

Abstract
The protein arginine deiminases (PADs) are known to play a crucial role in the onset and progression of multiple inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, and cancer. However, it is not known how each of the five PAD isozymes contributes to disease pathogenesis. As such, potent, selective, and bioavailable PAD inhibitors will be useful chemical probes to elucidate the specific roles of each isozyme. Because d-amino amino acids often possess enhanced in cellulo stability, and perhaps unique selectivities, we synthesized a series of d-amino acid analogues of our pan-PAD inhibitor Cl-amidine, hypothesizing that this change would provide inhibitors with enhanced pharmacokinetic properties. Herein, we demonstrate that d-Cl-amidine and d-o-F-amidine are potent and highly selective inhibitors of PAD1. The pharmacokinetic properties of d-Cl-amidine were moderately improved over those of l-Cl-amidine, and this compound exhibited similar cell killing in a PAD1 expressing, triple-negative MDA-MB-231 breast cancer cell line. These inhibitors represent an important step in our efforts to develop stable, bioavailable, and highly selective inhibitors for all of the PAD isozymes.
Keywords: d-amino acid, protein arginine deiminases, in cellulo efficacy, pharmacokinetic properties
Proteins undergo a host of post-translational modifications, including acetylation, phosphorylation, methylation, ubiquitination, and citrullination.1 Catalyzed by the protein arginine deiminase (PAD) family of enzymes, citrullination or deimination is the hydrolysis of peptidyl-arginine to peptidyl-citrulline. There are five PAD isozymes in humans and other mammals (PAD 1–4 and 6), and all five PADs are calcium-dependent enzymes.2,2b The PADs are distributed throughout several tissue types, and while all of the isozymes are cytoplasmic, only PAD2 and PAD4 are expressed in both the cytoplasm and the nucleus,2−3d where they alter gene transcription via their ability to citrullinate histones H3 and H4.4 The PADs are particularly intriguing given their role in a host of autoimmune and inflammatory diseases, including rheumatoid arthritis (RA), ulcerative colitis, cancer, and Alzheimer's disease, among others.2,2b
We and others have developed a number of halo-acetamidine-based PAD inhibitors;5−5g the halo-acetamidine warhead covalently modifies the active site cysteine in the PADs to irreversibly inhibit enzyme activity. Of the first generation PAD inhibitors, Cl-amidine has proven most useful.5e This pan-PAD inhibitor reduces disease severity in the collagen-induced arthritis (CIA) model of RA,6 as well as in a mouse model of ulcerative colitis.7 Furthermore, Cl-amidine was used to confirm that PAD4 plays an essential role in neutrophil extracellular trap (NET) formation; Cl-amidine prevents histone citrullination, chromatin decondensation, and subsequent NET formation.8,8b Further testing and characterization of these inhibitors led to the identification of a PAD3 selective inhibitor, Cl-4-amidine.5c With the goal of producing more potent inhibitors, second generation Cl-amidine analogues were synthesized, using the principles of rational design and structure–activity relationships. This work determined that incorporation of an ortho-carboxylate on the benzoyl moiety of Cl-amidine dramatically increased the potency (by up to 65-fold) of these inhibitors.5
Although these compounds are potent PAD inhibitors, their l-amino acid-based structures leave them potentially susceptible to proteolysis.9 For this reason, they have relatively short in vivo half-lives and lose cellular potency quite rapidly. Given the precedent of utilizing d-amino acids in peptide-based compounds to improve stability,9−10c we hypothesized that next generation Cl-amidine analogues incorporating d-amino acids would produce PAD inhibitors with improved stability and unique selectivities. To this end, a series of eight d-amino acid-based Cl-amidine analogues were synthesized and tested against PADs 1–4 to characterize their potency and selectivity. The best inhibitors from this series are d-Cl-amidine and d-o-F-amidine, as they exhibit enhanced selectivity for PAD1 over PADs 2–4 and improved in cellulo efficacy in MDA-MB-231 cells and show a moderate increase in stability. Herein, we also report for Cl-amidine, d-Cl-amidine, and d-o-F-amidine the maximum tolerable dose (MTD) and the results of in cellulo and pharmacokinetic studies.
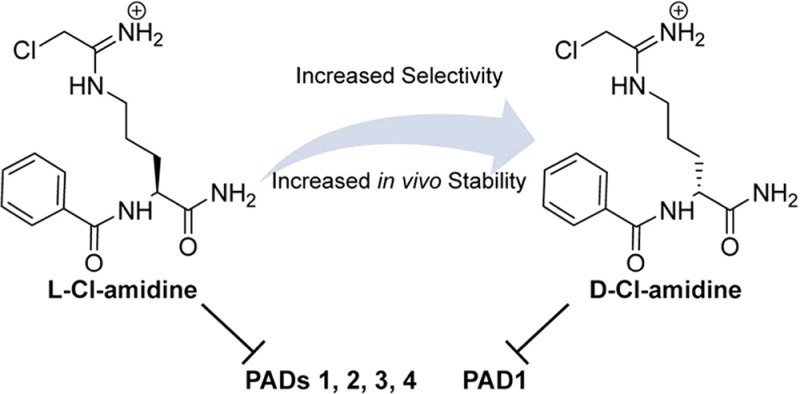
Given that F-amidine, Cl-amidine, o-F-amidine, and o-Cl-amidine have been the most useful PAD inhibitors,5,5e we initially synthesized their direct derivatives using d-ornithine, as opposed to l-ornithine, as the starting material (Figure 1 and Scheme S1 in the Supporting Information). These compounds are designated d-F-amidine, d-Cl-amidine, d-o-F-amidine, and d-o-Cl-amidine; the “d” indicates the altered stereochemistry. To evaluate their in vitro potency, kinact/KI values were determined for each compound with all four active PAD isozymes, that is, PADs 1–4 (Figure 2). Note that PAD6 was not tested because it shows no activity in vitro. On the basis of this analysis, d-Cl-amidine and d-o-F-amidine are the most potent and selective compounds from this initial series. For example, these compounds preferentially inhibit PAD1, by ≥10-fold, with kinact/KI values of 13500 and 12100 M–1 min–1, respectively (Figure 2). Given the altered stereochemistry and preference for PAD1, we used dialysis experiments and competitive activity-based protein profiling (ABBP) to confirm that they irreversibly inhibit this isozyme (Figures S1 and S2 in the Supporting Information, respectively). No enzymatic activity was observed postdialysis, and the compounds effectively competed labeling by rhodamine-conjugated Cl-amidine (RCA), an active site-directed ABPP. It should also be noted that d-Cl-amidine and d-o-F-amidine exhibited sigmoidal inhibition curves when the inhibition experiments were performed with PAD1 (Figure S3 in the Supporting Information). Although this type of inhibition has not been observed for any other previously reported PAD inhibitor, the PADs are known to dimerize,11 and it is possible that these compounds induce intrasubunit cooperativity, via a conformational change upon inhibition of one subunit of the dimer, that makes the second subunit more susceptible to inhibition. Further studies are required to confirm this hypothesis.
Figure 1.

Structures of d-series compounds.
Figure 2.
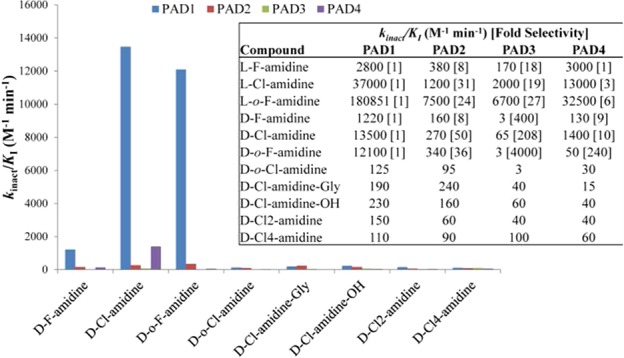
Inhibitor selectivity. kinact/KI values, determined with purified enzyme, are displayed graphically and in table format (inset) for the d-series compounds, as well as certain l-amino acid analogues, indicating potency and selectivity for PADs 1–4.
Although d-Cl-amidine and d-o-F-amidine are generally less potent than their l-counterparts, the d-series compounds exhibit enhanced PAD1 selectivity versus their l-amino acid counterparts (Figure 2). For example, l-Cl-amidine is only 3-fold selective for PAD1 over PAD4, whereas d-Cl-amidine shows roughly 10-fold selectivity between these two isozymes. The increased selectivity for PAD1 is not restricted to PAD4, as the inversion of stereochemistry also boosts selectivity against PADs 2 and 3 to 50- and 200-fold, respectively. Similar selectivity trends are observed with d-F-amidine, albeit with reduced potency, consistent with the data for l-F-amidine; the kinact/KI for PAD1 is only 1220 M–1 min–1. Interestingly, d-o-Cl-amidine and d-o-F-amidine displayed the opposite potency trends, with d-o-Cl-amidine showing limited inhibition toward any of the PADs, whereas d-o-F-amidine was the second most potent PAD1 inhibitor tested among the d-analogues. Even though chloride is a better leaving group than fluoride, this result is not unprecedented (e.g., Thr-Asp-F-amidine shows higher potency and selectivity than Thr-Asp-Cl-amidine)5b and likely reflects the steric constraints of the active site favoring the smaller leaving group, as a result of the inversion of stereochemistry. More significantly, d-o-F-amidine preferentially inhibits PAD1 by 35-, 3900-, and 225-fold versus PADs 2, 3, and 4, respectively, making it the most selective PAD1 inhibitor ever discovered (Figure 2).
Previous studies demonstrated that the linker length between the peptide backbone and the halo-acetamidine warhead lends inhibitors unique selectivity.5c As such, the d-amino acid versions of Cl2-amidine (two methylene linker) and Cl4-amidine (four methylene linker) were also synthesized (Figure 1). However, these compounds were quite poor inhibitors (kinact/KI < 150 M–1 min–1) for all of the PADs tested (Figure 2). This stresses the importance of linker length in the design of PAD inhibitors, that is, the distance between the halo-acetamidine warhead, which reacts with the active site cysteine, and the peptide backbone is critical for potent inhibition. Given that the carboxylate moiety of l-o-F-amidine and l-o-Cl-amidine interacts favorably with Trp347 in PAD45 and that inversion of the stereocenter for the d-amino acid inhibitors may flip the orientation of the inhibitor in the enzyme active site, we hypothesized that a negative charge on the C terminus of the d-amino acid inhibitors may increase the potency of these inhibitors. For this reason, d-Cl-amidine-OH and d-Cl-amidine-Gly were synthesized (Figure 1). However, these compounds were also poor PAD inhibitors (Figure 2; kinact/KI < 230 M–1 min–1). Although this result could suggest that the inhibitors do not bind to the enzyme in the opposite orientation, relative to their l-amino acid counterparts, structural studies will be necessary to confirm their true binding modes. While d-Cl-amidine-OH turned out to be a rather poor inhibitor for all of the PADs, it is interesting to note that for PAD1 the amide isostere is 60-fold more potent. Thus, if hydrolysis of the C-terminal amide occurs at an appreciable amount during the initial breakdown of these compounds, the hydrolysis of this bond in vivo would result in a significant loss of potency, with consequent effects on in vivo efficacy. The fact that L-Cl-amidine-OH is also a poor PAD inhibitor (Figure S4 in the Supporting Information) suggests that a similar breakdown pathway would decrease the in vivo efficacy of Cl-amidine. Given that core structure of L-Cl-amidine is similar to benzoyl-arginine amide, a classical substrate for multiple arginine directed proteases, we hypothesize that the C-terminal amide may be proteolyzed in vivo.
Having identified d-Cl-amidine and d-o-F-amidine as the most promising PAD1 inhibitors in this series, we examined the pharmacokinetics and pharmodynamics of these two compounds. l-Cl-amidine was used as a reference. Initially, we determined the MTD to examine the physiological effects of these compounds on mice when given orally. The total body weight changes, as well as observable changes in physiology of mice that received varying doses of these compounds, given per os, are shown in Tables S1–S3 in the Supporting Information. Many of the mice in these studies exhibited signs of toxicity (e.g., soft and bloody stool) at higher concentrations of inhibitor, with the most significant observable toxicity being in group 6 of l-Cl-amidine, where mice were notably shaking, anxious, hunching, and breathing heavily from 1 to 4 h after treatment. Thereafter, such signs and symptoms dissipated, and these mice developed diarrhea or soft stool that lasted 2–3 days. Mice receiving 100 mg/kg (group 5) and 150 mg/kg (group 6) l-Cl-amidine had significant weight loss 4 days (96 h) after consuming l-Cl-amidine (Figure 3). However, mice given d-Cl-amidine or d-o-F-amidine maintained healthy weight gains, similar to those mice given no compound at all (Figure 3). Given the definition of the MTD (i.e., the highest dose of a compound that does not cause weight loss or significant sign of toxicity), we conclude from this study that the MTD of l-Cl-amidine in mice is 75 mg/kg and the MTD of the two d-series compounds tested is greater than 150 mg/kg, the highest dose tested. Although unclear why compounds synthesized from d-amino acids have higher MTD values, it is possible that metabolism of l-Cl-amidine generates a toxic metabolite that is not produced as rapidly for the d-series compounds. We note that 100 mg/kg of l-Cl-amidine has been given to mice daily by ip for up to 56 days in our previous studies with no observable signs of toxicity.6,7
Figure 3.
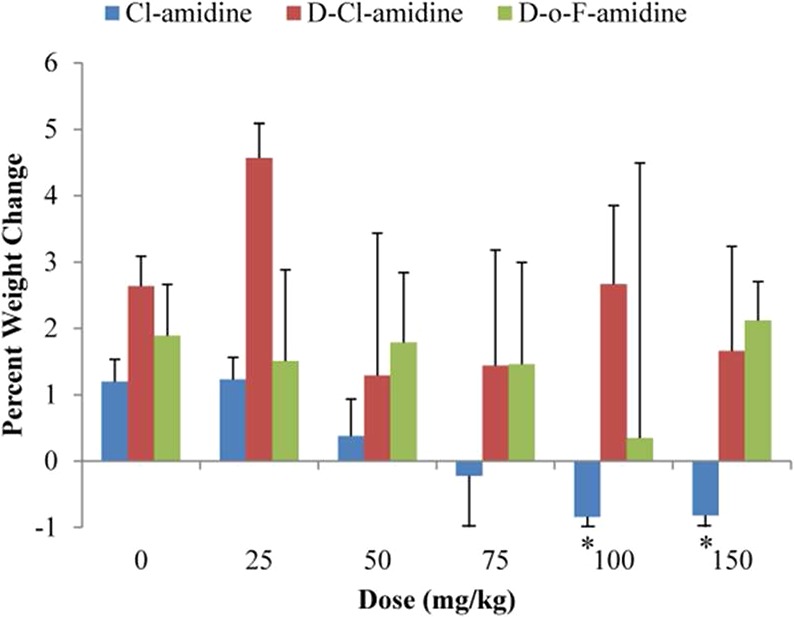
MTD studies. d-Series compounds are better tolerated in vivo than l-Cl-amidine in MTD studies. Graph of percent change in body weight of mice (n = 10) given varying doses of l-Cl-amidine, d-Cl-amidine, and d-o-F-amidine. A significant decrease (*P value <0.05) in weight is seen at higher doses of l-Cl-amidine but not with the d-compounds tested.
The efficacy of the two most potent inhibitors, d-Cl-amidine and d-o-F-amidine, were next tested in MDA-MB-231 cells, a triple negative breast cancer cell line. l-Cl-amidine was again used as the reference compound. MDA-MB-231 cells were chosen for these studies because these cells overexpress PAD1 (Figure S5 in the Supporting Information). All compounds were tested at varying concentrations (i.e., 100, 200, and 400 μM), and various measures of drug efficacy were evaluated (i.e., percent cell viability, cell number, and caspase 3 activity; Figure 4) 96 h after administration. Both l-Cl-amidine and d-Cl-amidine were effective in significantly decreasing cell viability at concentrations of 200 and 400 μM (P value <0.01; Table S2 in the Supporting Information). Similarly, both compounds significantly decreased cell number at a concentration of 400 μM (P value <0.05; Table S2 in the Supporting Information). Unfortunately, the most selective compound in vitro, d-o-F-amidine, had little efficacy in cellulo; we observed no effect on cell viability and only a small decrease in cell number at a concentration of 400 μM (P value <0.05; Table S2 in the Supporting Information). The negatively charged carboxylate likely limits cellular uptake.
Figure 4.

Cellular efficacy studies. l- and d-Cl-amidine show equal potency against PAD1 overexpressing MDA-MB-231 cells, in decreasing cell viability (A) and cell count (B). (C) Both l- and d-Cl-amidine increase caspase 3 activity, indicating that inhibition of PAD1 leads to an increase in apoptosis (*P value <0.05; **P value <0.01). No significant effects are observed with d-o-F-amidine.
To elucidate whether these inhibitors were inducing apoptosis and/or inhibiting proliferation, caspase 3 and Ki-67 activity was monitored in MDA-MB-231 cells. Increased caspase 3 activity is a hallmark of apoptosis,12 whereas Ki-67 is required for maintaining cellular proliferation, and a decrease in Ki-67, only seen during G0 phase, is indicative of cell cycle arrest.13 All of the compounds tested (i.e., l-Cl-amidine, d-Cl-amidine, and d-o-F-amidine) had no significant effect on Ki-67 levels (Figure S6 in the Supporting Information). Only increased amounts of tunicamycin, a positive control of cell cycle arrest, resulted in a decrease in Ki-67 levels. However, both l-Cl-amidine and d-Cl-amidine increased caspase 3 activity, similarly, in a dose-dependent manner, with a significant increase in activity at a concentration of 400 μM (Figure 4C; P value <0.01; Table S1 in the Supporting Information). Following the trend seen in cell number and viability studies, d-o-F-amidine treatment did not result in an increase in caspase 3 activity.
Taken together, these data indicate that while d-o-F-amidine is modestly potent and highly selective in vitro, it has poor efficacy in cellulo. The lack of in cellulo efficacy with this compound is likely due to poor cellular uptake, which we hypothesize is caused by the presence of the negatively charged carboxylate and lack of hydrophobic character. The data also indicate that although l-Cl-amidine and d-Cl-amidine have modestly different in vitro potencies toward PAD1 (kinact/Ki = 37000 and 13500 M–1 s–1, respectively), these compounds have relatively equal in cellulo potencies. The fact that d-Cl-amidine is more selective than l-Cl-amidine suggests that this compound will be useful for studying in vivo PAD1 activity because its use will minimize off target effects.
Because the stability of several l-amino acid-based inhibitors has been improved by synthesizing d-amino acid based mimics, we next compared the stability and pharmacokinetic properties of l- and d-Cl-amidine. We focused on d-Cl-amidine because it is the most potent in cellulo PAD1 inhibitor from the d-series of compounds. Initially, the stability of l- and d-Cl-amidine was tested in a murine hepatic microsome stability assay. This assay utilizes liver microsomes, which possess many of the enzymes responsible for drug metabolism in vivo, and are a common initial predictor of drug clearance properties.14 The results from this assay indicate that both l- and d-Cl-amidine have similar half-lives (37 and 33 min, respectively) (Figure 5A), indicating that inversion of the stereocenter did not improve the stability of Cl-amidine in this assay. The pharmacokinetic properties of both l- and d-Cl-amidine were then examined in mice by both intravenous (iv) and intraperitoneal (ip) injection methods (Figure 5B). These data indicate that l-Cl-amidine, when administered iv at a dose of 10 mg/kg, is completely degraded within 2 h. However, d-Cl-amidine, administered by iv at a dose of 2.5 mg/kg, was still detected after 2 h in serum at a concentration of ∼21 nM and at 4 h at ∼10 nM. l-Cl-amidine, administered by ip at a dose of 10 mg/kg, was completely degraded within 4 h. Similar to the results for iv, d-Cl-amidine was still observed in the blood serum at a concentration of ∼10 nM at 4 h when administered by ip at a dose of 10 mg/kg. These data indicate that d-Cl-amidine is significantly more stable within a mouse model as compared to l-Cl-amidine. This increase in stability is possibly due to decreased proteolysis of the inverted stereocenter.
Figure 5.
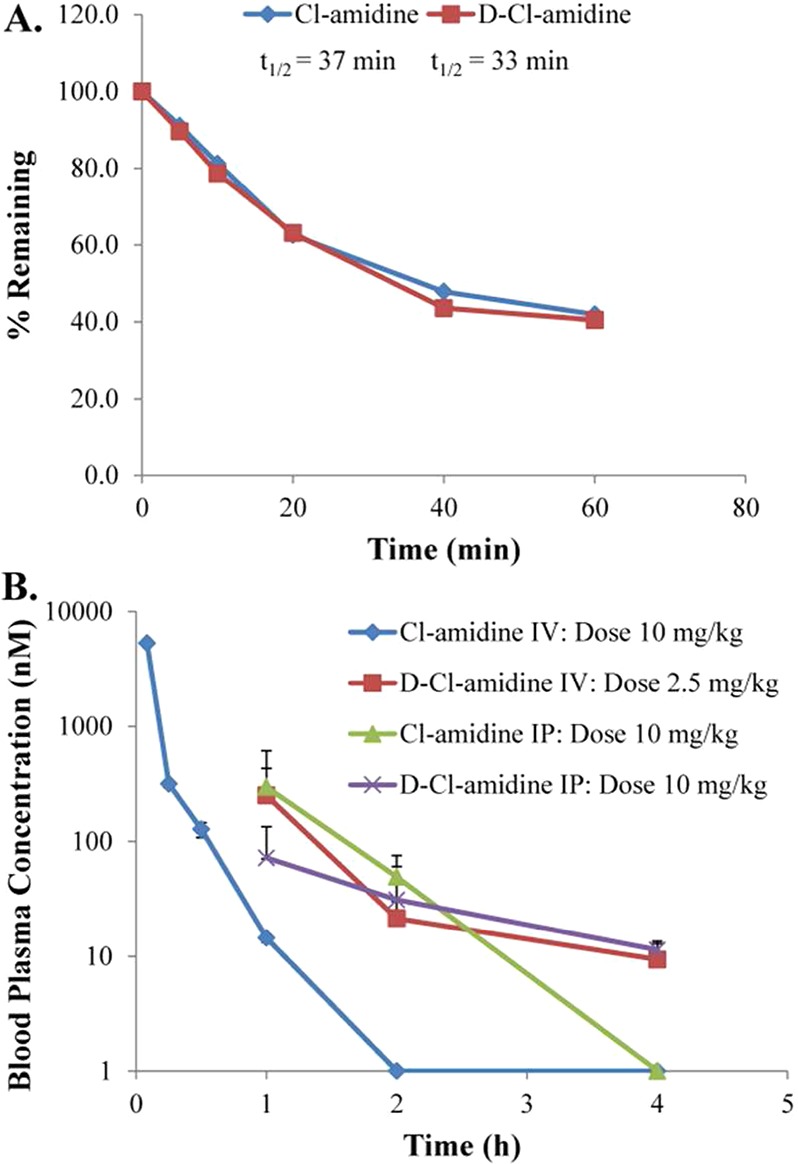
Stability and pharmacokinetic studies. (A) The stability of d-Cl-amidine in mouse hepatic microsomes is similar to Cl-amidine. (B) Mean plasma levels of Cl-amidine and d-Cl-amidine in mice over time, administered by iv and ip injection.
Taken together, these data represent a new class of PAD inhibitors with increased selectivity for PAD1 and increased in vivo stability and tolerance. Although found in the skin where it plays an undefined role in the cornification of the skin, little else is known regarding the normal and pathological roles of PAD1.2 Thus, these PAD1 selective inhibitors will be important tool compounds that can be used to gain insight into the function of this isozyme. Herein, we have shown that of the eight d-series compounds synthesized, d-Cl-amidine and d-o-F-amidine, are moderately potent and highly selective for PAD1 in vitro. MTD studies indicate that these compounds are tolerated better in animal models than l-Cl-amidine, perhaps due to their inherent proteolytic stability. Furthermore, we show that d-Cl-amidine, although less potent in vitro, is equally potent in cellulo as compared to l-Cl-amidine and shows better pharmacokinetics. These properties make d-amino acid-based PAD inhibitors a viable option for improving upon future classes of compounds. Current efforts are focused on expanding on these findings to increase the half-life, bioavailability, and cell permeability of both l- and d-amino acid-based PAD inhibitors.
Glossary
Abbreviations
- PAD
protein arginine deiminase
- MTD
maximum tolerable dose
- iv
intravenous
- ip
intraperitoneal
Supporting Information Available
Synthetic procedures, experimental details, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Financial support for this work was provided by NIH Grants GM079357 (P.R.T.), CA151304 (P.R.T. and L.J.H.), and TSRI.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem., Int. Ed. 2005, 44457342–7372. [DOI] [PubMed] [Google Scholar]
- Jones J. E.; Causey C. P.; Knuckley B.; Slack-Noyes J. L.; Thompson P. R. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discovery Dev. 2009, 125616–627. [PMC free article] [PubMed] [Google Scholar]
- Vossenaar E. R.; Zendman A. J. W.; van Venrooij W. J.; Pruijn G. J. M. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. BioEssays 2003, 25111106–1118. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Bolt M.; Guertin M. J.; Chen W.; Zhang S.; Cherrington B. D.; Slade D. J.; Dreyton C. J.; Subramanian V.; Bicker K. L.; Thompson P. R.; Mancini M. A.; Lis J. T.; Coonrod S. A. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc. Natl. Acad. Sci. U.S.A. 2012, 10.1073/pnas.1203280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington B. D.; Zhang X.; McElwee J. L.; Morency E.; Anguish L. J.; Coonrod S. A. Potential Role for PAD2 in Gene Regulation in Breast Cancer Cells. PLoS One 2012, 77e41242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K.; Hagiwara T.; Ishigami A.; Nagata S.; Asaga H.; Kuramoto M.; Senshu T.; Yamada M. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3). J. Biol. Chem. 1999, 2743927786–27792. [DOI] [PubMed] [Google Scholar]
- Cherrington B. D.; Morency E.; Struble A. M.; Coonrod S. A.; Wakshlag J. J. Potential Role for Peptidylarginine Deiminase 2 (PAD2) in Citrullination of Canine Mammary Epithelial Cell Histones. PLoS ONE 2010, 57e11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson P. R.; Fast W. Histone Citrullination by Protein Arginine Deiminase: Is Arginine Methylation a Green Light or a Roadblock?. ACS Chem. Biol. 2006, 17433–441. [DOI] [PubMed] [Google Scholar]
- Causey C. P.; Jones J. E.; Slack J. L.; Kamei D.; Jones L. E.; Subramanian V.; Knuckley B.; Ebrahimi P.; Chumanevich A. A.; Luo Y.; Hashimoto H.; Sato M.; Hofseth L. J.; Thompson P. R. The Development of N-α-(2-Carboxyl)benzoyl-N5-(2-fluoro-1-iminoethyl)-l-ornithine Amide (o-F-amidine) and N-α-(2-Carboxyl)benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine Amide (o-Cl-amidine) As Second Generation Protein Arginine Deiminase (PAD) Inhibitors. J. Med. Chem. 2011, 54196919–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. E.; Slack J. L.; Fang P.; Zhang X.; Subramanian V.; Causey C. P.; Coonrod S. A.; Guo M.; Thompson P. R. Synthesis and Screening of a Haloacetamidine Containing Library To Identify PAD4 Selective Inhibitors. ACS Chem. Biol. 2011, 71160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B.; Causey C. P.; Jones J. E.; Bhatia M.; Dreyton C. J.; Osborne T. C.; Takahara H.; Thompson P. R. Substrate Specificity and Kinetic Studies of PADs 1, 3, and 4 Identify Potent and Selective Inhibitors of Protein Arginine Deiminase 3. Biochemistry 2010, 49234852–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B.; Causey C. P.; Pellechia P. J.; Cook P. F.; Thompson P. R. Haloacetamidine-Based Inactivators of Protein Arginine Deiminase 4 (PAD4): Evidence that General Acid Catalysis Promotes Efficient Inactivation. ChemBioChem 2010, 112161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Arita K.; Bhatia M.; Knuckley B.; Lee Y.-H.; Stallcup M. R.; Sato M.; Thompson P. R. Inhibitors and Inactivators of Protein Arginine Deiminase 4: Functional and Structural Characterization. Biochemistry 2006, 453911727–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone E. M.; Schaller T. H.; Bianchi H.; Person M. D.; Fast W. Inactivation of Two Diverse Enzymes in the Amidinotransferase Superfamily by 2-Chloroacetamidine: Dimethylargininase and Peptidylarginine Deiminase. Biochemistry 2005, 444213744–13752. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li P.; Wang S.; Hu J.; Chen X. A.; Wu J.; Fisher M.; Oshaben K.; Zhao N.; Gu Y.; Wang D.; Chen G. Anticancer PAD inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J. Biol. Chem. 2012, 10.1074/jbc.M112.375725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis V. C.; Gizinski A. M.; Banda N. K.; Causey C. P.; Knuckley B.; Cordova K. N.; Luo Y.; Levitt B.; Glogowska M.; Chandra P.; Kulik L.; Robinson W. H.; Arend W. P.; Thompson P. R.; Holers V. M. N-α-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-l-Ornithine Amide, a Protein Arginine Deiminase Inhibitor, Reduces the Severity of Murine Collagen-Induced Arthritis. J. Immunol. 2011, 18674396–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanevich A. A.; Causey C. P.; Knuckley B. A.; Jones J. E.; Poudyal D.; Chumanevich A. P.; Davis T.; Matesic L. E.; Thompson P. R.; Hofseth L. J. Suppression of colitis in mice by Cl-amidine: A novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 3006G929–G938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Li M.; Stadler S.; Correll S.; Li P.; Wang D.; Hayama R.; Leonelli L.; Han H.; Grigoryev S. A.; Allis C. D.; Coonrod S. A. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 1842205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmers S.; Teijaro J. R.; Arandjelovic S.; Mowen K. A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 2011, 67e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. Comparison of the proteolytic susceptibilities of homologous l-amino acid, d-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 1995, 35120–32. [Google Scholar]
- Tugyi R.; Uray K.; Iván D.; Fellinger E.; Perkins A.; Hudecz F. Partial d-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. U.S.A. 2005, 1022413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle M.; Bernkop-Schnürch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 304351–367. [DOI] [PubMed] [Google Scholar]
- Adessi C.; Soto C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 99963–978. [DOI] [PubMed] [Google Scholar]
- Liu Y.-L.; Chiang Y.-H.; Liu G.-Y.; Hung H.-C. Functional Role of Dimerization of Human Peptidylarginine Deiminase 4 (PAD4). PLoS ONE 2011, 66e21314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnemri E. S.; Livingston D. J.; Nicholson D. W.; Salvesen G.; Thornberry N. A.; Wong W. W.; Yuan J. Human ICE/CED-3 Protease Nomenclature. Cell 1996, 872171. [DOI] [PubMed] [Google Scholar]
- Scholzen T.; Gerdes J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 1823311–322. [DOI] [PubMed] [Google Scholar]
- Baranczewski P.; Stanczak A.; Sundberg K.; Svensson R.; Wallin A.; Jansson J.; Garberg P.; Postlind H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 583453–472. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.