

Abstract
Heart disease remains the major cause of death in males and females, emphasizing the need for novel strategies to improve patient treatment and survival. A therapeutic approach, still in its infancy, is the development of site-specific drug-delivery systems. Nanoparticle-based delivery systems, such as liposomes, have evolved into robust platforms for site-specific delivery of therapeutics. In this review, the clinical impact of cardiovascular disease and the pathophysiology of different subsets of the disease are described. Potential pathological targets for therapy are introduced, and promising advances in nanotherapeutic cardiovascular applications involving liposomal platforms are presented.
Keywords: liposomes, cardiovascular disease, therapeutics, nanoparticles, nanomedicine
Introduction
Cardiovascular disease remains the largest cause of mortality in the general population. As a harrowing statistic, every 39 seconds someone dies due to cardiovascular disease, claiming more lives than cancer in the United States.1 Prevention and the National Vital Statistics System reported that deaths from “Diseases of Heart” outnumbered those from “Malignant Neoplasms” in both 2010 and 2011.2 It is estimated that 82 million American adults have one or more cardiovascular diseases (ie, hypertension, coronary artery disease, etc). In 2008 alone, the American Heart Association reported that more than 811,940 people died from cardiovascular disease, representing 33% of total deaths in the United States.1
Several significant advancements have resulted in a more than two-fold decline in cardiovascular-related deaths from the decades spanning 1975 to 2005,3 due mostly to improvements in diagnostics and surgical interventions, as well as awareness and consequent lifestyle changes. While this decrease in patient mortality has had a significant impact, deaths from cardiovascular disease remain unacceptably high, warranting more effective strategies to improve patient outcomes. Currently, the use of therapeutic agents in heart disease has become indispensable for the treatment and prevention of cardiovascular disease. As an example, statins have emerged as powerful therapeutics for the treatment of increased cholesterol levels that contribute to atherosclerosis,4 the most widely used being atorvastatin (trade name Lipitor). Medications for high blood pressure now comprise a number of distinct antihypertensive drugs, all shown to act preventively against a variety of cardiovascular events, such as myocardial infarction.5 Importantly, the application of drug-based interventions for cardiovascular conditions such as restenosis and heart failure ameliorate but fail to bring about significant improvements in patient outcomes. The treatment of end-stage patients with chronic heart failure relies principally on palliative strategies to combat the onset of disease, leaving heart transplantation as the only viable option to cure the disease.6 However, this therapeutic modality is not sustainable, given the poor availability of donor hearts and their suitability for transplantation.6 With regards to in-stent restenosis, therapeutic options are limited to the implantation of drug-eluting stents in hopes of preventing proliferation of smooth-muscle cells and eventual narrowing of the blood vessels.7 However, a recent study has demonstrated that minimal improvements are observed after implantation of drug-eluting stents compared to nondrug-eluting stents in patients with restenosis.7 Hence, while giant strides have been made regarding drug therapy in cardiovascular disease, their greatest success comes in the form of disease prevention.
While eventually all-encompassing, conditions such as heart failure, myocardial infarction, atherosclerosis, and in-stent restenosis initially comprise highly site-specific cardiovascular diseases. Myocardial infarction is located within the ischemic area, heart failure is initially confined to the left ventricle, and atherosclerosis and in-stent restenosis comprise plaque accumulation and neointimal narrowing, respectively, in specific arteries. Hence, in order to effectively treat the condition, therapeutics must accumulate site-specifically in the region of interest. This site-specific targeting is not feasible with several clinical formulations of drugs, mainly because of their nonspecific distribution upon administration, resulting in decreased bioavailability of drugs at the site and increased risk of patient side effects. Moreover, there are several biological barriers present that impede adequate delivery of drugs to specific sites of the body.8
Cancer represents another disease condition that requires site-specific, targeted delivery of therapeutics in order to maximize efficacy and minimize patient morbidity. Cancer has recently benefited from the emergence of nanomedicine, or the application of several nanoscale platforms for diagnosis and treatment, specifically within the realm of drug delivery for chemotherapy.9 Nanoparticle drug-delivery platforms such as liposomes significantly extend the circulation half-life of drugs, protect the drug from degradation, and result in increased accumulation of active agents within tumors, due in large part to the enhanced permeability and retention (EPR) effect.10 The best example of a Food and Drug Administration-approved nanoparticle platform currently used in clinics is liposomal doxorubicin (trade name Doxil), which improved patient outcomes by increasing the half-life of doxorubicin from minutes to hours, and more importantly minimized cardiotoxicity associated with the drug.11
Given the significant impact that nanoparticle-based drug delivery has had on the management and treatment of cancer, the question remains as to whether several of these platforms can be translated towards cardiovascular disease. This review will focus on innovative strategies involving liposomal-based drug-delivery applications in cardiovascular disease. A brief overview is given on the pathophysiology of several manifestations of cardiovascular disease, followed by a description of liposomal platforms for drug delivery. It is now well known that various characteristics of the tumor microenvironment, such as the EPR effect, enhance tumor accumulation.12 The presence of several overexpressed receptors on their vasculature enables the addition of targeting moieties on the surface for increased site-specific accumulation.13 This review compares and contrasts the tumor microenvironment with cardiovascular disease environments, in hopes of finding correlates that enable the potential use of nanomedicine platforms in cardiovascular disease. Several innovative examples of liposomal technologies are then highlighted and applied towards cardiovascular disease treatment.
Pathophysiology of cardiovascular diseases
Heart disease
Heart disease primarily refers to hypertension and coronary artery disease, which are commonly associated with other cardiovascular diseases, such as atherosclerosis, myocardial infarction, angina pectoris, arrhythmias (irregular heartbeat), and chronic heart failure.
Primary hypertension is a chronic cardiovascular condition due to increased blood pressure within the arteries.14 While some of the etiological and pathophysiological aspects of hypertension remain unknown, it has been correlated with genetic factors, obesity, insulin resistance, high alcohol intake, high salt intake, and aging.15 The initial treatment for a patient with hypertension is lifestyle modification, followed by pharmacological therapy if warranted. Hypertension is considered a risk factor for many cardiovascular diseases, including coronary artery disease, myocardial infarction, arrhythmias, left ventricular hypertrophy, chronic heart failure, and hypertensive cardiomyopathy, all of which emphasize the need to screen patients and start therapy at an early stage in the disease.
Coronary artery disease is an important risk factor for heart disease and is defined as plaque buildup, or atherosclerosis, in the coronary arteries that supply the heart muscle with an oxygen-rich blood supply. Currently, coronary artery disease is visualized by coronary arteriography or computed tomography angiography. Therapy includes either pharmacological or invasive measures, depending on the severity of the disease. Coronary artery disease is a common risk factor for cardiomyopathies, arrhythmias, myocardial infarction, and congestive heart failure.
Atherosclerosis
Atherosclerosis is the buildup of arterial plaque, comprised of calcium, cholesterol, fat, and inflammatory components, along the vascular walls. The presence of lipoproteins, oxidized phospholipids, and platelet adhesion induces changes in resident endothelial cells.16–18 The inflammatory reaction that ensues includes expression of various adhesion molecules and proinflammatory cytokines, such as vascular cell adhesion molecule 1 and macrophage colony-stimulating factor. These changes lead to the development of an endothelial lining that is procoagulant and permeable in nature. Persistence of the endothelial response causes proliferation of a lesion in the lumen of the vessel wall, known as plaque.19,20 Plaque accumulates in the vasculature throughout the lifetime of a patient. As plaque accumulates, it hardens and may limit blood flow, causing stable angina, or it may rupture, producing either temporary occlusion (unstable angina) or permanent occlusion (myocardial infarction). Current pharmacological treatments for treating cholesterol production include statins, inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, fibric acid derivatives, and bile acid sequestrants.
Myocardial infarction
Myocardial infarction, commonly known as a heart attack, is an irrevocable necrosis of cardiac tissue, caused by the rupture of an atherosclerotic plaque. During coronary artery disease, the arteries supplying blood to the heart become narrowed or blocked by atheroma rupture and blood clots. When an artery of the heart is completely blocked, lack of blood flow causes insufficient oxygen and nutrients, causing ischemia and a subsequent myocardial infarction. This creates muscular damage in the affected regions.21 Creatine kinase-MB and troponin are two biomarkers that are upregulated and are used in the diagnosis and prognosis of myocardial infarction. Cardiac troponin is released by myocytes and has been shown to correlate directly with the size of the infarct.22
Treatment of myocardial infarction involves restoration of perfusion (ie, reperfusion) to recover the damaged myocardium by either pharmacological or mechanical means, such as percutaneous coronary intervention or coronary artery bypass graft. After reperfusion, there is an increase in the expression of chemotactic factors, including complement component 5a, monocyte chemoattractant protein-1, transforming growth factor beta (TGF-β), interleukin (IL)-1, and intercellular adhesion molecule-1. These factors lead to infltration of neutrophils and monocytes to the threatened myocardium, provoking an inflammatory response that initiates repair, fibrosis, and remodeling of the myocardial tissue.23–26 An additional cytokine, IL-8, is involved in neutrophil activation. Inhibition of IL-8 has been shown to decrease necrosis in arabbit model of myocardial ischemia/reperfusion.27
Heart failure
Each year, there are 400,000 new cases of heart failure and approximately 280,000 deaths resulting from the disease, thus representing the leading cause of hospitalization in the United States.1 Heart failure has a diverse range of etiologies, clinical features, and subsets, with its origins lying in several diseases, including diabetes, hypertension, and myocardial infarction.28 Cardiomyopathy is a diseased state of the heart muscle, which interferes with its ability to pump blood in an efficient manner to satisfy the metabolic needs of the body. Heart failure is the final stage of both ischemic and nonischemic cardiomyopathies. The most common cause of heart failure is coronary artery disease.29 During coronary artery disease, blood clots produce ischemic regions that end in extensive muscular damage. Many pathophysiological manifestations that lead to heart failure go unrecognized, and if the damage affects large regions of muscular tissue, the heart’s ability to pump blood is severely compromised.28
The principal characteristics of heart failure are left ventricular dilation (diastolic heart failure) or hypertrophy (systolic heart failure), generally resulting from dysfunction or damage to cardiac myocytes or their molecular components. This leads to neurohormonal and circulatory abnormalities and clinical symptoms such as fluid retention, shortness of breath, and fatigue on exertion.28 As stated previously, cytokines and other inflammatory biomarkers play an important role in the pathogenesis and progression of heart failure. The specific impact of proinfammatory cytokines on cardiac tissue is presented in Table 1. Inflammation has detrimental effects on heart disease, and cytokines have been proposed as targets for clinical intervention. As an example, tumor necrosis factor alpha (TNF-α) is suggested to have a major role in cardiac dysfunction, particularly in ischemic heart disease.30 TNF-α depresses myocardial function and induces apoptosis and necrosis, and also has a role in ventricular remodeling. One of the functional consequences of elevated TNF-a is increased calcium levels in neonatal cardiac myocytes.31,32 Therapeutic reduction of proinflammatory cytokines may therefore minimize cardiac cell damage. Unfortunately, clinical trials involving anti-TNF-α therapy, including Research into Etanercept Cytokine Antagonism in Ventricular Dysfunction (RECOVER), Randomized Etanercept North American Strategy to Study Antagonism of Cytokines (RENAISSANCE), and Anti-TNF Therapy Against Congestive Heart Failure (ATTACH), have yielded modest patient outcomes.33
Table 1.
Cardiac responses to proinfammatory biomarkers
| TGF-β | Stimulates fibroblasts and increases fibrous tissue deposition;102 monocyte chemoattractant24 |
| TNF-α | Induces cellular remodeling, apoptosis, inflammation, and hypertrophy103 |
| IL-1 | Promotes cardiac hypertrophy and myocyte apoptosis and decreases cardiac contractility104 |
| IL-6 | Associated with left ventricular hypertrophy and dysfunction105 |
| IL-17 | Regulates cardiac fibrosis106 |
| IL-18 | Mediates fibrosis and has been found to induce osteopontin107 |
Abbreviations: TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor alpha; IL, interleukin.
Potential cellular targets for therapy include cardiac myocytes, endothelial cells, and myofibroblasts. Therapeutic goals include reducing secretion of proinflammatory cytokines,34–36 reducing formation of extracellular matrix and fibrosis (potentially through downregulation of TGF-β1),37 and restoration of calcium transport to improve muscular function, with recent focus on expression of genes such as cardiac sarcoplasmic reticulum Ca2+ adenosine triphos-phatase a.38
In-stent restenosis
Stenosis is the narrowing of a blood vessel, impeding the flow of blood. In-stent restenosis is a reduction in lumen diameter after stenting due to arterial damage. This mechanical injury creates an infammatory response, which in turn leads to elevated C-reactive protein and plasminogen activator inhibitor type 1; proliferation of vascular smooth muscle cells and extracellular matrix formation; and neointimal thickening.39–41 Gradual renarrowing of the stented segment occurs 3-12 months after stent placement and can occur in 20%–40% of cases, with determinants being age, condition, and lesion complexity42 Often, restenosis appears as a recurrent stable angina, but can also present as an acute myocardial infarction. In-stent restenosis is managed by repeat percutaneous revascularization.
Drug-eluting stents releasing sirolimus or paclitaxel have successfully prevented in-stent restenosis, as opposed to bare-metal stents. Unfortunately, these alternative treatments have failed to demonstrate a benefit over bare-metal stents in overall mortality, due to the increased risk of stent thrombosis.7 Drugs released from drug-eluting stents cause distinct stimuli that affect biological processes at the site of injury, such as activation of signal-transduction pathways and inhibition of proliferation.43 While these drugs prevent vascular smooth-muscle cell proliferation and migration, they also impair, or slow, the reendothelialization process, leading to delayed arterial healing and induced expression of tissue factors that create a prothrombogenic environment.43
Nanomedicine and liposomal technologies: enabling drug delivery
The application of nanotechnology to medicine has led to the development of several innovative treatments, predominately in the realm of cancer therapy. The design and fabrication of nanoscale particles for delivery of therapeutics offers new mechanisms for selective transport to tissues of interest. Site-specifc targeting of tissues may result in higher efficacy and reduced toxicity.44,45
Nearly 50 years have passed since the first description of liposomes by Horne et al.46 Liposomes possess immense versatility with regard to physicochemical properties, allowing adaptation of the particle to ft the specific biological application.47 Liposomes are spherical platforms that use natural lipids and/or synthetic polymers and cholesterol as building blocks to form the bilayer structure. They possess a hydrophilic core surrounded by a hydrophobic membrane,48 and span in size from 50 nm to several microns.47 The introduction of poly(ethylene glycol) (PEG) has been shown to increase the circulation half-life of liposomes, assisting in evasion of phagocytic cells of the reticuloendothelial system (RES).49 With regard to chemistry, inclusion of cationic lipids may be advantageous for specific situations, such as enhanced cellular uptake.50 Liposomes can also be conjugated with proteins or peptides for enhanced targeting to specific tissues, or to induce biological effects.47,49 The liposomal drug Doxil was the first nanotherapeutic drug to be used in the clinic. Originally used in the treatment of Kaposi’s sarcoma, more than a decade after its approval it is still used in the treatment of several cancers, including ovarian and metastatic breast cancer.44,50
Currently, there are no liposomal formulations approved for human use for the treatment of cardiovascular disease. However, attempts have been made to exploit the potential benefits of this approach. In 2003, Hedman et al initiated a clinical trial in which a plasmid encoding vascular endothelial growth factor was delivered by means of liposomes and an adenoviral vector during angioplasty and stenting for the treatment of chronic myocardial ischemia.51 The main objective was to prevent postangioplasty and in-stent restenosis. Gene transfer using liposomes was demonstrated to be feasible and well tolerated; however, the treatment failed to alter the incidence of restenosis. In 2007, another clinical trial to treat in-stent restenosis was carried out using albumin-bound paclitaxel nanoparticles (trade name Abraxane). Treatment was well tolerated at 10 or 30 mg/m2, with no major complications. Additional studies are under way to explore the efficacy of this formulation when delivered by intravenous and intracoronary administration.52,53
The cardiac microenvironment and opportunities for nanomedicine
A healthy heart involves multiple cellular components interacting with one another to maintain electrical, chemical, biomechanical, and structural integrity.54 The majority of cells in the heart are cardiac myocytes (30%–40%) (Figure 1A and B), with fibroblasts, endothelial cells (Figure 1C), and vascular smooth-muscle cells making up the bulk of the remaining cells.54 The heart functions as a giant muscle, with actin–myosin interactions within the sarcomere of the myofbril, generating the force needed for cardiac contractions. A major complication in heart disease is loss of myocardial contractile function and membrane disruption due to loss of calcium homeostasis.55–58 Insufficient contractile force leads to pathological cardiac remodeling, known as cardiomyopathy (dilated or hypertrophic).55 The prevalence of terminally differentiated cardiomyocytes, and the limited presence of stem cells,59 favors therapies for cardiac dysfunction that rely on rescuing injured or susceptible cells.
Figure 1.

(A–C) Scanning electron micrographs show cell populations within the heart. (A) Cross-section of a human cardiac muscle. (B) Cardiac muscle is comprised of bundles of myocytes surrounded by basement membrane. The inset displays a single myocyte, as would be seen beneath the basement membrane. The surrounding sarcolemma contains evenly spaced invaginations, or T-tubules, running longitudinally across the unit. Within the myocyte is an organized array of myoflaments, composed primarily of myosin and actin. (C) A scanning electron micrograph of cardiac tissue excised from the free wall of the left ventricle showing a capillary surrounded by fibrosis and cardiac muscle.
Currently, there are two distinct targeting strategies to enhance nanoparticle accumulation in tumors: passive and active. In passive targeting, nanoparticles of specific size accumulate nonspecifically within the tumor due to the EPR effect. Defects in the endothelial lining and local fenestrations of basement membrane, caused by chronic inflammation, angiogenesis, and hypoxia, increase permeation of nanoparticles into the tumor, with defective lymphatic drainage enhancing retention.60 Active targeting involves conjugation or incorporation of specific targeting ligands on nanoparticles. Ligands include monoclonal antibodies and their fragments, aptamers, peptides, and low-molecular-weight compounds, which selectively bind to biomarkers on target cells. Examples of targeting biomarkers include αVβ3 and αVβ5 integrins, overexpressed on tumor microcapillary endothelial cells.61 The arginine–glycine–aspartic acid (RGD) tripeptide binds to αVβ3 and αVβ5 integrins, providing a ligand frequently used to target nanoparticles to the tumor.61
In cardiovascular disease, each stage of the disease presents novel biomarkers and tissue architecture that can be exploited for targeting, akin to cancer. As an example, myocardial infarction causes blood vessels in the left ventricle to become leaky, potentially favoring nanoparticle penetration into the tissue.62 Cellular injury and inflammation induce activation of platelets, mast cells, monocytes and mac-rophages, and vascular cells, leading to the release of vascular endothelial growth factor, thrombin, and histamine.62 Similar to the tumor microenvironment, these propermeability agents cause paracellular vascular leakage, reducing endothelial cell-to-cell interactions mediated by adhesion molecules and adherens junctions. For example, TNF-α disrupts vascular endothelial cadherin complexes.63 Gaps in the vascular barrier expose the underlying basal lamina and lead to recruitment of platelets and leukocytes.62 Adhesion molecules used to recruit cells can also serve as targets for nanoparticle engagement. However, engagement of specific endothelial cell adhesion molecules also triggers increased permeability. For example, engagement of intercellular adhesion molecule-1 (which is upregulated by TNF-α) induces Src and focal adhesion-related kinase activation, leading to vascular endothelial cadherin phosphorylation64 and reduced cell-to-cell adhesion. Thus, while cell-surface markers present opportunities for targeting, the biological consequences of their engagement need to be considered.
The expression of targetable moieties and the presence of the EPR effect have been observed not only in oncology but also in other diseases that present chronic inflammation, hypoxia, and infection.65 Thus, the concepts of passive and active targeting of therapeutic nanoparticles can be applied efficiently to other types of focal pathological processes and tissues with similar physiological presentations. Cardiovascular disease resembles some of the key vascular pathophysiological characteristics of cancer. For example, microvascular and myocardial cell injury are associated with expression of injury markers,66,67 chronic inflammation is associated with increased microvascular permeability,68 and reactive hyperemia contributes to enhanced delivery of nano-carriers due to increased blood flow to the area of interest.69
Heart-disease manifestations are typically localized to discrete regions of the heart (eg, atherosclerosis, restenosis,and inflammation), making site-specific delivery of therapeutic molecules attractive. Images of the most common presentations of cardiovascular disease and corresponding pathophysiological events are shown in Figure 2.
Figure 2.

(1) Pathophysiological presentations and progression of coronary artery disease. (A) Atherosclerosis plaque buildup in the artery and blood clot occluding blood fow; (B) stent compressing the plaque, with widening of the artery; (C) in-stent restenosis decreasing and occluding the blood flow. (2) Myocardial infarction highlighting a localized area of ischemia and dead heart muscle.
Liposomal applications in cardiovascular disease
Significant advances in the application of liposomal platforms for the treatment of cardiovascular disease have been made. Specific pathophysiological presentations and their exploitation for the delivery of nanoparticle-based therapeutics are presented in Figure 3. The following sections present recent advances in liposomal technology designed to target specific cardiovascular diseases, including myocardial infarction, atherosclerosis, and in-stent restenosis.
Figure 3.
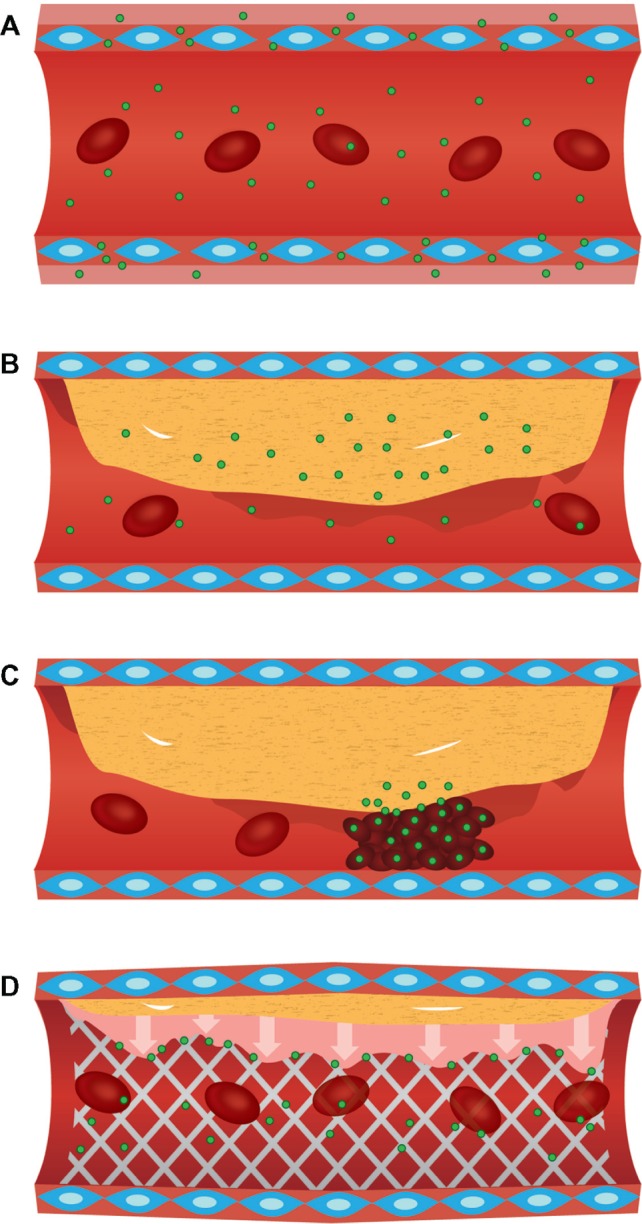
(A) Enhanced extravasation and accumulation of nanoparticles in cardiac regions with increased microvascular endothelial permeability. (B) Accumulation of nanoparticles in metabolically active atherosclerotic plaques, due to increased macrophage recruitment. (C) Nanoparticle delivery to hyperplasic smooth-muscle cells during in-stent restenosis. (D) Platelet-based delivery of nanoparticles to acute pathological events, such as atheroma ruptures.
Vasculature targeting
Benefiting from the abundance of research performed on vascular targeting within the tumor microenvironment,70,71 molecular features associated with the vasculature of patients with cardiovascular disease are already being explored for targeting liposomes to afflicted regions of the cardiovascular system. Heart vasculature expresses a unique array of molecular markers (eg, vascular zip codes) compared to endothelial cells comprising organs. In 2005, Zhang et al discovered five peptides with high affinity and specificity to heart vasculature.72 One peptide with an arginine-rich sequence (CRPPR) displayed a greater than 300-fold selectivity for heart endothelial cells compared with endothelial cells or other organs in a murine model. The cellular target of the CRPPR peptide, identified as cysteine-rich protein 2, is enriched in cell membrane, and although it possesses no discernible membrane-spanning domain, it binds to the submembranous protein tyrosine phosphatase PTP-BL.73 Zhang et al successfully conjugated 18F-CRPPR to a variety of liposomal nanocarriers ranging in size from 71 to 110 nm. Using positron emission tomography imaging, the group successfully targeted healthy cardiac vasculature in a murine model.74
Liposomes with a 6 mol% coating of CRPPR were located in the heart within 100 seconds following intravenous injection. The average radioisotope density in the heart was 44% of the injected dose (per gram of tissue).
Myocardial infarction
In 2012, Zhang et al used the CRPPR peptide to successfully target surface-modified liposomes to coronary endothelia in ischemia/reperfusion and myocardial infarction models.75 The CRPPR-conjugated nanoconstructs achieved a 47-fold increase in accumulation in the surrounding vasculature of injured tissue compared to nontargeted liposomes. CRPPR-conjugated liposomes had a size of 143 ± 12 nm, while the nontargeted liposomes were 116 ± 8 nm. It is also noteworthy that there was less accumulation in the vasculature of healthy tissue.
Dvir et al demonstrated the fabrication of a fluorescent PEGylated liposomal system capable of delivering therapeutics to the infarcted heart.59 The technology relies on both passive and active targeting. Passive targeting was the result of ischemic dysfunctional blood vessels in the left ventricle after myocardial infarction,62 as depicted in Figure 3A. The liposomal vehicle had a mean size of 142 ± 8 nm. Active targeting was based on the integration of ligands in the liposomal carrier targeting the overexpressed angiotensin II type 1 receptor in the heart following myocardial infarction.76 Significantly higher levels of targeted liposomes were found in damaged tissue on days 1, 4, and 7 following injection in a murine model of myocardial infarction compared to nontargeted liposomes.
Cell-based targets for therapy include macrophages that exist at the infarction site. Harel-Adar et al engineered liposomes that present phosphatidylserine (PS) on their surface. PS, a ligand exposed on the outer membrane leafet of apoptotic cells, triggers anti-inflammatory responses in macrophages.77 The liposomes had a diameter of 1.2 ± 0.3 μm and a zeta potential of -98.6 ± 11.3 mV Macrophage engulfment of the PS liposomes resulted in secretion of high levels of anti-inflammatory cytokines (TGF-β and IL-10), upregulation of CD206, and concomitant downregulation of TNF-α and CD86, both in vitro and in vivo. Magnetic resonance imaging or iron oxide-entrapped liposomes supported targeting of PS liposomes to acute myocardial infarction present in a rat model. Similar to macrophage responses to apoptotic cells, the PS liposomes were able to successfully reduce inflammatory responses by macrophages, reducing collateral damage to adjacent healthy tissue after myocardial infarction.
Atherosclerosis
Walton et al78 designed a liposomal platform that attracts activated macrophages in metabolically active atheroma components, as shown in Figure 3B. The liposomes, with an overall negative surface potential, contained rhodamine, nano-gold, and lipoprotein-associated phospholipase A2, the latter an inflammatory biomarker expressed primarily by activated macrophages. The group demonstrated positive targeting of macrophages in lesions in Watanabe heritable hyperlipidemic rabbits. High concentrations of liposomes were found in shoulder regions of plaque located in advanced atheromas. The study also used transmission electron microscopy to confirm accumulation of liposomes within lipid-laden areas of the atheromas based on detection of the nanogold component. This study supports the use of lipoprotein-associated phospholipase A2-modified liposomes for targeting therapeutics to metabolically active regions of plaque.
An immunoliposomal formulation containing fasudil, a potent Rho-kinase inhibitor, and a ligand-decorated surface to target the lectin-like oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1) on carotid plaque lesions was investigated by Saito et al.79 Liposomes were conjugated with an anti-LOX-1 antibody on their surface and tested in a model of carotid intimal hypertrophy induced by balloon injury in rats. On the seventh day of intravenous treatment, targeted liposomes were prominently observed in the lesion, resulting in reduced expression of matrix metalloproteinase-9 and a reduced intimal thickness compared with control groups.
One approach for treating atherosclerosis is to solubilize cholesterol in atherosclerotic plaques. Cho et al80 created liposomes containing phosphatidylcholine (PC) with a goal of enriching high-density lipoprotein (HDL) at the lesion. In the atherosclerotic process, LDL molecules accumulate within the artery walls, where they are oxidized and taken up by foam cells, leading in the progression and formation of atheromas. HDL removes cholesterol from the foam cells, inhibiting oxidation of LDL molecules, thereby limiting the inflammatory process that leads to atherosclerosis.81 Cholesterol-fed rabbits were infused once a week with liposomal formulations. After 5 weeks of treatment, there was a significant reduction in the atherosclerotic plaque volume and decreased cholesterol content in the aortic walls, suggesting that liposomal PC could be an attractive therapy for atherosclerotic plaque regression.
Arterial intimal hyperplasia and in-stent restenosis
Joner et al82 created a novel 100 nm liposomal formulation, TMR-484, consisting of prednisolone. Prednisolone has a high binding affinity for chondroitin sulfate proteoglycans, enriched at the site of injury, illustrated in Figure 3C. When tested in a stented rabbit model of atheroma, there was a 100-fold increase in the concentration of liposomes at the lesion compared with nonstented arteries 24 hours after the administration. There was a significant reduction in the percentage of stenosis compared with control groups, and adverse side effects were reduced as compared to the free drug. The cost-effective use of this therapy for the prevention of in-stent restenosis remains to be evaluated.
Other approaches for the treatment of in-stent restenosis have been explored, including positively charged echogenic liposomes loaded with argon and nitric oxide. Huang et al83 evaluated the synthetic dimyristoylphosphatidylcholine liposomal formulation of this antiproliferative agent in vitro using smooth-muscle cells and found a sevenfold higher uptake of nitric oxide-argon liposomes compared to nonencapsulated nitric oxide. Using an in vivo model of hyperplasia based on balloon-injured carotid arteries in rabbits, the liposomes promoted regression of lesions in cholesterol-fed rabbits, demonstrating attenuated hyperplasic development of smooth-muscle cells and reduced arterial wall thickening by 41%.
Activated-platelet adhesion and aggregation therapy
The processes of platelet activation, adhesion, and aggregation in injured sites are primary events in the cascade of diseases such as myocardial infarction, atherosclerosis, thrombosis, and restenosis.84,85 Thus, platelet targeting is a promising therapeutic approach in acute cardiovascular disease, as illustrated in Figure 3D. Lestini et al86 independently incorporated oligodextran surfactants and RGD into liposomes with the goals of reducing RES uptake and achieving targeted delivery, respectively. Oligosaccharide surfactants mimic the cell glycocalyx, which has limited nonspecific protein adsorption (opsonization), leading to reduced RES uptake.87 RGD targets integrin GPIIb-IIIa on activated platelets, and was found to cause greater platelet labeling compared to control liposomes. Their study supported the camoufage of liposomes to reduce opsonization and RES clearance and the use of peptides for guiding liposomes to targeted receptors. Oligosaccharide-modified liposomes ranged in size from 104 to 190 nm, with increases in vesicle size correlating with increases in clearance rate.
Srinivasan et al88 also demonstrated enhanced liposome targeting of platelets using RGD peptide-labeled liposomes (150 nm) in a model of restenosis in rats induced by catheter-mediated injury of the carotid artery. Scanning electron microscopy confirmed association of the targeted liposomes to activated human platelets, suggesting that this platform may be useful for treating certain types of cardiovascular disease.
Recent work by Zhu et al89 demonstrated that negatively charged glycoliposomes, with vesicle size of 113 nm, successfully target P-selectin on activated platelets. The delivery system was fabricated to mimic the structure of P-selectin glycoprotein ligand 1 on activated leukocytes, in order to facilitate the specific binding of targeted liposomes to the P-selectin receptor on activated platelets. The model was tested in vitro with fluorescent microscopy techniques, supporting liposome binding to activated platelets.
Biodistribution and cellular uptake of liposomes
The biodistribution of liposomes in vivo is influenced by mechanical filtration, serum proteins, and the physicochemical properties of liposomes. Opsonization, which increases with increasing particle size,90 alters uptake by specifc cell populations, with complement and immunoglobulin increasing binding and uptake by macrophages.91 Researchers using liposomal platforms to decrease drug accumulation in the heart have shown that drug delivery using positive liposomes reduces in vivo drug uptake in normal murine cardiac tissue compared to negative liposomes and free drug.92 However, cationic liposomes are preferentially taken up by angiogenic vessels in tumors and at sites of chronic infammation,93,94 indicating that their biodistribution in diseased cardiovascular tissue may differ from that of normal tissue. The endocytic capacity of cells within the heart and kidney has been described as low compared to liver, spleen, and certain tumors, with macrophages in the liver and spleen being the main source of liposome uptake.95,96 Changes in the cellular milieu, as well as altered properties of resident cells at sites of pathology within the cardiovascular system, are likely to alter liposomal accumulation. Both cationic and anionic liposomal platforms have demonstrated success in targeting, with the presence of targeting ligands having the greatest impact on cardiac accumulation at sites of pathology.
The disadvantage of using liposomes is the same as applies to all categories of nanoparticles: high levels of accumulation within the reticular endothelial organs. However, the lack of inherent toxicity of liposomes and its biodegradation offers significant benefits over other types of nanoparticles, specifically ultrafine and metallic nanoparticles, shown to have cardiac toxicity due to enhanced inflammation, oxidative stress, altered vasoconstriction and vasorelaxation, acute endothelial disruption, and changes in autonomic outfow.97101
Conclusion
While still in their infancy, liposomal-based strategies to treat cardiovascular disease have demonstrated successful targeting and accumulation at pathological sites. This is primarily due to several pathophysiological phenomena present during cardiovascular disease that aid in nanoparticle accumulation via active and passive targeting. In this regard cardiovascular disease shares several characteristics with cancer, such as the presence of vessel fenestrations and overexpressed receptors. The innovative platforms presented herein show promise in exploiting several of these characteristics. With further increases in our understanding of the microenvironment and pathophysiology of cardiovascular disease, the creation of novel platforms for the treatment of diseases, such as heart failure and atherosclerosis, will continue to mature.
Key points
• Cardiovascular disease is one of the leading causes of morbidity and mortality.
• The anatomical/pathological characteristics and expression of molecules, such as integrins and cell adhesion molecules, during different cardiovascular conditions provide opportunities for the development of site-specific drug-delivery platforms.
• Nanomedicine provides tools to package drugs and biomolecules and to target select sites, with the potential for increased therapeutic efficacy, reduced systemic toxicity, and high versatility.
• Liposomes have evolved as robust multifunctional platforms for drug delivery, with several innovative examples currently being examined in preclinical models.
Acknowledgment
MF is grateful for support from Ernest Cockrell Jr in the form of a Distinguished Chair Endowment.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics - 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoyert D, XU J. Deaths: Preliminary Data for 2011. National Vital Statistics Reports Hyattsville, MD. National Center for Health Statistics. 2012;61:1–65. [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 4.Lewington S, Whitlock G, Clarke R, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 5.Law M, Wald N, Morris J. Lowering blood pressure to prevent myocardial infarction and stroke: a new preventive strategy. Health Technol Assess. 2003;7:1–94. doi: 10.3310/hta7310. [DOI] [PubMed] [Google Scholar]
- 6.McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 7.Stettler C, Wandel S, Allemann S, et al. Outcomes associated with drugeluting and bare-metal stents: a collaborative network meta-analysis. Lancet. 2007;370:937–948. doi: 10.1016/S0140-6736(07)61444-5. [DOI] [PubMed] [Google Scholar]
- 8.Michor F, Liphardt J, Ferrari M, Widom J. What does physics have to do with cancer? Nat Rev Cancer. 2011;11:657–670. doi: 10.1038/nrc3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 10.Blanco E, Hsiao A, Mann A P, Landry MG, Meric-Bernstam F, Ferrari M. Nanomedicine in cancer therapy: innovative trends and prospects. Cancer Sci. 2011;102:1247–1252. doi: 10.1111/j.1349-7006.2011.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamilton A, Biganzoli L, Coleman R, et al. EORTC 10968: a phase I clinical and pharmacokinetic study of polyethylene glycol liposomal doxorubicin (Caelyx, Doxil) at a 6-week interval in patients with metastatic breast cancer. European Organization for Research and Treatment of Cancer. Ann Oncol. 2002;13:910–918. doi: 10.1093/annonc/mdf157. [DOI] [PubMed] [Google Scholar]
- 12.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 13.Holgado MA, Martin-Banderas L, Alvarez-Fuentes J, Fernandez-Arevalo M, Arias JL. Drug targeting to cancer by nanoparticles surface functionalized with special biomolecules. Curr Med Chem. 2012;19:3188–3195. doi: 10.2174/092986712800784720. [DOI] [PubMed] [Google Scholar]
- 14.Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000;101:329–335. doi: 10.1161/01.cir.101.3.329. [DOI] [PubMed] [Google Scholar]
- 15.Sever PS, Poulter NR. A hypothesis for the pathogenesis of essential hypertension: the initiating factors. J Hypertens Suppl. 1989;7:S9–S12. doi: 10.1097/00004872-198902001-00004. [DOI] [PubMed] [Google Scholar]
- 16.Skalen K, Gustafsson M, Rydberg EK, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 17.Leitinger N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr Opin Lipidol. 2003;14:421–430. doi: 10.1097/00041433-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Massberg S, Brand K, Gruner S. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 20.Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colonystimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A. 1995;92:8264–8268. doi: 10.1073/pnas.92.18.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horie T, Sekiguchi M, Hirosawa K. Coronary thrombosis in pathogenesis of acute myocardial infarction. Histopathological study of coronary arteries in 108 necropsied cases using serial section. Br Heart J. 1978;40:153–161. doi: 10.1136/hrt.40.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antman EM, Tanasijevic MJ, Thompson B, et al. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med. 1996;335:1342–1349. doi: 10.1056/NEJM199610313351802. [DOI] [PubMed] [Google Scholar]
- 23.Dreyer WJ, Michael LH, Nguyen T, et al. Kinetics of C5a release in cardiac lymph of dogs experiencing coronary artery ischemiareperfusion injury. Circ Res. 1992;71:1518–1524. doi: 10.1161/01.res.71.6.1518. [DOI] [PubMed] [Google Scholar]
- 24.Birdsall HH, Green DM, Trial J, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95:684–692. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins HK, Entman ML, Zhu JY, et al. Acute inflammatory reaction after myocardial ischemic injury and reperfusion. Development and use of a neutrophil-specific antibody. Am J Pathol. 1996;148:1957–1969. [PMC free article] [PubMed] [Google Scholar]
- 26.Youker KA, Hawkins HK, Kukielka GL. Molecular evidence for induction of intracellular adhesion molecule-1 in the viable border zone associated with ischemia-reperfusion injury of the dog heart. Circulation. 1994;89:2736–2746. doi: 10.1161/01.cir.89.6.2736. [DOI] [PubMed] [Google Scholar]
- 27.Boyle EM, Jr, Kovacich JC, Hebert CA, et al. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 1998;116:114–121. doi: 10.1016/S0022-5223(98)70249-1. [DOI] [PubMed] [Google Scholar]
- 28.Lindenfeld J, Albert NM, Boehmer J P. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010;16:e1–e194. doi: 10.1016/j.cardfail.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Fox K F, Cowie MR, Wood DA. Coronary artery disease as the cause of incident heart failure in the population. Eur Heart J. 2001;22:228–236. doi: 10.1053/euhj.2000.2289. [DOI] [PubMed] [Google Scholar]
- 30.Shames BD, Barton HH, Reznikov LL, et al. Ischemia alone is sufficient to induce TNF-alpha mRNA and peptide in the myocardium. Shock. 2002;17:114–119. doi: 10.1097/00024382-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Bick RJ, Liao J P, King T W, LeMaistre A, McMillin JB, Buja LM. Temporal effects of cytokines on neonatal cardiac myocyte Ca2+ transients and adenylate cyclase activity. The Am J Physiol. 1997;272:H1937–H1944. doi: 10.1152/ajpheart.1997.272.4.H1937. [DOI] [PubMed] [Google Scholar]
- 32.Bick RJ, Wood DE, Poindexter B, et al. Cytokines increase neonatal cardiac myocyte calcium concentrations: the involvement of nitric oxide and cyclic nucleotides. J Interferon Cytokine Res. 1999;19:645–653. doi: 10.1089/107999099313794. [DOI] [PubMed] [Google Scholar]
- 33.Coletta A P, Clark AL, Banarjee P, Cleland JG. Clinical trials update: RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail. 2002;4:559–561. doi: 10.1016/s1388-9842(02)00121-6. [DOI] [PubMed] [Google Scholar]
- 34.Torre-Amione G, Kapadia S, Lee J, Durand JB, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–711. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]
- 35.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–1153. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 36.Flores-Arredondo JH, Garcia-Rivas G, Torre-Amione G. Immune modulation in heart failure: past challenges and future hopes. Curr Heart Fail Rep. 2011;8:28–37. doi: 10.1007/s11897-010-0044-2. [DOI] [PubMed] [Google Scholar]
- 37.Lijnen PJ, Petrov V V, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-beta (1) Mol Genet Metab. 2000;71:418–435. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 38.Kawase Y, Ly HQ, Prunier F, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 39.Ferrante G, Niccoli G, Biasucci LM, et al. Association between C-reactive protein and angiographic restenosis after bare metal stents: an updated and comprehensive meta-analysis of 2747 patients. Cardiovasc Revasc Med. 2008;9:156–165. doi: 10.1016/j.carrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Christ G, Nikfardjam M, Huber-Beckmann R, et al. Predictive value of plasma plasminogen activator inhibitor-1 for coronary restenosis: dependence on stent implantation and antithrombotic medication. J Thromb Haemost. 2005;3:233–239. doi: 10.1111/j.1538-7836.2004.01062.x. [DOI] [PubMed] [Google Scholar]
- 41.Shah PK. Inflammation, neointimal hyperplasia, and restenosis: as the leukocytes roll, the arteries thicken. Circulation. 2003;107:2175–2177. doi: 10.1161/01.CIR.0000069943.41206.BD. [DOI] [PubMed] [Google Scholar]
- 42.Höher M, Hombach V, Kochs M, Eggeling T, Haerer W. Angioplasty using high-frequency energy in coronary stenosis. Herz. 1990;15:245–252. German. [PubMed] [Google Scholar]
- 43.Lüscher TF, Steffel J, Eberli FR, et al. Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation. 2007;115:1051–1058. doi: 10.1161/CIRCULATIONAHA.106.675934. [DOI] [PubMed] [Google Scholar]
- 44.Lammers T, Kiessling F, Hennink WE, Storm G. Nanotheranostics and image-guided drug delivery: current concepts and future directions. Mol Pharm. 2010;7:1899–1912. doi: 10.1021/mp100228v. [DOI] [PubMed] [Google Scholar]
- 45.Barenholz Y. Doxil(R)-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Horne RW, Bangham AD, Whittaker V P. Negatively Stained Lipoprotein Membranes. Nature. 1963;200:1340. doi: 10.1038/2001340a0. [DOI] [PubMed] [Google Scholar]
- 47.Mufamadi MS, Pillay V, Choonara YE, et al. A review on composite liposomal technologies for specialized drug delivery. J Drug Deliv. 2011;2011:939851. doi: 10.1155/2011/939851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maurer N, Fenske DB, Cullis PR. Developments in liposomal drug delivery systems. Expert Opin Biol Ther. 2001:923–947. doi: 10.1517/14712598.1.6.923. [DOI] [PubMed] [Google Scholar]
- 49.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 50.Torchilin V P. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 51.Hedman M, Hartikainen J, Syvanne M. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT) Circulation. 2003:2677–2683. doi: 10.1161/01.CIR.0000070540.80780.92. [DOI] [PubMed] [Google Scholar]
- 52.Margolis J, McDonald J, Heuser R, et al. Systemic nanoparticle paclitaxel (nab-paclitaxel) for in-stent restenosis I (SNAPIST-I): a first-in-human safety and dose-finding study. Clin Cardiol. 2007;30:165–170. doi: 10.1002/clc.20066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDowell G, Slevin M, Krupinski J. Nanotechnology for the treatment of coronary in stent restenosis: a clinical perspective. Vasc Cell. 2011;3:8. doi: 10.1186/2045-824X-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 55.Fatkin D, McConnell BK, Mudd JO. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Semsarian C, Ahmad I, Giewat M, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boixel C, Gonzalez W, Louedec L, Hatem SN. Mechanisms of L-type Ca(2+) current downregulation in rat atrial myocytes during heart failure. Circ Res. 2001;89:607–613. doi: 10.1161/hh1901.096702. [DOI] [PubMed] [Google Scholar]
- 58.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 59.Dvir T, Bauer M, Schroeder A, et al. Nanoparticles targeting the infarcted heart. Nano Lett. 2011;11:4411–4444. doi: 10.1021/nl2025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ali I, Rahis U, Salim K, Rather MA, Wani WA, Haque A. Advances in nano drugs for cancer chemotherapy. Curr Cancer Drug Targets. 2011;11:135–146. doi: 10.2174/156800911794328493. [DOI] [PubMed] [Google Scholar]
- 61.Vermorken JB, Guigay J, Mesia R, et al. Phase I/II trial of cilengitide with cetuximab, cisplatin and 5-fuorouracil in recurrent and/or metastatic squamous cell cancer of the head and neck: findings of the phase I part. Br J Cancer. 2011;104:1691–1696. doi: 10.1038/bjc.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol. 2008;15:243–249. doi: 10.1097/MOH.0b013e3282f97d86. [DOI] [PubMed] [Google Scholar]
- 63.Menon C, Ghartey A, Canter R, Feldman M, Fraker DL. Tumor necrosis factor-alpha damages tumor blood vessel integrity by targeting VE-cadherin. Annals of surgery. 2006;244:781–791. doi: 10.1097/01.sla.0000231723.81218.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 65.Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuliresponsive nanocarriers for drug and gene delivery. J Control Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 66.Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72:561–566. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parish RC, Evans JD. Infammation in Chronic Heart Failure. Ann Pharmacother. 2008;42:1002–1016. doi: 10.1345/aph.1K272. [DOI] [PubMed] [Google Scholar]
- 68.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 69.Viau S, Fontaine É, Véronneau M, Jasmin G, Dumont L. Myocardial reactive hyperemia in experimental chronic heart failure: Evidence for the role of K+ adenosine triphosphate-dependent channels and cyclooxygenase activity. J Card Fail. 1997;3:207–215. doi: 10.1016/s1071-9164(97)90017-8. [DOI] [PubMed] [Google Scholar]
- 70.Hu Z, Sun Y, Garen A. Targeting tumor vasculature endothelial cells and tumor cells for immunotherapy of human melanoma in a mouse xenograft model. Proc Natl Acad Sci. 1999;96:8161–8166. doi: 10.1073/pnas.96.14.8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans. 2004;32:397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 72.Zhang L, Hoffman JA, Ruoslahti E. Molecular profling of heart endothelial cells. Circulation. 2005;112:1601–1611. doi: 10.1161/CIRCULATIONAHA.104.529537. [DOI] [PubMed] [Google Scholar]
- 73.van Ham M, Croes H, Schepens J, Fransen J, Wieringa B, Hendriks W. Cloning and characterization of mCRIP2, a mouse LIM-only protein that interacts with PDZ domain IV of PTP-BL. Genes Cells. 2003;8:631–644. doi: 10.1046/j.1365-2443.2003.00660.x. [DOI] [PubMed] [Google Scholar]
- 74.Zhang H, Kusunose J, Kheirolomoom A, et al. Dynamic imaging of arginine-rich heart-targeted vehicles in a mouse model. Biomaterials. 2008;29:1976–1988. doi: 10.1016/j.biomaterials.2007.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Li N, Sirish P. The cargo of CRPPR-conjugated liposomes crosses the intact murine cardiac endothelium. J Control Release. 2012;163:10–17. doi: 10.1016/j.jconrel.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Molavi B, Chen J, Mehta JL. Cardioprotective effects of rosiglitazone are associated with selective overexpression of type 2 angiotensin receptors and inhibition of p42/44 MAPK. Am J Physiol Heart Circ Physiol. 2006;291:H687–H693. doi: 10.1152/ajpheart.00926.2005. [DOI] [PubMed] [Google Scholar]
- 77.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci U S A. 2011;108:1827–1832. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walton BL, Leja M, Vickers KC, et al. Delivery of negatively charged liposomes into the atheromas of Watanabe heritable hyperlipidemic rabbits. Vasc Med. 2010;15:307–313. doi: 10.1177/1358863X10374118. [DOI] [PubMed] [Google Scholar]
- 79.Saito A, Shimizu H, Doi Y. Immunoliposomal drug-delivery system targeting lectin-like oxidized low-density lipoprotein receptor-1 for carotid plaque lesions in rats. J Neurosurg. 2011;115:720–727. doi: 10.3171/2011.5.JNS10227. [DOI] [PubMed] [Google Scholar]
- 80.Cho BH, Park JR, Nakamura MT, Odintsov BM, Wallig MA, Chung BH. Synthetic dimyristoylphosphatidylcholine liposomes assimilating into high-density lipoprotein promote regression of atherosclerotic lesions in cholesterol-fed rabbits. Exp Biol Med (Maywood) 2010;235:1194–1203. doi: 10.1258/ebm.2010.009320. [DOI] [PubMed] [Google Scholar]
- 81.Barter P. The role of HDL-cholesterol in preventing atherosclerotic disease. Eur Heart J Suppl. 2005;7:F4–F8. [Google Scholar]
- 82.Joner M, Morimoto K, Kasukawa H, et al. Site-specifc targeting of nanoparticle prednisolone reduces in-stent restenosis in a rabbit model of established atheroma. Arterioscler Thromb Vasc Biol. 2008;28:1960–1966. doi: 10.1161/ATVBAHA.108.170662. [DOI] [PubMed] [Google Scholar]
- 83.Huang SL, Kee PH, Kim H, et al. Nitric oxide-loaded echogenic liposomes for nitric oxide delivery and inhibition of intimal hyperplasia. J Am Coll Cardiol. 2009;54:652–659. doi: 10.1016/j.jacc.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–453. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 85.Becker RC. Thrombosis and the role of the platelet. Am J Cardiol. 1999;83:3E–6E. doi: 10.1016/s0002-9149(99)00083-1. [DOI] [PubMed] [Google Scholar]
- 86.Lestini BJ, Sagnella SM, Xu Z, et al. Surface modifcation of liposomes for selective cell targeting in cardiovascular drug delivery. J Control Release. 2002;78:235–247. doi: 10.1016/s0168-3659(01)00505-3. [DOI] [PubMed] [Google Scholar]
- 87.Holland NB, Qiu Y, Ruegsegger M, Marchant RE. Biomimetic engineering of non-adhesive glycocalyx-like surfaces using oligosaccharide surfactant polymers. Nature. 1998;392:799–801. doi: 10.1038/33894. [DOI] [PubMed] [Google Scholar]
- 88.Srinivasan R, Marchant RE, Gupta AS. In vitro and in vivo platelet targeting by cyclic RGD-modifed liposomes. J Biomed Mater Res A. 2010;93:1004–1015. doi: 10.1002/jbm.a.32549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu J, Xue J, Guo Z, Zhang L, Marchant RE. Biomimetic glycoliposomes as nanocarriers for targeting P-selectin on activated platelets. Bioconjug Chem. 2007;18:1366–1369. doi: 10.1021/bc700212b. [DOI] [PubMed] [Google Scholar]
- 90.Liu D, Liu F, Song YK. Recognition and clearance of liposomes containing phosphatidylserine are mediated by serum opsonin. Biochim Biophys Acta. 1995;1235:140–146. doi: 10.1016/0005-2736(95)00005-n. [DOI] [PubMed] [Google Scholar]
- 91.Reid KB. Activation and control of the complement system. Essays Biochem. 1986;22:27–68. [PubMed] [Google Scholar]
- 92.Rahman A, Kessler A, More N, et al. Liposomal protection of adriamycin-induced cardiotoxicity in mice. Cancer Res. 1980;40:1532–1537. [PubMed] [Google Scholar]
- 93.Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561–567. doi: 10.1002/ijc.11108. [DOI] [PubMed] [Google Scholar]
- 94.Thurston G, McLean J W, Rizen M, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic infammation in mice. J Clin Invest. 1998;101:1401–1413. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gregoriadis G, Wills EJ, Swain C P, Tavill AS. Drug-carrier potential of liposomes in cancer chemotherapy. Lancet. 1974;1:1313–1316. doi: 10.1016/s0140-6736(74)90682-5. [DOI] [PubMed] [Google Scholar]
- 96.Segal AW, Wills EJ, Richmond JE, Slavin G, Black CD, Gregoriadis G. Morphological observations on the cellular and subcellular destination of intravenously administered liposomes. Br J Exp Pathol. 1974;55:320–327. [PMC free article] [PubMed] [Google Scholar]
- 97.Mann EE, Thompson LC, Shannahan JH, Wingard CJ. Changes in cardiopulmonary function induced by nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4:691–702. doi: 10.1002/wnan.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cuevas AK, Liberda EN, Gillespie PA, Allina J, Chen LC. Inhaled nickel nanoparticles alter vascular reactivity in C57BL/6 mice. Inhal Toxicol. 2010;22(Suppl 2):100–106. doi: 10.3109/08958378.2010.521206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Legramante JM, Valentini F, Magrini A, et al. Cardiac autonomic regulation after lung exposure to carbon nanotubes. Hum Exp Toxicol. 2009;28:369–375. doi: 10.1177/0960327109105150. [DOI] [PubMed] [Google Scholar]
- 100.Pacurari M, Qian Y, Fu W, et al. Cell permeability, migration, and reactive oxygen species induced by multiwalled carbon nanotubes in human microvascular endothelial cells. J Toxicol Environ Health A. 2012;75:112–128. doi: 10.1080/15287394.2011.615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu L, Chen B, Teng F, et al. Effect of Fe(3)O(4)-magnetic nanoparticles on acute exercise enhanced KCNQ(1) expression in mouse cardiac muscle. Int J Nanomedicine. 2010;5:109–116. [PMC free article] [PubMed] [Google Scholar]
- 102.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ueland T, Yndestad A, Dahl C P, Gullestad L, Aukrust P. TNF revisited: osteoprotegerin and TNF-related molecules in heart failure. Curr Heart Fail Rep. 2012;9:92–100. doi: 10.1007/s11897-012-0088-6. [DOI] [PubMed] [Google Scholar]
- 104.Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz) 2009;57:165–176. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lai NC, Gao MH, Tang E, et al. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab Invest. 2012;92(11):1518–1526. doi: 10.1038/labinvest.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feng W, Li W, Liu W, Wang F, Li Y, Ya n W. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol Pathol. 2009;87:212–218. doi: 10.1016/j.yexmp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 107.Yu Q, Vazquez R, Khojeini E V, Patel C, Venkataramani R, Larson D F. IL-18 induction of osteopontin mediates cardiac fbrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol. 2009;297:H76–H85. doi: 10.1152/ajpheart.01285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]