the way for HR-based therapeutic strategies (Cathomen and Joung, 2008). These artificial nucleases can increase the likelihood of HR at the mutation site up to 10,000 fold. This technology has been successfully applied to iPSCs, providing a more efficient strategy for gene-targeting as compared to conventional techniques (Zou et al., 2009; Hockemeyer et al., 2009).
Figure 2.
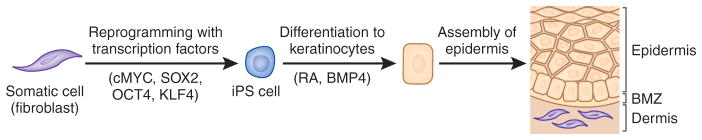
Schematic steps of reprogramming somatic cells, such as fibroblasts, to induced pluripotent stem (iPS) cells, and their differentiation into epidermal keratinocytes capable of forming skin-like structures. The reprogramming process is initiated by introduction of transcription factors (cMYC, SOX2, OCT4 and KLF4) into the somatic cells by transduction of expression vectors, synthetic mRNA or recombinant protein. The iPS cells have characteristic features that allow their identification and enrichment. The iPS cells can then be differentiated into keratinocytes under specific culture conditions, e.g., medium supplemented with retinoic acid (RA) and bone morphogenic protein-4 (BMP-4). BMZ, basement membrane zone. (Adapted from Uitto, 2011b).
Another option for mutation-free iPSC derivation from patients with skin disorders is taking advantage of natural gene therapy in the form of revertant mosaicism. This is a phenomenon wherein a subpopulation of cells re-acquires the wild-type phenotype through a naturally occurring second-site mutation or gene crossover/conversion (Lai-Cheong et al., 2011). Revertant mosaicism has been observed in several human genetic disorders (Klein et al., 1992; Wada et al., 2004), including several types of EB (Jonkman et al., 1997; Lai-Cheong et al., 2011). Since revertant skin in the case of EB is both visible and easy to obtain via skin biopsy, it may serve as a natural, patient-specific source of gene-corrected cells. Taken together, these observations suggest that generation of gene-corrected PS-iPSCs can be achieved, enabling development of gene-targeted stem cell-based therapies for inherited human diseases.
A recent study demonstrated another possible challenge with iPSCs related to their immunogenicity (Zhao et al., 2011a). It was shown that autologous mouse iPSC-derived teratomas were immune-rejected in syngeneic recipient mice with T cell infiltration. Although this study only suggested that tumor immunity, and not transplantation immunity, may be induced by undifferentiated iPSCs, elimination of the other populations from differentiated cell pools should be achieved for future clinical use of iPSCs. We recently demonstrated that both normal iPSCs and RDEB PS-iPSCs can be directly differentiated into functional keratinocytes, one of the relevant cell types for EB treatment (Itoh et al., 2011). Moreover, the utility of keratinocyte-specific surface markers has been also shown to purify iPSC-derived keratinocytes (Itoh et al., 2011), allowing for enrichment of keratinocyte lineage cells. In sum, iPSCs hold great promise to provide an unlimited and autologous source of cells for regenerative therapies for heritable skin diseases.
NOVEL THERAPEUTICS FOR KERATIN DISORDERS
The last two decades have seen tremendous progress in unravelling the molecular basis of keratinizing skin disorders – a large heterogeneous group of genodermatoses characterized by fragility and/or over-proliferation of epithelial tissues, typically affecting various regions of the epidermis and its appendages (Irvine and McLean, 1999; McLean and Irvine, 2007). The keratinopathies – disorders caused by mutations in genes encoding keratin intermediate filament proteins – are a good example of such conditions affecting epidermis, and mutations in 23 of the 54 human keratin genes have now been linked to human disorders of keratinization (Omary et al., 2004). The archetypal keratin disease is epidermolysis bullosa simplex (EBS), which is caused by mutations in either of the genes encoding the basal keratinocyte-specific keratins K5 or K14 (Bonifas et al., 1991; Coulombe et al., 1991; Lane et al., 1992). Heterozygous missense or in-frame insertion/deletion mutations in K5 or K14 act via dominant-negative interference to weaken the structural integrity of the intermediate filament cytoskeleton within the basal cell compartment of the epidermis. Functionally, this cytoplasmic filament network is responsible for maintaining the structural integrity of the basal cells when exposed to the high levels of mechanical trauma experienced by the skin in everyday life. Thus, failure of this system leads to skin blistering confined to the basal layer of the epidermis resulting in the clinical and histopathologic hallmarks of EBS (Irvine and McLean, 1999).
Most keratin mutations act through a dominant-negative pathomechanism because of the highly polymeric nature of the keratin cytoskeleton whereby the mutant keratin monomers interact with the normal keratin to destabilize the entire keratin network within the cell. The great challenge in developing therapy for the common, dominantly inherited forms of EBS, and most other autosomal dominant keratinopathies, is to inhibit expression of the mutant allele without silencing the wild-type allele. Proof of concept for allele-specific gene silencing therapy comes from the study of unusual EBS families where homozygous loss-of-expression (typically nonsense or frameshift) mutations in K14 lead to recessively inherited EBS. In these kindreds, the affected individuals have complete absence of K14 in their basal keratinocytes, leading to cell fragility via a recessive, loss-of-function pathomechanism (Rugg et al., 1994). Interestingly, the heterozygous carriers of these loss-of-function mutations, who only express one K14 allele, have clinically normal skin. Thus, if a means of silencing a dominant-negative mutant keratin allele could be found, these data strongly suggest that such a therapeutic approach would be likely to succeed (Kaspar, 2005; Lewin et al., 2005).
RNA-interference Technology
Great strides towards therapy for keratin disorders have been made in recent years by application of RNA-interference (RNAi) technology, in the form of short interfering RNA (siRNA), to address the problem of allele-specific inhibition (Bumcrot et al., 2006). siRNAs are 19-mer double-stranded RNA molecules with 2-nucleotide overhangs at either end, that are a perfect match for the mRNA that is to be silenced. Within the cell, these molecules are incorporated into the RNA-induced silencing complex (RISC) where the antisense strand is used to scan mRNA species. When an exact match is found, endonuclease components of RISC degrade the target message. For a given point mutation, there are 19 possible positions where 19-mer siRNA can be positioned relative to the single mutant base. Reporter gene systems, combined with a sequence walk methodology, facilitate the systemic determination of both potency and specificity of each of the 19 possible siRNAs to identify positions where the mutant reporter gene is strongly inhibited with minimal or negligible effect on the wild-type reporter. This methodology has been applied successfully to specific point mutations in keratins K6a causing pachyonychia congenita (PC) (Hickerson et al., 2008), as well as EBS-causing mutations in K5 (Atkinson et al., 2011).
PC is a rare genetic skin disorder that can be caused by dominant mutations in any one of the genes encoding keratins K6a, K6b, K6c, K16 or K17 (McLean et al., 2011). These keratins are strongly expressed in the palmoplantar epidermis (Swensson et al., 1998), and therefore, a major clinical feature of PC is painful and highly debilitating focal keratoderma, particularly affecting the soles of the feet (McLean et al., 2011). The PC patient advocacy organization PC Project (www.pcproject.org), has been a key driving force in the development and implementation of siRNA therapy for this condition, in close collaboration with an international network of researchers and a small biotech start-up company, Transderm Inc. This work has recently led to a first-in-human double-blinded phase 1b clinical trial where an siRNA specific for the missense mutation N171K in the K6a protein was targeted (Leachman et al., 2009). The therapeutic siRNA was delivered into hyperkeratotic plantar lesions by intradermal injection, which was both painful and highly inefficient. Nevertheless, significant clinical improvement was seen in the lesion that received the siRNA whereas no difference was observed in the lesion that received vehicle only. This study provides proof-of-concept for allele-specific siRNA therapy for dominant-negative skin diseases, however, an efficacious and non-invasive delivery method still needs to be developed (Figure 3). Cutaneous siRNA delivery is currently the focus of much attention in the keratin and RNAi fields where a number of potential delivery systems are being explored including chemical modification of the siRNA, topical formulation chemistry, nanoparticle technology and physical methods such as microneedle arrays.
Figure 3.

siRNA strategies for autosomal dominant keratin 6a disorders by targeting either mutant or both mutant/wild-type alleles. (a) In normal keratinocytes, synthesis of K6a (blue), K6b (red) and K6c (green) occurs; (b) in PC keratinocytes with a heterozygous missense mutation in KRT6A there is dominant-negative interference between the wild-type and mutant K6a protein that perturbs the keratin network and compromises cell integrity, leading to skin blistering as a result of minor trauma; (c) one siRNA approach is to target the mutant KRT6A allele to leave only residual wild-type KRT6A allele expression; (d) an alternative siRNA strategy is to silence all KRT6A, both mutant and wild-type – blistering does not occur in the absence of K6a because of functional redundancy with K6b and K6c, allowing normal intermediate filament network integrity.
One disadvantage of the allele-specific gene silencing approach is that the FDA and related agencies internationally may consider each mutation-specific siRNA as a separate entity requiring individual toxicological studies and regulatory approval. With this in mind, an alternative approach to treatment of certain keratin disorders, notably in the keratodermas such as PC, is a gene-specific approach where both wild-type and mutant alleles of the target gene is silenced. In palmoplantar epidermis in particular, there is significant keratin gene redundancy. For example, humans possess three versions of the K6 gene – encoding K6a, K6b and K6c, all of which are expressed in thick skin. The mouse genome has orthologs for only K6a and K6b. Single knock out of either of these muring genes produces a negligible phenotype (Wojcik et al., 2001; Wong et al., 2000). Only when both K6a and K6b genes are ablated is an epithelial fragility phenotype produced. This shows that there is considerable gene redundancy in certain epithelial tissues, such as palmoplantar epidermis. To this end, highly potent gene-specific siRNAs for human K6 genes have been developed and may be the weapon of choice in future clinical trials in PC (Smith et al., 2008). In particular, most PC patients carry mutations in the K6a gene (Wilson et al., 2011), and therefore, most could be treated with a single K6a-specific inhibitor (Smith et al., 2008) (Figure 3).
Small Molecule Library Screen
With the increasing availability of chemical compound libraries and falling costs of robotic liquid handling technologies, recent years have seen considerable developments in academic drug discovery, which previously was solely within the domain of the pharmaceutical industry (Kozikowski et al., 2006). Accordingly, increasing numbers of drugs entering clinical use have their origins in academic laboratories. Again, this technology has been applied to PC, where small molecules that reduce the expression of the PC-related keratins, such as K6a, have been sought through high-throughput screening using cell-based reporter gene assay systems. This has led to the discovery that the statins, drugs in common use for cholesterol control, have a modest inhibitory effect on K6a and certain other keratins (Zhao et al., 2011b). This work has led to on-going clinical trials in a well-studied and genetically confirmed PC case series, coordinated by the International PC Research Consortium. Although this type of therapy may not be curative, it may help alleviate symptoms until more potent and specific gene silencing therapies enter the clinic.
Overall, the development of therapies from basic discoveries in genetics has been slow and gene therapy has been disappointingly slow to enter common clinical application. However, with the advent of RNAi, academic drug discovery and greater industry-academia partnerships (Vallance et al., 2010), the landscape is changing rapidly, giving hope for patients with devastating inherited skin disorders.
Acknowledgments
The authors thank the numerous colleagues who contributed to the original studies cited in this overview. Carol Kelly assisted in preparation of this manuscript. The authors’ original work was supported by National Institutes of Health, Dermatology Foundation, DebRA International, the UK Technology Strategy Board, the Wellcome Trust, and PC Project.
References
- Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–6. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson SD, McGilligan VE, Liao H, et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Invest Dermatol. 2011 doi: 10.1038/jid.2011.169. (in press) [DOI] [PubMed] [Google Scholar]
- Aumailley M, Has C, Tunggal L, et al. Molecular basis of inherited skin-blistering disorders and therapeutic implications. Exp Rev Mol Med. 2006;8:1–21. doi: 10.1017/S1462399406000123. [DOI] [PubMed] [Google Scholar]
- Badiavas EV, Falanga V. Treatment of chronic wounds with bone marrow-derived cells. Arch Dermatol. 2003;139:510–6. doi: 10.1001/archderm.139.4.510. [DOI] [PubMed] [Google Scholar]
- Badiavas EV, Abedi M, Butmarc J, et al. Participation of bone marrow derived cells in cutaneous wound healing. J Cell Physiol. 2003;196:245–50. doi: 10.1002/jcp.10260. [DOI] [PubMed] [Google Scholar]
- Bonifas JM, Rothman AL, Epstein EH. Epidermolysis bullosa simplex: evidence in two families for keratin gene abnormalities. Science. 1991;254:1202–5. doi: 10.1126/science.1720261. [DOI] [PubMed] [Google Scholar]
- Bruckner-Tuderman L, McGrath JA, Robinson EC, et al. Animal models of epidermolysis bullosa: Update 2010. J Invest Dermatol. 2010;130:1485–8. doi: 10.1038/jid.2010.75. [DOI] [PubMed] [Google Scholar]
- Bumcrot D, Manoharan M, Koteliansky V, et al. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–9. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomen T, Joung JK. Zinc-finger nucleases: the next generation emerges. Mol Ther. 2008;16:1200–7. doi: 10.1038/mt.2008.114. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Mauviel A, Ryynänen J, et al. Type VII collagen gene expression by human skin fibroblasts and keratinocytes in culture: Influence of donor age and cytokine responses. J Invest Dermatol. 1994;102:205–9. doi: 10.1111/1523-1747.ep12371763. [DOI] [PubMed] [Google Scholar]
- Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173:803–14. doi: 10.2353/ajpath.2008.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano AM, Uitto J. Molecular diagnosis of inherited skin diseases: The paradigm of dystrophic epidermolysis bullosa. Adv Dermatol. 1996;11:199–213. [PubMed] [Google Scholar]
- Chung HJ, Uitto J. Type VII collagen: The anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:93–105. doi: 10.1016/j.det.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conget P, Rodriguez F, Kramer S, et al. Replenishment of type VII collagen and re-epithelialization ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12:429–31. doi: 10.3109/14653241003587637. [DOI] [PubMed] [Google Scholar]
- Coulombe PA, Hutton ME, Letai A, et al. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: Genetic and functional analysis. Cell. 1991;66:1301–11. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- Feramisco JD, Sadreyev RI, Murray ML, et al. Phenotypic and genotypic analyses of genetic skin disease through the Online Mendelian Inheritance in Man (OMIM) database. J Invest Dermatol. 2009;129:2628–36. doi: 10.1038/jid.2009.108. [DOI] [PubMed] [Google Scholar]
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the third international consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Fritsch A, Loeckermann S, Kern JS, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest. 2008;118:1669–79. doi: 10.1172/JCI34292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Abe R, Inokuma D, et al. Bone marrow transplantation restores epidermal basement membrane protein expression and rescues epidermolysis bullosa model mice. Proc Natl Acad Sci U S A. 2010;107:14345–50. doi: 10.1073/pnas.1000044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–3. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- Hickerson RP, Smith FJD, Reeves RE, et al. Single-nucleotide-specific siRNA targeting in a dominant-negative skin model. J Invest Dermatol. 2008;128:594–605. doi: 10.1038/sj.jid.5701060. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–7. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci U S A. 2011;108:6537–42. doi: 10.1073/pnas.1103388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine AD, McLean WHI. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br J Dermatol. 1999;140:815–28. doi: 10.1046/j.1365-2133.1999.02810.x. [DOI] [PubMed] [Google Scholar]
- Itoh M, Kiuru M, Cairo MS, et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2011;108:8797–802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang QJ, Uitto J. Animal models of epidermolysis bullosa – targets for gene therapy. J Invest Dermatol. 2005;124:xi–xiii. doi: 10.1111/j.0022-202X.2005.23652.x. [DOI] [PubMed] [Google Scholar]
- Jonkman MF, Scheffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88:543–51. doi: 10.1016/s0092-8674(00)81894-2. [DOI] [PubMed] [Google Scholar]
- Kaspar RL. Challenges in developing therapies for rare diseases including pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:62–6. doi: 10.1111/j.1087-0024.2005.10208.x. [DOI] [PubMed] [Google Scholar]
- Kern JS, Loeckermann S, Fritsch A, et al. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol Therapy. 2009;17:1605–15. doi: 10.1038/mt.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuru M, Itoh M, Cairo MS, et al. Bone marrow stem cell Therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:371–82. doi: 10.1016/j.det.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Coovert DD, Bulman DE, et al. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am J Hum Genet. 1992;50:950–9. [PMC free article] [PubMed] [Google Scholar]
- Kozikowski AP, Roth B, Tropsha A. Why academic drug discovery makes sense. Science. 2006;313:1235–6. doi: 10.1126/science.313.5791.1235c. [DOI] [PubMed] [Google Scholar]
- Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–8. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane EB, Rugg EL, Navsaria H, et al. A mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature. 1992;356:244–6. doi: 10.1038/356244a0. [DOI] [PubMed] [Google Scholar]
- Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther. 2009;18:442–6. doi: 10.1038/mt.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin AS, Glazer PM, Milstone LM. Gene therapy for autosomal dominant disorders of keratin. J Investig Dermatol Symp Proc. 2005;10:47–61. doi: 10.1111/j.1087-0024.2005.10207.x. [DOI] [PubMed] [Google Scholar]
- Li CF, MacDonald JR, Wei RY, et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated aggressive fibromatosis, is absent in the mouse. BMC Genomics. 2007;8:92. doi: 10.1186/1471-2164-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Frank M, Thisse CI, et al. Zebrafish: A model system to study heritable skin diseases. J Invest Dermatol. 2011;131:565–71. doi: 10.1038/jid.2010.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: Zebrafish swim into view. Nat Rev Genet. 2007;8:353–67. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- McLean WH, Hansen CD, Eliason MJ, et al. The phenotypic and molecular genetic features of pachyonychia congenita. J Invest Dermatol. 2011;131:1015–7. doi: 10.1038/jid.2011.59. [DOI] [PubMed] [Google Scholar]
- McLean WH, Irvine AD. Disorders of keratinisation: from rare to common genetic diseases of skin and other epithelial tissues. Ulster Med J. 2007;76:72–82. [PMC free article] [PubMed] [Google Scholar]
- Nagy N, Almaani N, Tanaka A, et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:1771–4. doi: 10.1038/jid.2011.85. [DOI] [PubMed] [Google Scholar]
- Natsuga K, Shinkuma S, Nishie W, et al. Animal models of epidermolysis bullosa. Dermatol Clin. 2010;28:137–42. doi: 10.1016/j.det.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–53. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- Omary MB, Coulombe PA, McLean WHI. Intermediate filament proteins and their associated diseases. N Engl J Med. 2004;351:2087–100. doi: 10.1056/NEJMra040319. [DOI] [PubMed] [Google Scholar]
- Petrova A, Ilic D, McGrath JA. Stem cell therapies for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2010;163:1149–56. doi: 10.1111/j.1365-2133.2010.09981.x. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Ringpfeil F, Uitto J. Progress in heritable skin diseases: Molecular bases and clinical implications. J Am Acad Dermatol. 2002;47:91–104. doi: 10.1067/mjd.2002.120601. [DOI] [PubMed] [Google Scholar]
- Rugg EL, McLean WHI, Lane EB, et al. A functional “knock-out” for human keratin 14. Genes Dev. 1994;8:2563–73. doi: 10.1101/gad.8.21.2563. [DOI] [PubMed] [Google Scholar]
- Sercu S, Poumay Y, Herphelin F, et al. Functional redundancy of extracellular matrix protein 1 in epidermolysis differentiation. Br J Dermatol. 2007;157:771–5. doi: 10.1111/j.1365-2133.2007.08114.x. [DOI] [PubMed] [Google Scholar]
- Smith FJD, Hickerson RP, Sayers JM, et al. Development of therapeutic siRNAs for pachyonychia congenita. J Invest Dermatol. 2008;128:50–8. doi: 10.1038/sj.jid.5701040. [DOI] [PubMed] [Google Scholar]
- Sonawane M, Carpio Y, Geisler R, et al. Zebrafish penner/lethal giant larvae 2 functions in hemidesmosome formation, maintenance of cellular morphology and growth regulation in the developing basal epidermis. Development. 2005;132:3255–65. doi: 10.1242/dev.01904. [DOI] [PubMed] [Google Scholar]
- Stanley JR, Rubinstein N, Klaus-Kovtun V. Epidermolysis bullosa acquisita antigen is synthesized by both human keratinocytes and human dermal fibroblasts. J Invest Dermatol. 1985;85:542–5. doi: 10.1111/1523-1747.ep12277377. [DOI] [PubMed] [Google Scholar]
- Swensson O, Langbein L, McMillan JR, et al. Specialized keratin expression pattern in human ridged skin as an adaptation to high physical stress. Br J Dermatol. 1998;139:767–75. doi: 10.1046/j.1365-2133.1998.02499.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tamai K, Yamazaki T, Chino T, et al. PDGFR -positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci U S A. 2011;108:6609–14. doi: 10.1073/pnas.1016753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Ishida-Yamamoto A, Riddle M, et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113:1167–74. doi: 10.1182/blood-2008-06-161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Xia L, Riddle MJ, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;131:848–856. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Christiano AM. Molecular genetics of the cutaneous basement membrane zone. Perspectives on epidermolysis bullosa and other blistering skin diseases. J Clin Invest. 1992;90:687–92. doi: 10.1172/JCI115938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, McGrath JA, Rodeck U, et al. Progress in epidermolysis bullosa research: Toward treatment and cure. J Invest Dermatol. 2010;130:1778–84. doi: 10.1038/jid.2010.90. [DOI] [PubMed] [Google Scholar]
- Uitto J. Cell-based therapy for RDEB: How does it work? J Invest Dermatol. 2011a;131:1597–9. doi: 10.1038/jid.2011.125. [DOI] [PubMed] [Google Scholar]
- Uitto J. Regenerative medicine for skin diseases: iPS cells to the rescue. J Invest Dermatol. 2011b;131:812–14. doi: 10.1038/jid.2011.2. [DOI] [PubMed] [Google Scholar]
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: Progress in diagnostics and research towards treatment: Summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155:1517–26. doi: 10.1002/ajmg.a.34067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P, Williams P, Dollery C. The future is much closer collaboration between the pharmaceutical industry and academic medical centers. Clin Pharmacol Ther. 2010;87:525–7. doi: 10.1038/clpt.2010.29. [DOI] [PubMed] [Google Scholar]
- Venugopal SS, Yan WF, Frew JW, et al. First double-blind randomized clinical trial of intradermal allogeneic fibroblast therapy for recessive dystrophic epidermolysis bullosa randomized against placebo injections resulted in similar wound healing that is independent of type VII collagen expression. J Invest Dermatol. 2010;130 (Suppl 2):S67. [Google Scholar]
- Wada T, Schurman SH, Jagadeesh GJ, et al. Multiple patients with revertant mosaicism in a single Wiskott-Aldrich syndrome family. Blood. 2004;104:1270–2. doi: 10.1182/blood-2004-03-0846. [DOI] [PubMed] [Google Scholar]
- Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–39. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NJ, Leachman SA, Hansen CD, et al. A large mutational study in pachyonychia congenita. J Invest Dermatol. 2011;131:1018–24. doi: 10.1038/jid.2011.20. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Longley MA, Roop DR. Discovery of a novel murine keratin 6 (K6) isoform explains the absence of hair and nail defects in mice deficient for K6a and K6b. J Cell Biol. 2001;154:619–30. doi: 10.1083/jcb.200102079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Colucci-Guyon E, Takahashi K, et al. Introducing a null mutation in the mouse K6alpha and K6beta genes reveals their essential structural role in the oral mucosa. J Cell Biol. 2000;150:921–8. doi: 10.1083/jcb.150.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179–89. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Krueger GG, Jorgensen CM, et al. Normal and gene-corrected dystrophic epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Invest Dermatol. 2003;121:1021–8. doi: 10.1046/j.1523-1747.2003.12571.x. [DOI] [PubMed] [Google Scholar]
- Yan WF, Murrell DF. Fibroblast-based cell therapy strategy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:367–70. doi: 10.1016/j.det.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011a;474:212–5. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Gartner U, Smith FJ, et al. Statins downregulate K6a promoter activity: a possible therapeutic avenue for pachyonychia congenita. J Invest Dermatol. 2011b;131:1045–52. doi: 10.1038/jid.2011.41. [DOI] [PubMed] [Google Scholar]
- Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27:2667–74. doi: 10.1002/stem.201. [DOI] [PubMed] [Google Scholar]
- Zou J, Maeder ML, Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]