

Abstract
The fragile X-associated tremor/ataxia syndrome (FXTAS) is an adult-onset neurodegenerative disorder caused by CGG trinucleotide repeat expansions in the fragile X mental retardation 1 (FMR1) gene. The neuropathological hallmark of FXTAS is the presence of ubiquitin-positive intranuclear inclusions in neurons and in astroglia. Intranuclear inclusions have also been reported in the neurons of male CGG KI mice carrying an expanded CGG trinucleotide repeat and used to model FXTAS, but no study has been carried out quantifying inclusions in female CGG KI mice heterozygous for the fragile X premutation. We used histologic and immunocytochemical methods to determine the pathological features of intranuclear inclusions in astroglia and neurons. In female CGG KI mice, ubiquitin-positive intranuclear inclusions were found in neurons and astroglia throughout the brain in cortical and subcortical regions. These inclusions increased in number and became larger with advanced age and increasing CGG repeat length, supporting hypotheses that these pathologic features are progressive across the lifespan. The number of inclusions in neurons was reduced by ~25% in female CGG KI mice compared to male CGG KI mice, but not so low as the 50% predicted. These data emphasize the need to evaluate the neurocognitive and pathological features in female carriers of the fragile X premutation with and without FXTAS symptomatology is warranted, as this population shows similar neuropathological features present in male FXTAS patients.
Keywords: Fragile X premutation, CGG KI mouse, Trinucleotide repeat disorder, Intranuclear inclusions
1. Introduction
Fragile X-associated tremor ataxia syndrome (FXTAS) is a neurodegenerative disorder resulting from a 55–200 long CGG trinucleotide repeat expression (fragile X premutation) within the 5′ untranslated region of the X-linked FMR1 gene. Beyond 200 CGG repeat expansions, CpG island methylation of the FMR1 promoter and the CGG repeat usually occurs (Kenneson et al., 2001; Tassone et al., 2000, 2004, 2008; Tassone et al., 2012). This silences FMR1 gene transcription, leading to Fragile X Syndrome (FXS, Hagerman and Hagerman, 2008; Raske and Hagerman, 2009; Tassone and Berry-Kravis, 2010). Approximately 40% of male fragile X premutation carriers over the age of 50 develop motor features associated with FXTAS. These motor features include kinetic, intention, and/or postural tremors, cerebellar gait and limb ataxia, Parkinsonism, as well as peripheral neuropathy and progressive cognitive impairments (Hamlin et al., 2011; Leehey et al., 2011); neurologically, the hallmark pathological feature is ubiquitin-positive intranuclear inclusions throughout the brain and white matter disease on T2 weighted magnetic resonance imaging (MRI; Greco et al., 2002, 2006, 2008; Jacquemont et al., 2003).
Female fragile X premutation carriers also present with symptoms but far less frequently than do male carriers, and with somewhat less profound symptomatology (Berry-Kravis et al., 2005; Karmon and Gadoth, 2008). Approximately 8–16% of female premutation carriers older than 50 years of age present with FXTAS, and a few cases have been described within the literature (Berry-Kravis et al., 2005; Coffey et al., 2008; Leehey et al., 2011; Roberts et al., 2009; Tassone et al., 2012; Yachnis et al., 2010). One explanation for the less frequent presentation of FXTAS in female carriers is X-inactivation silences transcription of one of the FMR1 alleles, one carrying the premutation allele and the other unaffected by the premutation (i.e., CGG repeat length <45 repeats). X-inactivation silences transcription of one of the two copies of the X chromosome, and patterns of X-inactivation are normally thought to be random with no preference for inactivation of paternal or maternal alleles. However, it has also been suggested that in FXTAS skewed (or non-random) X-inactivation may occur in females where a ‘stronger’ allele dominates over a ‘weaker’ allele (cf., Berry-Kravis et al., 2005; Tassone et al., 2012). Therefore, X-inactivation in females heterozygous for the FMR1 premutation may favor the unmutated allele, leading to suppression of the FXTAS pathologic phenotype (cf., Tassone et al., 2012). This type of selective action has been described for a number of other X-linked genetic disorders (Plenge et al., 2002; Yonath et al., 2011).
It is difficult to correlate human neuropathologies with the dosage of molecular measures such as CGG repeat length due to the lack of available human tissue. Therefore, an Fmr1 premutation CGG knock-in mouse model (CGG KI) was developed in Rotterdam and is a useful model of the fragile X premutation and FXTAS. Willemsen and colleagues (Brouwer et al., 2007, 2008; Willemsen et al., 2003) found that intranuclear inclusions were present in the brain of these CGG KI mice. Wenzel and colleagues reported astroglia contrained inclusions in this same line of CGG KI mice on a congenic C57BL/6J background (Wenzel et al., 2010). Previous studies correlating neuropathological features of FXTAS with repeat length have focused primarily on male mice (Brouwer et al., 2007, 2008); however, there are presently no studies which have examined these features in female CGG KI mice heterozygous for the fragile X premutation. Therefore, this study was carried out to identify the presence and distribution of ubiquitin positive intranuclear inclusions, the hallmark neuropathological feature of FXTAS, throughout the brains of female CGG KI mice heterozygous for the fragile X premutation.
The brains of female CGG KI mice heterozygous for CGG repeat expansions between 71 and 326 were formalin-fixed, sectioned, and examined using histological, immunoperoxidase, and immunofluorescent staining methods. Ubiquitin positive intranuclear inclusions were identified in neurons and astroglia throughout the brain of female CGG KI mice. Further analysis of these findings also suggest that intranuclear inclusion density correlates with increasing dosage of the CGG repeat length, that the number of inclusions increases with age, and that these factors may interact. This study points to the potential for a relationship between ubiquitin positive intranuclear inclusions observed in FXTAS and the pathologic phenotype associated with the fragile X premutation.
2. Results
2.1. General histological results
As has been reported previously, there were no gross histological differences among all groups of mice. On H&E, cresyl violet, and thionin stained sections there were no obvious differences in cell distribution, cell packing, cortical thickness, or overall brain morphology (Entezam et al., 2007; Greco et al., 2002, 2006, 2008; Tassone et al., 2012; Willemsen et al., 2003; Wenzel et al., 2010). Luxol fast blue stains counter-stained with neutral red demonstrated that the CGG KI mice did not show any white matter pallor or reduced integrity of white matter tracts throughout the brain, at least that can be identified at the light microscopic level. More specifically, no differences were observed between wildtype and CGG KI mice for white matter integrity in periventricular regions, brainstem, cerebellar peduncles, internal capsule, or corpus callosum. Subcortical white matter pathways (e.g., fimbria/fornix, anterior commissure) were free of neuropathological features in CGG KI mice.
2.2. Nuclear pathology in neurons of female CGG KI mice and female FXTAS patients
The presence of intranuclear inclusions in neurons and astroglia is a pathological hallmark of FXTAS. Thus far, similar intranuclear inclusions have not been reported in oligodendroglia or microglia in human FXTAS or CGG KI mice. Fig. 1 shows typical intranuclear inclusions in the hippocampus of a female premutation carrier without FXTAS symptomatology at 76 years of age presented as Case 1 in Tassone et al. (2012). These inclusions in a female patient are shown for comparison with intranuclear inclusions found in the brains of female CGG KI mice shown in Fig. 2 that provides examples of inclusions in the granule cell and inner molecular layer of the hippocampal dentate gyrus and in the posterior nucleus of the amygdala of female CGG KI mice. In both cases inclusions appeared as well-delineated 2–3 μm diameter spherical bodies, were intensely eosinophilic, and hyaline in appearance with H&E staining. Identification of cell types in human tissue was based on methods reported previously (Greco et al., 2002, 2006; Tassone et al., 2012; Wenzel et al., 2010).
Fig. 1.
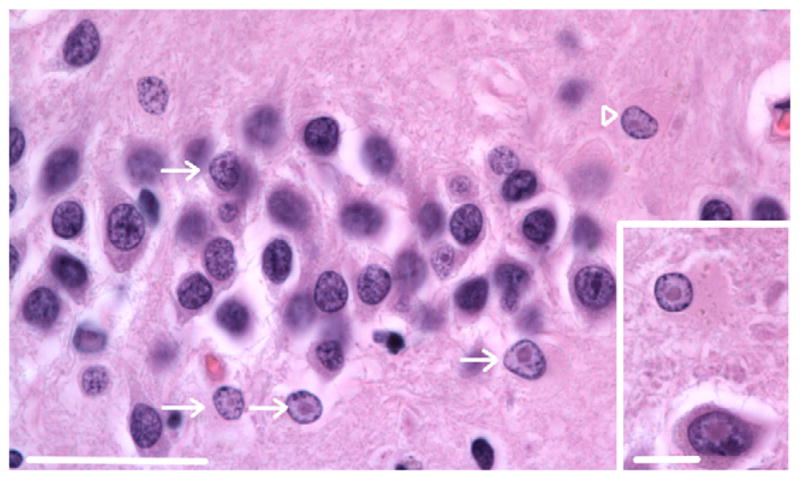
Intranuclear inclusions in the dentate gyrus of a human female carrier of the fragile X premutation. Inclusions are present in the granule cell layer (arrow) and an astroglial cell in the hilus (arrowhead) of a 76 year old female premutation carrier that did not show FXTAS symptoms (Case 1 from Tassone et al., 2012). Magnification 1000 ×. Scale bar = 50 μm. H&E stain. Inset: inclusions in a pyramidal neuron (lower) and astroglial cell (upper) in the hilus. Original magnification 1000 ×. Scale bar = 10 μm. H&E stain.
Fig. 2.
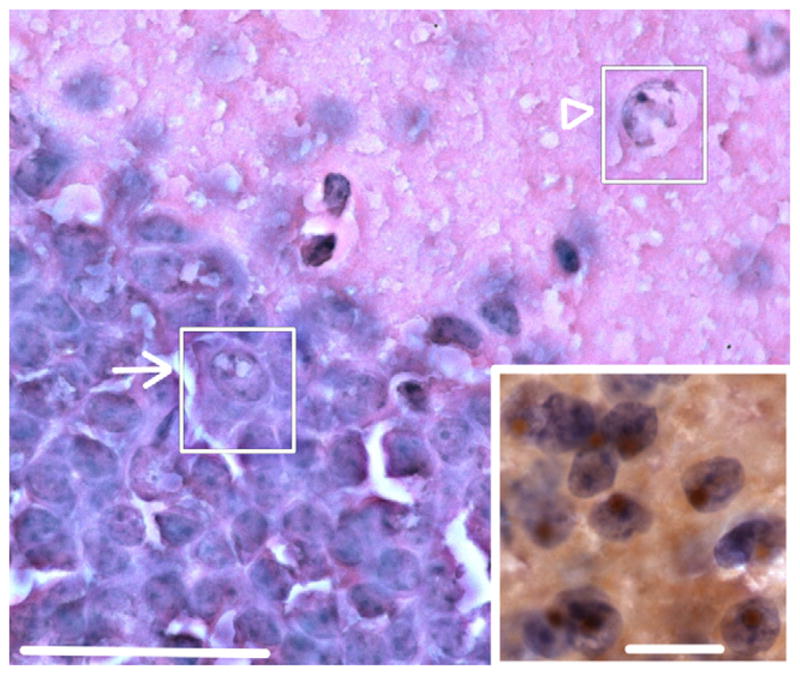
Intranuclear inclusions in dentate gyrus of female CGG KI mice. Hematoxylin and eosin (H&E) stained section of the dentate gyrus showing inclusions in granule cells (arrow; inset) and putative GABAergic cells in the inner molecular layer (arrowhead). Original magnification 1000 ×. Scale bar = 50 μm. Inset: numerous brown intranuclear inclusions by immunoperoxidase staining in hematoxylin stained cells of the posterior nucleus of the amygdala of a female CGG KI mouse. Original magnification 1000 ×. Scale bar = 10 μm. Main figure H&E stain, inset imunoperoxidase stain for ubiquitin. These images are from a female CGG KI mouse, 48 weeks of age with 12 repeats in the Fmr1 gene one X chromosome and 128 on the second Fmr1 gene. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The results of the semiquantitative analysis for the distribution of intranuclear inclusions are presented in Table 1 (cf., Fig. 2 for examples of inclusions in the granule cell and inner molecular layer of the dentate gyrus (H&E stain) and in the posterior nucleus of the amygdala (immunoperoxidase stain) of female CGG KI mice). Inspection of the table shows a clear developmental course to the development of the inclusions, such that in nearly all cases mice at 48–52 weeks of age showed a greater number of inclusions than mice aged 24–28 weeks of age. Similarly, a dosage effect for the mutation was identified such that mice with 70–110 CGG repeats showed much fewer inclusions than mice with 140–200 CGG repeats.
Table 1.
emiquantitative analysis of the presence and relative distribution of intranuclear inclusions in female CGG KI mouse brains in H&E stained parasagittal sections. n = 5 female CGG KI mice were counted for each repeat length and age group. Not included in this table are 16 wildtype littermate mice, which never showed intranuclear inclusions. The numbers of counted intranuclear inclusions across mice is given, along with the median number of inclusions counted per 200 cells. Regions of interest were determined at 20 × magnification in wildtype mice and subsequently standardized across all mice.
| 24–28 weeks of age
|
48–52 weeks of age
|
||||
|---|---|---|---|---|---|
| 70–110 CGG | 140–200 CGG | 70–110 CGG | 140–200 CGG | ||
| Prelimbic cortexa | Values (of 200) | 0,1,1,1,1 | 5,5,7,9,9 | 3,8,11,11,14 | 11,14,26,27,36 |
| Median+/−sd | 1+/−0.44 | 7+/−2.00 | 11+/−4.12 | 26+/−10.23 | |
| Motor cortexa | Values (of 200) | 0,0,1,3,3 | 0,3,5,6,8 | 0,1,4,4,6 | 2,6,9,10,15 |
| Median+/−sd | 1+/−1.51 | 5+/−3.04 | 4+/−2.45 | 9+/−4.88 | |
| Posterior parietal cortexa | Values (of 200) | 0,0,0,1,3 | 2,3,7,11,13 | 4,6,6,9,13 | 4,11,13,13,26 |
| Median+/−sd | 0+/−1.30 | 7+/−4.82 | 6+/−3.51 | 13+/−7.96 | |
| Visual cortexa | Values (of 200) | 0,0,0,0,1 | 0,3,4,4,6 | 0,1,3,5,5 | 1,2,7,8,11 |
| Median+/−sd | 0+/−0.45 | 4+/−2.19 | 3+/−2.28 | 7+/−4.21 | |
| Anterior cingulate cortexa | Values (of 200) | 0,1,3,3,7 | 3,5,14,14,19 | 4,7,8,10,11 | 10,19,23,25,26 |
| Median+/−sd | 3+/−2.68 | 14+/−6.75 | 8+/−2.74 | 23+/−5.60 | |
| Piriform cortexb | Values (of 200) | 0,1,2,2,5 | 4,11,15,17,23 | 9,13,16,16,22 | 25,35,37,39,44 |
| Median+/−sd | 2+/−1.87 | 15+/−7.07 | 16+/−4.76 | 37+/−7.00 | |
| Entorhinal cortex | |||||
| Ventromedial areaa | Values (of 200) | 0,0,1,1,3 | 4,4,8,9,11 | 15,20,21,24,30 | 20,28,35,36,39 |
| Median+/−sd | 1+/−1.51 | 8+/−3.11 | 21+/−5.52 | 35+/−7.64 | |
| Dorsolateral areaa | Values (of 200) | 0,0,0,0,1 | 1,1,3,5,6 | 4,5,7,7,13 | 9,9,11,14,15 |
| Median+/−sd | 0+/−0.45 | 3+/−2.28 | 7+/−3.49 | 11+/−2.79 | |
| Hippocampusb | |||||
| CA1 | Values (of 200) | 2,2,5,5,8 | 5,6,9,11,11 | 8,9,13,16,17 | 15,22,27,28,31 |
| Median+/−sd | 5+/−2.51 | 9+/−2.79 | 13+/−4.04 | 27+/−6.27 | |
| CA3 | Values (of 200) | 0,0,1,1,1 | 0,0,1,1,3 | 0,0,2,2,5 | 2,3,8,9,12 |
| Median+/−sd | 1+/−0.55 | 1+/−1.51 | 2+/−2.04 | 8+/−4.21 | |
| DG | Values (of 200) | 5,10,10,11,11 | 15,33,34,37,40 | 23,31,47,50,61 | 50,57,76,83,105 |
| Median+/−sd | 10+/−2.51 | 34+/−9.78 | 47+/−15.26 | 76+/−21.86 | |
| Subiculum | Values (of 200) | 0,0,2,2,3 | 2,2,6,7,9 | 7,11,13,14,19 | 11,20,28,30,39 |
| Median+/−sd | 2+/−1.34 | 6+/−3.11 | 13+/−4.38 | 28+/−10.60 | |
| Amygdalar nuclei | |||||
| Central nucleusd | Values (of 200) | 1,1,2,2,2 | 9,9,11,13,18 | 3,9,9,14,15 | 12,15,17,18,22 |
| median+/−sd | 2+/−0.55 | 11+/−3.74 | 9+/−4.80 | 17+/−3.70 | |
| Basolateral nucleusc | Values (of 200) | 2,3,7,7,10 | 7,9,15,16,16 | 21,40,41,46,50 | 40,44,58,59,65 |
| Median+/−sd | 7+/−3.27 | 15+/−4.28 | 41+/−11.15 | 58+/−10.66 | |
| Posterior nucleus | Values (of 200) | 9,13,19,22,26 | 31,39,43,44,52 | 48,50,51,55,73 | 70,94,100,105,117 |
| Median+/−sd | 19+/−6.83 | 43+/−7.66 | 51+/−10.16 | 100+/−17.40 | |
| Septal nuclei | |||||
| Lateral septumg | Values (of 200) | 0,0,1,1,2 | 2,2,3,3,7 | 4,6,6,8,11 | 3,7,8,11,12 |
| Median+/−sd | 1+/−0.84 | 3+/−2.07 | 6+/−2.65 | 8+/−3.56 | |
| Medial septum | Values (of 200) | 0,0,0,1,1 | 1,1,1,3,4 | 2,2,4,4,5 | 2,2,5,6,6 |
| Median+/−sd | 0+/−0.55 | 1+/−1.41 | 4+/−1.34 | 5+/−2.05 | |
| Lateral geniculate nucleus | Values (of 200) | 0,0,5,9,10 | 6,14,20,21,24 | 17,19,22,24,27 | 30,46,49,51,55 |
| Median+/−sd | 5+/−4.76 | 20+/−7.14 | 22+/−3.96 | 49+/−9.62 | |
| Inferior colliculusf | Values (of 200) | 0,1,3,3,4 | 2,3,15,18,19 | 11,19,30,37,45 | 37,49,61,66,70 |
| Median+/−sd | 3+/−1.64 | 15+/−8.26 | 30+/−13.63 | 61+/−13.50 | |
| Superior colliculusf | Values (of 200) | 0,0,0,1,1 | 0,0,2,2,3 | 0,4,5,6,9 | 5,11,16,20,21 |
| Median+/−sd | 0+/−0.55 | 2+/−1.34 | 5+/−3.27 | 16+/−6.66 | |
| Medial vestibular nucleus | Values (of 200) | 0,1,1,2,2 | 5,11,12,14,17 | 7,13,15,15,21 | 13,22,24,26,29 |
| Median+/−sd | 1+/−0.83 | 12+/−4.44 | 15+/−5.02 | 24+/−6.06 | |
| Cerebellum | |||||
| Cerebellar lobule IIIe | |||||
| PCL | Values (of 200) | 0,0,0,0,0 | 0,0,0,0,0 | 0,0,0,0,0 | 0,0,0,1,1 |
| Median+/−sd | 0+/−0 | 0+/−0 | 0+/−0 | 0+/−55 | |
| GCL | Values (of 200) | 0,0,0,0,0 | 0,0,1,1,2 | 0,0,2,2,5 | 5,6,11,11,13 |
| Median+/−sd | 0+/−0 | 1+/−0.83 | 2+/−2.05 | 11+/−3.49 | |
| Flocculuse | |||||
| PCL | Values (of 200) | 0,0,0,0,0 | 0,0,0,0,0 | 0,0,0,0,0 | 0,0,0,0,1 |
| Median+/−sd | 0+/−0 | 0+/−0 | 0+/−0 | 0+/−0.45 | |
| GCL | Values (of 200) | 4,5,7,8,12 | 8,11,12,16,19 | 6,6,11,12,17 | 14,17,19,19,25 |
| Median+/−sd | 7+/−3.11 | 12+/−4.32 | 11+/−4.62 | 19+/−4.02 | |
For cortical quantifications, cells were counted in layers II–V of the cortex starting with layer II and moving ventrally into layer V, layers I and VI were not sampled.
For hippocampus and piriform cortices, the primary cell layers (pyramidal and granule cell layers) were counted, the plexiform and molecular layers were not sampled. The hippocampus was sampled midway between the septal and temporal poles, sampling the dorsal half of the hippocampus. The piriform cortex was sampled at the most rostral aspect in the most lateral sections in which it appeared.
The posterior part of the basolateral amygdalar nucleus was sampled.
The medial part of the central amygdalar nucleus was sampled.
For the Cerebellum, the granule cell layer was counted in the most distal portion of each folia. The Purkinje cells were sampled along the length of the folia surrounding the granule cell layer sampled. The molecular layer was not sampled, and putative Bergmann Glia containing intranuclear inclusion bodies were not counted, although rarely identified.
For the superior and inferior colliculi, the dorsal cortex was sampled.
The rostroventral area of the lateral septal nucleus was sampled. Nomenclature for regions of interest followed the conventions used by the Allen Mouse Brain Atlas [Internet] (2009). Findings were corroborated with adjacent sections immunostained for ubiquitin that were not explicitly quantified.
To more fully characterize this pattern, the number of inclusions counted was related to CGG repeat length in both the young and old mice. Table 2 contains the Pearson’s correlation coefficients that were calculated for each brain region and associated probability (p) values. In Fig. 3 the number of inclusions are shown plotted against CGG repeat length for six brain regions for mice older than 6 months (circles) or younger than 60 months (diamonds) of age. The most intriguing pattern of results that is apparent in the top row of plates in Fig. 3 is that the slope of the relationship is similar for young and old mice across a number of regions. The list of regions showing similar correlation slopes across age include: motor cortex, posterior parietal cortex, visual cortex, anterior cingulate cortex, dorsolateral and ventromedial entorhinal cortex, CA1 and the dentate gyrus in the hippocampus, all amygdalar nuclei, septal nuclei, anterior thalamus, vestibular nucleus, and the granule cell layer of the flocculus.
Table 2.
Association between CGG repeat length and intranuclear inclusion number on young and old female CGG KI mice.
| 48–52 Weeks of Age
|
24–28 Weeks of Age
|
||||
|---|---|---|---|---|---|
| Pearson’s rho | Slope | Pearson’s rho | Slope | Difference | |
| Prelimbic cortex | ρ = 0.87; R2 = 0.77, p<0.01 | 0.194 | ρ = 0.97; R2 = 0.95, p<0.01 | 0.07 | 0.12, p<0.05 |
| Motor cortex | ρ = 0.91; R2 = 0.82, p<0.01 | 0.09 | ρ = 0.91; R2 = 0.82, p<0.05 | 0.06 | 0.03, ns |
| Posterior parietal cortex | ρ = 0.80; R2 = 0.64, p<0.01 | 0.07 | ρ = 0.97; R2 = 0.94, p<0.01 | 0.09 | 0.02, ns |
| Visual cortex | ρ = 0.92; R2 = 0.85, p<0.05 | 0.07 | ρ = 0.96; R2 = 0.93, p<0.05 | 0.06 | 0.01, ns |
| Anterior cingulate cortex | ρ = 0.98; R2 = 0.97, p<0.01 | 0.17 | ρ = 0.97; R2 = 0.95, p<0.05 | 0.14 | 0.03, ns |
| Piriform cortex | r = 0.99; R2 = 0.98, p<0.01 | 0.14 | ρ = 0.98; R2 = 0.97, p<0.01 | 0.26 | 0.12, p<0.01 |
| Entorhinal cortex | |||||
| Ventromedial area | ρ = 0.95; R2 = 0.91, p<0.05 | 0.16 | r = 0.96; R2 = 0.93, p<0.01 | 0.11 | 0.05, ns |
| Dorsolateral area | ρ = 0.93; R2 = 0.86, p<0.01 | 0.07 | ρ = 0.96; R2 = 0.93, p<0.01 | 0.08 | 0.01, ns |
| Hippocampus | |||||
| CA1 | ρ = 0.96; R2 = 0.92, p<0.01 | 0.16 | ρ = 0.89; R2 = 0.80, p<0.01 | 0.12 | 0.04, ns |
| CA3 | ρ = 0.91; R2 = 0.83, p<0.05 | 0.09 | ρ = 0.14; R2 = 0.02, p = 0.17 | 0.001 | 0.09, p<0.01 |
| DG | ρ = 0.91; R2 = 0.84, p<0.01 | 0.50 | ρ = 0.99; R2 = 0.99, p<0.001 | 0.43 | 0.07, ns |
| Subiculum | ρ = 0.94; R2 = 0.88, p<0.05 | 0.20 | ρ = 0.90; R2 = 0.81, p<0.01 | 0.06 | 0.14, p<0.01 |
| Amygdalar nuclei | |||||
| Central nucleus | ρ = 0.88; R2 = 0.79, p<0.01 | 0.09 | ρ = 0.98; R2 = 0.97, p<0.001 | 0.11 | 0.02, ns |
| Basolateral nucleus | ρ = 0.75; R2 = 0.55, p<0.01 | 0.15 | ρ = 0.91; R2 = 0.83, p<0.001 | 0.11 | 0.04, ns |
| Posterior nucleus | ρ = 0.99; R2 = 0.79, p = 0.055 | 0.57 | ρ = 0.98; R2 = 0.97, p<0.05 | 0.48 | 0.09, ns |
| Septal nuclei | |||||
| Lateral septum | ρ = 0.82; R2 = 0.67, p = 0.061 | 0.02 | ρ = 0.90; R2 = 0.81, p<0.05 | 0.04 | 0.02, ns |
| Medial septum | ρ = 0.67; R2 = 0.45, p<0.05 | 0.02 | ρ = −0.77; R2 = 0.60, p<0.05 | 0.02 | 0, ns |
| LGN | ρ = 0.99; R2 = 0.99, p<0.001 | 0.31 | ρ = 0.96; R2 = 0.9,2 p<0.01 | 0.17 | 0.14, p<0.01 |
| Inferior colliculus | ρ = 0.90; R2 = 0.82, p<0.01 | 0.38 | ρ = 0.99; R2 = 0.99, p<0.05 | 0.16 | 0.22, p<0.01 |
| Superior colliculus | ρ = 0.95; R2 = 0.90, p<0.05 | 0.14 | ρ = 0.94; R2 = 0.89, p = 0.053 | 0.02 | 0.12, p<0.05 |
| Medial vestibular nucleus | ρ = 0.96; R2 = 0.93, p<0.01 | 0.14 | ρ = 0.99; R2 = 0.98, p<0.01 | 0.13 | 0.01, ns |
| Cerebellum | |||||
| Cerebellar lobule III | |||||
| PCL | N/A | N/A | N/A | N/A | |
| GCL | ρ = 0.97; R2 = 0.94, p<0.01 | 0.10 | ρ = 0.83; R2 = 0.70, p<0.05 | 0.008 | 0.09, p<0.05 |
| Flocculus | |||||
| PCL | N/A | N/A | N/A | N/A | |
| GCL | ρ = 0.95; R2 = 0.90, p<0.01 | 0.12 | ρ = 0.94; R2 = 0.89, p<0.01 | 0.10 | 0.12, ns |
Pearson’s correlation coefficients were calculated comparing CGG repeat length with intranuclear inclusion number. The differences in slopes of the correlation between young and old mice were compared. All p values have been FDR adjusted to control for false discovery rate (cf., Diep et al., 2012; Hunsaker et al., 2011c).
Fig. 3.

Example correlations between CGG repeat length and intranuclear inclusion number in female CGG KI mice. Top row: the slopes of the correlations are similar between young and old mice (p>1), suggesting in these brain regions age may be a major factor in the progression of inclusion formation. Bottom row: the slope of the correlations are dissimilar (p<0.05) between young and old mice, suggesting a combination of CGG repeat length and age may accelerate pathological features in these brain regions. Circles = female CGG KI mice older than 6 months of age; diamonds = female CGG KI mice younger than 6 months of age. These correlations are from the data presented in Table 1.
However, in other brain regions, older mice show a steeper slope of the correlation, suggesting age and CGG repeat dosage additively affect the progression of intranuclear inclusions. The few brain regions that showed a greater slope in older relative to young mice include the prelimbic cortex, piriform cortex, CA3 pyramidal cells and the subiculum in the hippocampus, the superior and inferior colliculus, and granule cell layer of lobe III in the cerebellum. Regions that showed near zero inclusions showed no relationship between inclusions number and repeat length, including the Purkinje cell layers of the cerebellum and the flocculus, and no correlations were computed for these regions. These data suggest that age and CGG repeat length may additively affect the progression of intranuclear inclusions in some, but not all brain regions.
In comparison to previously published results quantifying intranuclear inclusions in male CGG KI mice (Brouwer et al., 2007, 2008; Willemsen et al., 2003; Wenzel et al., 2010), the female CGG KI mice consistently showed 20–35% fewer inclusions, and surprisingly not the 50% fewer as we had predicted. Four female CGG KI mice with 250, 265, 290, and 326 CGG repeats at 50 weeks of age showed a similar pattern of intranuclear inclusion distribution as the CGG KI mice with 70–110 CGG repeats, but 10–15% fewer inclusions in each region (data not shown). These data are consistent with reports of reduced number of intranuclear inclusions in male CGG KI mice when CGG repeat expansions are much greater than 200.
Four female CGG KI mice with 154, 168, 172, and 188 CGG repeats examined at 3 months of age only rarely showed intranuclear inclusions in the granule cell layers in the hippocampus dentate gyrus, olfactory bulb, and cerebellum. This pattern also differed from male CGG KI mice that show a much greater number of intranuclear inclusions in these granule cell populations at 3 months of age (Hunsaker et al., 2009; and unpublished observations).
2.3. Nuclear pathology in astroglia of CGG KI mice
Previous studies of male CGG KI mice have identified intranuclear inclusions in astroglia of CGG KI mice (Wenzel et al., 2010). While such inclusions in astroglia are consistently observed in the human brain from FXTAS patients (cf., Greco et al., 2006; Tassone et al., 2012), fewer inclusions (by percentage) are present in astroglia in female FXTAS cases than in male FXTAS cases (Tassone et al., 2012). Because the CGG KI mouse models other neuropathological features of FXTAS (e.g., elevated FMR1 mRNA and slightly reduced FMRP levels), a major goal of the present study was to determine whether intranuclear inclusions could be demonstrated in the astroglia of female CGG KI mice (cf., Fig. 4 for examples of astroglia and Bergmann Glia with intranuclear inclusions in the flocculus).
Fig. 4.
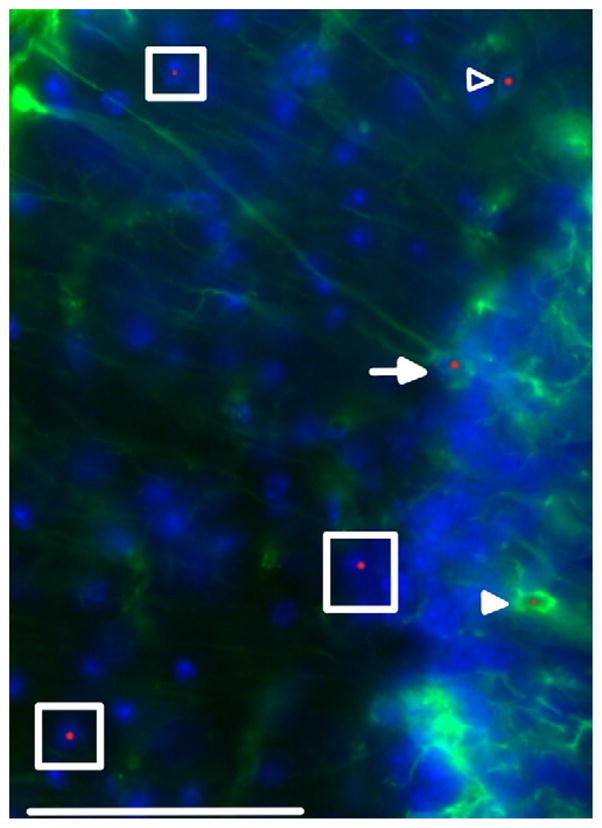
Intranuclear inclusion in astroglia and Bergmann glia. Protoplasmic (open arrowhead) and velate astroglial cells (filled arrowhead) stained for GFAP (green) and ubiquitin (red). Bergmann glial cell (arrow) stained for GFAP (green) and ubiquitin (red). Neurons negative for GFAP with intranuclear inclusions are shown with boxes. Cell nuclei stained with DAPI (blue). Arrowheads point to an astroglial cell in the inner molecular layer with an intranuclear inclusion as well as a velate astroglial cell in the granule cell layer with an intranuclear inclusion. Single plane confocal image. Original magnification 600 ×. Scale bar = 50 μm. This image came from a female CGG KI mouse, 55 weeks of age with 8 CGG repeats in the Fmr1 gene one X chromosome and 164 on the second Fmr1 gene. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Using the same methods as reported by Wenzel et al. (2010), we found that female CGG KI mice show inclusions in 4.9% (+/−0.83 standard error) of astroglial cells in layer I of the somatosensory and motor cortices in 48–52 week old female CGG KI mice with 140–200 CGG repeats, and 2.1% (+/−0.13) in 48–52 week old mice with 70–100 CGG repeats. Similarly, the percentage with inclusions were 3.3% (+/−0.09) in 24–28 week old mice with 140–200 CGG repeats, while inclusions were virtually undetectable in 24–28 week old mice with 70–100 CGG repeats. For the oldest female CGG KI mice with 140–200 repeats this quantification corresponds to roughly 50% of the frequency of inclusion in astroglia previously reported in the brains of male CGG KI mice as previously reported. Also, similarly to the male CGG KI mice, the pattern of astroglial intranuclear inclusions was unequally distributed in female CGG KI mice, with some fields of view having a greater density of inclusions than others.
2.4. Localization of intranuclear inclusions in Bergmann glia
Ubiquitin-positive intranuclear inclusions were found diffusely in Bergmann glia in the cerebellum of 48–52 week old female CGG KI mice with 140–200 CGG repeat expansions. Compared to male CGG KI mice, these inclusions were relatively rare, being found only about 25% as often in 48–52 week old female CGG KI mice with 140–200 CGG repeats, a 75% reduction compared to the male CGG KI mice. Bergmann glia in all other age and CGG repeat size groups of female CGG KI mice only rarely showed intranuclear inclusions. When identified, these inclusions were always clearly defined, round, and localized to the nucleus. Similar to what has been reported in male CGG KI mice, there was no evidence for reactive astroglia throughout the brain or Bergmann gliosis in the cerebellum of CGG KI mice as shown by GFAP and S100β staining in female CGG KI mice.
2.5. Pathologic features in microglia and oligodendroglia
Similar to male CGG KI mice, female CGG KI mice had ubiquitin positive masses with an amorphic or vacuolar appearance in the somal cytoplasm of approximately 10–15% of microglia examined in CGG KI mice and in under 1% of oligodendroglia examined (masses in microglia shown in Fig. 5). In the female CGG KI mice these masses were 3–10 μm in diameter at the widest point. Although similar cytoplasmic masses were present in wildtype mice, they were on only 2–5 μm in diameter and had a more rounded, less amorphic appearance. Also, these features were only present in 2–5% of the microglia examined in wildtype littermate mice to the female CGG KI mice. Similarly to what was reported by Wenzel et al. (2010), the majority of those microglial cytoplasmic masses in female CGG KI mice demonstrated autofluorescence in control sections in which the antibodies were omitted. In order to determine if this autofluorescence was due to accumulation of lipofuscin, sections were treated with CuSO4 (Schnell et al., 1999). This procedure did not affect autofluorescence, suggesting that it was not lipofuscin-related. The source of autofluorescence of these cytoplasmic inclusions remains to be determined.
Fig. 5.
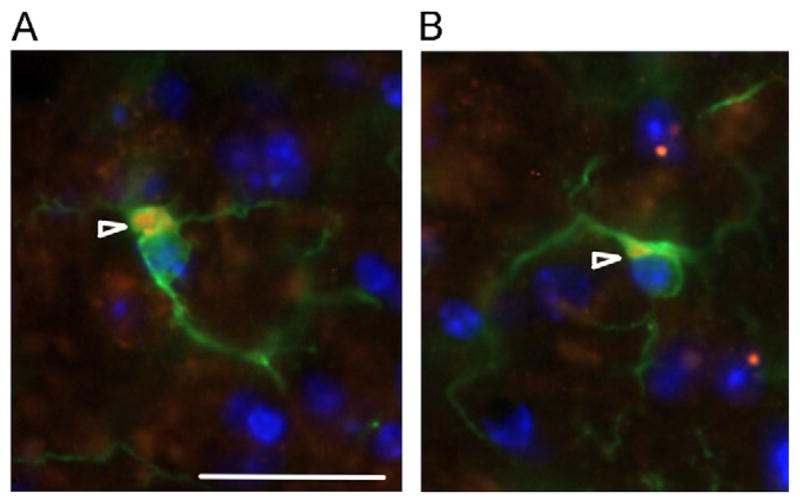
Intracellular masses in microglia. (A) Intracellular masses in CGG KI mice and (B) wildtype mice at 72 weeks of age. In microglia there appear to be intracellular, cytoplasmic masses (arrowheads) that stain positive for ubiquitin (red) as well as iba1 (green) in microglia. Nuclei stained blue with DAPI. These masses appear larger and less organized in wildtype mice (B) compared to female CGG KI mice (A). Original magnification 1000 ×. Scale bar = 10 μm. The image in A came from a female CGG KI mouse with 11 CGG repeats in the Fmr1 gene one X chromosome and 128 on the second Fmr1 gene and the image in B came from a wildtype littermate with both Fmr1 genes containing 10 CGG repeats. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Any role for these pathologic features remains unknown, but it is clear that there is an effect for age in the development of these glial pathologies, as the brains from CGG KI mice at 24–28 weeks of age only rarely contained these features, whereas the CGG KI mice at 48–52 weeks of age consistently showed these features throughout the brain, regardless of CGG repeat length. It is important to note that female wildtype mice also showed similar microglial features, but only after 60–70 weeks of age (cf., Fig. 5). This is similar to what was reported in male wildtype mice by Wenzel et al. (2010).
3. Discussion
Female CGG KI mice heterozygous for the fragile X premutation develop intranuclear inclusions in neurons and astroglia that are widely distributed throughout the brain. The numbers of inclusions identified in female CGG KI mice were compared to numbers in male CGG KI mice from Brouwer et al. (2008), Willemsen et al. (2003), and Wenzel et al. (2010). However, although widespread, the numbers of inclusions in neurons in female CGG KI mice were only approximately 25–30% fewer than that seen in male mice of similar age and CGG repeat length. This was somewhat unexpected because a reduction closer to 50% would be expected if random X-inactivation determined the degree of histopathology. More inclusions than expected in the brains of female CGG KI mice based on X-inactivation is consistent with the observation that intranuclear inclusions in the brains of female carriers of the Fragile X premutation are also more frequent than previously thought (Tassone et al., 2012). These findings are important because female carriers have been assumed to be largely unaffected cognitively and pathologically by the fragile X premutation. While the numbers of inclusions in neurons in female CGG KI mice were less than expected by X-inactivation, inclusions in astrocytes did approximate the predicted 50% of that seen in astrocytes of male CGG KI mice. These findings indicate that differences in the factors that influence inclusions formation in neurons versus astrocytes.
These data are important as in humans, female carriers of the premutation have been assumed largely cognitive and pathologically unaffected by the premutation (Hunter et al., 2010, 2011, 2012). FXTAS occurs in approximately 40% of male fragile X premutation carriers (Jacquemont et al., 2004a). In contrast, penetrance of FXTAS is reduced (range between 8% and 17%) in female premutation carriers compared to males (Chonchaiya et al., 2010a,b; Coffey et al., 2008; Jacquemont et al., 2005; Rodriguez-Revenga et al., 2009), and this has been thought to be due primarily to the protective effect of the normal allele on the second X chromosome (Berry-Kravis et al., 2005; Jacquemont et al., 2005).
Despite the reduced penetrance of FXTAS in female carriers of the premutation, it remains important to evaluate the pathologic features in both female FXTAS cases as well as female CGG KI mice used as a model of FXTAS. Such studies are necessary to inform research about the development and progression of disorders associated with the premutation leading to the full clinical manifestation of FXTAS. Contrary to the X-inactivation hypothesis, female CGG KI mice showed 75% the neuropathological features as male mice in neurons (25% reduction), but did show the predicted 50% the pathology in astroglia, replicating the finding in human female premutation carriers (Tassone et al., 2012).
Examination of H&E and luxol fast blue strained brain tissue section showed no gross pathological features and no evidence for white matter pathology in brain compared to wildtype mice, including no evidence for white matter pathology in periventricular regions or in the murine homolog of the middle cerebellar peduncle in female CGG KI mice (cf., Brunberg et al., 2002; Hagerman et al., 2004; Jacquemont et al., 2004a,b, 2005).
Although females are thought to have milder symptoms of FXTAS than males, those who do develop FXTAS may have neurological and psychiatric symptoms that are at least as severe as those of their male counterparts (Adams et al., 2007; Karmon and Gadoth, 2008). Moreover, and somewhat paradoxically, female premutation carriers with and without FXTAS symptomatology show a higher incidence of immunological disorders, such as thyroid disease and fibromyalgia, than male premutation carriers with FXTAS (Coffey et al., 2008; Hunsaker et al., 2011a; Leehey et al., 2011; Rodriguez-Revenga et al., 2009), as well as elevated prevalence of depression and anxiety (Hamlin et al., 2011; Roberts et al., 2009). Hypertension, seizures, and peripheral neuropathy also have higher prevalence in female premutation carriers with FXTAS compared to premutation carriers that do not how FXTAS symptomatology (Chonchaiya et al., 2010b), as has the co-occurrence of multiple sclerosis and Alzheimer’s disease (cf., Greco et al., 2008; Tassone et al., 2012). To date, these features have not been compared between genders, but preliminarily it appears these features are more prevalent in female over male FXTAS patients. Additionally, in a previous report evaluating pathological features in the autonomic nervous system and somatic organ systems of human FXTAS and CGG KI mice, female CGG KI mice and a female premutation carrier with FXTAS showed comparable cardiac pathology. Female CGG KI mice also show pathologic features in both the pineal and pituitary glands (Hunsaker et al., 2011b).
An important feature of female CGG KI mice heterozygous for the fragile X premutation is that they model a number of cognitive and behavioral features reported in human female premutation carriers (Al-Hinti et al., 2007; Goodrich-Hunsaker et al., 2011a,b,c; Hunsaker et al., 2010, 2011c). What remains unclear is if there are any relationships between performance on cognitive tasks and the increase in neuropathological features present in the female CGG KI mice similar to that suggested in male CGG KI mice (e.g., increase in the number intranuclear inclusions associated with worsening of spatial processing; cf., Hunsaker et al., 2009).
In the context of FXTAS, there are at least two consequences of inclusion formation. First, inclusions may be directly pathological and contribute to the cognitive deficits observed in the fragile X premutation and FXTAS. Alternatively, inclusions may not contribute to symptomatology and may be more simply formed as a reaction to the expanded CGG repeats on the FMR1 mRNA and serve to sequester expanded CGG repeat-containing RNA and associated RNA binding proteins. The latter possibility suggests a neuroprotective effect of inclusions similar to that proposed in Huntington’s disease (cf., Wenzel et al., 2010). In CGG KI female mice (Diep et al., 2012; Hunsaker et al., 2011a,c) and in male CGG KI mice there have been reports suggesting inclusion presence may correlate with cognitive deficits, but no causative relationship was elucidated (Hunsaker et al., 2009). More work must be done to establish the connection between inclusion formation and cognitive deficits.
In male CGG KI mice, the presence of intranuclear inclusions in granule cell populations was commonly observed from 3 months of age, whereas these pathological features were only rarely seen in female CGG KI mice at this age. These data support hypotheses that higher expression of the Fmr1 mRNA with expanded CGG repeats in male premutation carriers relative to female premutation carriers (due to X inactivation) results in more profound neurocognitive deficits and more numerous pathological features in males than females (Berry-Kravis et al., 2005; Jacquemont et al., 2005; Tassone et al., 2012). However, previous studies have found that X-inactivation patterns in patients with other X-linked mental retardation disorders preferentially inactivate the X-chromosome carrying the mutated or diseased allele (Plenge et al., 2002; Zhang et al., 2009).
It remains surprising that we observed less than 50% of the numbers of intranuclear inclusions formed in female CGG KI mice compared to male CGG KI mice. If X-inactivation patterns were completely random then we would expect to see a 50% reduction to the number of the inclusions in female mice compared to what is seen in male CGG KI mice. Alternatively, if X-inactivation patterns were skewed towards inactivation of the premutation Fmr1 gene then we would expect to see <50% inclusion formation. A study in two human sisters with FXTAS showed that X-inactivation could favor either X-chromosome, leading to the expression of disparate pathologic phenotypes (Berry-Kravis et al., 2005). Our results provide potential evidence that X-inactivation may favor the X-chromosome carrying the premutation, at least in the CGG KI mouse model of the fragile X premutation. Evaluating both male and female CGG KI mice across a wide range of CGG repeat lengths and ages may serve to elucidate the effects of genetic dosage for expression of the premutation allele on the premutation phenotype.
The present study, however, was not directly able to elucidate any direct relationships between X inactivation and intranuclear inclusion formation. Specific limitations include the fact that the inclusion formation rates are largely being compared across studies and are not necessarily from littermate controls or from the same laboratory. To minimize this limitation, we evaluated as many littermate controls as possible (including the mice included in Wenzel et al. (2010) that were littermates with a number of female mice included in the present study), but there were a number of female mice evaluated without corresponding male CGG KI comparison mice. Secondly, the altered inclusion formation rates could be cell autonomous or non-autonomous, which could explain any differences among CGG repeats and ages that we have observed, a mechanism similar to that proposed by Garden and La Spada (2012) for a number of neurodegenerative disorders. Third, the use of inclusion formation as readout for X inactivation is too disconnected to be used as an appropriate readout. Follow-up analyses directly quantifying X inactivation need to be performed and directly compared with inclusions number to determine any relationships.
In summary, this report provides new information on the prevalence and distribution of intranuclear inclusions in female CGG KI mice, as well as the relationship between frequency of inclusions, age, and CGG repeat length. Female CGG KI mice heterozygous for the fragile X premutation may serve to model a number of features of the fragile X premutation and FXTAS pathology that male CGG KI mice may not model as completely, including potential immunologic and psychiatric disturbances. These studies highlight the need to include female CGG KI mice, and female carriers of the fragile X premutation in future studies in view of evidence that pathology, but histopathological and neurobehavioral, are more severe than previously predicted.
4. Experimental procedures
4.1. Mice
Thirty-four female CGG KI mice heterozygous for the fragile X premutation (range 71–326 CGG repeats) at 3–13 months of age and 16 female wildtype mice of the same age range were included in this study. The included wildtype mice were littermates with one of the CGG KI mice described in the study. All female CGG KI mice were bred onto a congenic C57BL/6J background over 9 to >12 generations from founder mice originally on a mixed FVB/N x C57BL/6J background. Sections from the male mice reported by Hunsaker et al. (2009) and Wenzel et al. (2010) were evaluated as well for direct comparison with the female CGG KI mice, as these mice were littermates with the female CGG KI mice evaluated for the present report. Mice were housed in same sex, mixed genotype groups with three or four mice per cage in a temperature and humidity controlled vivarium on a 12 h light-dark cycle. Mice had ad libitum access to food and water throughout their lifespan. All experiments conformed to University of California, Davis IACUC approved protocols and all effort was taken to reduce stress in all mice.
4.2. Genotyping
As modest somatic instability of CGG repeats among tissues in the CGG KI mouse has been shown to be negligible (typically under 10 CGG repeats across tissues; cf., Berman and Willemsen, 2009; Willemsen et al., 2003), genotyping to verify CGG repeat length was carried out upon tail snips. Following a method kindly provided by Rob Willemsen (Brouwer et al., 2007, 2008; Willemsen et al., 2003) and modified in collaboration with the laboratories of Tassone and Hagerman (Saluto et al., 2005, personal communication), CGG repeat lengths were measured using the FastStart Taq DNA Polymerase, dNTP Pack (Roche Diagnostics; Manheim, Germany) DNA was extracted from mouse tails by incubating with 10 mg/mL Proteinase K (Fermentas, Inc.; Glen Burnie, MD) in 300 μL lysis buffer containing 50 mM Tris–HCl, pH 7.5, 10 mM EDTA, 150 mM NaCl, 1% SDS overnight at 55 °C. One hundred microliters (100 μL) saturated NaCl was then added, mixed and centrifuged. The supernatant was gently mixed with two volumes of 100% ethanol, and the DNA was pelleted by centrifugation. The DNA was washed and centrifuged in 500 μL 70% ethanol. The DNA was then dissolved in 100 μL milliQ-H2O. CGG repeat lengths were determined by PCR using solutions from the FastStart Taq DNA Polymerase, dNTP Pack (Roche Diagnostics). Briefly, approximately 500–700 ng of DNA was added to 20 μL of PCR mixture containing 0.5 μM/L of each primer, 250 μM/L of each dNTP (Roche Diagnostics ). 2.5 M Betaine (Sigma-Aldrich), 1× Buffer 2 and 0.05 U of FastStart Taq DNA Polymerase (Roche Diagnostics) The primers flank the CGG repeat region of Fmr1 gene, the forward primer was 5′-CGG GCA GTG AAG CAA ACG-3′ and the reverse primer was 5′-CCA GCT CCT CCA TCT TCT CG-3′ The CGG repeats were amplified using a 3′-step PCR with 10 min denaturation at 98 °C, followed by 35 cycles of 35 s denaturation at 98 °C, annealing for 35 s at 55 °C, and at the end each cycle elongation for 2 min at 72 °C. The last step of the PCR consisted in a 10 min elongation at 72 °C too. The sizes of CGG DNA amplicons were determined by running 20 μL of PCR reaction per sample and a molecular weight marker (O’GeneRuler 50 bp DNA ladder; Fermentas, Inc.) for 2 h at 150 V on a 2.5% agarose gel with 0.03 μL/ml ethidium bromide. The number of CGG repeats was calculated from pictures acquired with a GelDoc-It Imaging system (UVP, LLC Upland, CA) and using VisionWorks LS software (UVP, LLC Upland, CA) (Brouwer et al., 2008, 2007). For female CGG KI mice heterozygous for the fragile X premutation there were two bands present, one corresponding to the wildtype allele (8–12 CGG repeats), and another corresponding to the premutation allele (generally 70–200 CGG repeat length. This method can detect up to 358 CGG repeats from animals in the present mouse colony). For female wildtype mice, only the normal Fmr1 allele was present. Genotyping was performed twice on each mouse, once using tail snips taken at weaning and again on tail snips and/or brain tissue collected at sacrifice. In all cases the genotypes matched. For CGG repeat lengths ≫200 repeats, the genotypes were evaluated a third time, and always matched the previous two genotype results.
4.3. General tissue preparation
CGG KI and wildtype mice were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.; Euthasol, Virbac AH, Inc., Fort Worth, TX), then intracardially perfused with 12 mL isotonic heparinized saline (1000 U heparin/mL saline) over 1 min, followed by 60 mL of a solution of freshly prepared, slightly chilled 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (PB; pH 7.4) via gravity feed over 20 min. The brains were immediately removed from the skull and placed in the same fixative for 1 h at 4 °C with gentle agitation on a shaker table. After post-fixation, the brains were rinsed twice in 0.1 M PB, cryoprotected in 10% sucrose in 0.1 M PB for 1 h, followed by 30% sucrose in 0.1 M PB for 24 h at 4 °C, then frozen on dry ice for 1 h. Frozen brains were stored at −80 °C to await sectioning. Thirty-micrometer (30 μm) parasagittal serial sections were cut on a sliding microtome equipped with a freezing stage and collected into series of every fifth section directly into 30% sucrose, flash frozen on dry ice for 30 min, and stored at −80 °C until further processing.
Single sets of sections were selected for further processing, which included cresyl violet, thionin, and/or H&E staining for general histological evaluation, and luxol fast blue staining with a neutral red counterstain to evaluate white matter integrity. Immunohistochemistry for neuronal and glial cell markers was used to identify cell type as well as for ubiquitin to visualize intranuclear inclusions in different cell classes. Ubiquitin stained intranuclear inclusions are the hallmark pathology of FXTAS patients and are also features of CGG KI mice (Brouwer et al., 2007, 2008; Entezam et al., 2007; Greco et al., 2002, 2006, 2008; Hunsaker et al., 2009, 2011a,b; Tassone et al., 2012; Wenzel et al., 2010; Willemsen et al., 2003).
For the human tissue reported, formalin-fixed brain postmortem tissue was processed for paraffin sections, and histological and immunohistological staining in standard fashion as previous reported (Greco et al., 2002, 2006, 2008; Hunsaker et al., 2011a,b; Tassone et al., 2012). Tissue blocks of frontal cortex and hippocampus from a patient with FXTAS were sectioned in 7 μm sections. The case reported is Case 1 from Tassone et al. (2012) and all clinical information on this case is provided in that manuscript. This case was obtained in accordance to UC Davis approved IRB protocols.
4.4. Immunohistochemistry
Immunocytochemical and immunofluorescence techniques were used to visualize the occurrence and distribution of intranuclear and cytoplasmic inclusions, with a focus on their presence or absence in glial cells (i.e., astroglia, oligodendroglia, and microglial cells) of female wildtype and CGG KI mice. Subsets of alternate sections were processed for immunohistochemistry using a modification of the avidin–biotin complex (ABC)–peroxidase technique as previously described (Hunsaker et al., 2009, 2011b,c; Wenzel et al., 2010). Briefly, free-floating sections were rinsed in 0.1 M phosphate buffer PB (pH 7.4), and pretreated with 0.1% sodium borohydride for 15 min followed by treatment with 0.5–2% H2O2 in PB for 90 min to inactivate endogenous peroxidases. Sections were then treated with 3% goat, horse, or swine serum as appropriate (Sigma-Aldrich; DAKO, Inc., Carpinteria, CA) and 0.3% Triton X-100 (TX-100) in 0.01 M PB with 0.15 M NaCl, pH 7.4 (PBS) for 1 h to reduce nonspecific staining. Sections were rinsed in 0.01 M PBS for 30 min and incubated for 48–72 h at 4 °C in the various antibodies and dilutions: mouse monoclonal or polyclonal anti-glial fibrillary acidic protein (GFAP; ICN Biomedicals; Irvine, CA; DAKO, Inc.), 1:4000 (1:1000 for immunofluorescence (IF)); rabbit polyclonal anti-S100 β (Abcam, Inc., Cambridge, MA), 1:1000; mouse monoclonal anti-myelin basic protein (MBP; ICN Biochemicals), 1:500; rabbit polyclonal anti-Iba1 (ionized calcium binding adapter molecule 1; Wako Chemicals USA, Inc., Richmond, VA), 1:2000 (1:1000 for IF); and rabbit polyclonal and mouse monoclonal antibodies against ubiquitin (DAKO, Inc.; Abcam, Inc.) 1:2000 and 1:1000, respectively (1:1000 for IF), in PBS containing 1% goat, horse or swine serum, 2% BSA, and 0.3% TX-100. Following rinses for 2 h in PBS, sections were incubated in biotinylated goat or swine anti-rabbit IgG or horse anti-mouse IgG (DAKO, Inc.; Vector Laboratories, Burlingame, CA), diluted 1:500 for 24 h at 4 °C, rinsed 2 h in PBS and then incubated in ABC (Elite ABC Kit, Vector Laboratories), diluted 1:500 in 1% goat or horse serum, 2% BSA, 0.3% TX-100, and PBS for 24 h at 4 °C. Sections were rinsed thoroughly in PB (pH 7.4), then transferred to Tris–HCl buffers (TB; pH 7.4, 7.6), and then incubated for 15 min in 0.025% 3,3′-diaminobenzidine (DAB, Sigma) in TB (pH 7.6). After reacting for 5–10 min in fresh DAB with 0.003% H2O2 until staining was optimized on a positive control section, sections were rinsed in TB, followed by PB, and mounted on gelatinized slides. In some experiments the DAB included 0.05% nickel ammonium sulfate to enhance the visibility of the DAB reaction product after counterstaining with hematoxylin. For all experiments female CGG KI and wildtype mice were run in parallel.
4.5. Immunofluorescence staining
For single- and double-immunofluorescent labeling of ubiquitin co-localized with neuronal/glial cell markers, sections were transferred into 10% sucrose in 0.1 M PB, then rinsed in 0.1 M PB, and treated with 0.1% sodium borohydride for 15 min. Thereafter, sections were rinsed again with 0.1 M PB and then permeabilized with 0.5% H2O2 in 0.1 M PB for 15 min followed by rinses in 0.1 M PB and 0.01 M PBS. Free-floating sections were treated with 10% goat or horse serum in 0.01 M PBS containing 0.3% TX-100 (vehicle) for 1 h and then incubated overnight at 4 °C in vehicle containing different combinations of mouse monoclonal/rabbit polyclonal antibodies of different IgG isotypes (see above). After rinses in 0.01 M PBS and 10% goat or horse serum, sections were incubated in isotype-specific Alexa-conjugated secondary antibodies (1:2000): Alexa 568-labeled goat anti-rabbit IgG and/or Alexa 488-labeled goat anti-mouse IgG (Invitrogen, Carlsbad, CA) for 1 h. Following rinses in 10% goat or horse serum followed by 0.01 M PBS, sections were mounted on gelatin-coated slides and coverslipped with mounting medium containing DAPI (4′,6-diamidino-2-phenyindole di-lactate) for nuclear staining (Vectashield “Hard Set”, Vector Laboratories).
To differentiate between specific immunofluorescent labeling and nonspecific autofluorescence resulting from accumulations of lipofuscin in aging brains, sections were first treated with 10 mM copper sulfate solution in 50 mM ammonium acetate buffer (pH 5.0) for 20 min (Schnell et al., 1999). This treatment reduced autofluorescence but did not significantly affect the intensity of specific immunofluorescent labeling, although immunofluorescent staining for ubiquitin within the neuronal cytoplasm was subtly enhanced, similar to previous reports in male CGG KI mice and human tissues (Hunsaker et al., 2011b,c; Wenzel et al., 2010). For all experiments female CGG KI and wildtype mice were run in parallel.
4.6. Cell identification and evaluation of intranuclear inclusions
Stained tissue sections were analyzed using either a Nikon ECLIPSE E600 microscope with epifluorescence attachment and/or a Zeiss LSM 510 META confocal microscope. In the case of the confocal microscope, single plane confocal images were collected. Images were analyzed to verify the presence of ubiquitin-positive intranuclear inclusions in different cell types identified with various neuronal and glial cell markers in brains of female CGG KI and wild-type mice.
Cresyl violet, thionin, and/or H&E-stained sets of serial sections from wild-type and CGG KI mouse brains at different ages were used for comparison and evaluation of gross anatomical differences. Identification of different cell types in the brain was carried out based on standard morphological criteria using H&E, cresyl violet, or thionin staining, and immunochemistry using specific neuronal and glial cell markers (Wenzel et al., 2010). Neurons were identified by their size, large round nuclei, single or multiple nucleoli or heterochromatin, and their abundant cytoplasm, as well as by using specific neuronal markers. Astroglia were identified by their round/ovoid nuclei with light euchromatin, and absence of nucleoli and cytoplasm. In addition, GFAP and S100β immunohistochemistry and/or immunofluorescence were used to identify subpopulations of astroglia based on their differing immunoreactivities (i.e., protoplasmic, velate, and/or fibrous astroglia) in different brain regions.
To estimate the percentage of astroglia with intranuclear inclusions, the number of astroglia and intranuclear inclusions was counted manually in 10 high power (i.e., 400 ×) microscopic fields on two GFAP- and ubiquitin-immunoreacted sections from 20 CGG KI mice within layer I of the neocortex (including sensory and motor areas) using an optical imaging system (StereoInvestigator, MBF Biosciences; Williston, VT). This method precisely replicated the methods used in male CGG KI mice (Wenzel et al., 2010). Wildtype mice were similarly analyzed, though intranuclear inclusions were never observed in wildtype mice. Notably, a similar number of glial cells were counted in wildtype mice as CGG KI mice, suggesting a similar number of astroglia in CGG KI mice and wildtype mice, at least in layer I of the motor and somatosensory neocortex.
Oligodendroglia were identified based on their typically small, round, hyperchromatic nuclei (which did not allow differentiation between nucleus, nucleoli, or cytoplasm), localization in white and gray matter, and confirmed by immunohistochemistry for myelin basic protein (MBP). Microglia were identified primarily from Iba1 immunostaining, which displayed small cell bodies with a round nucleus and fine, ramified processes that are characteristic of resting microglia, as well as on cellular morphology from H&E stained tissues. Microglial cells with retracted and/or hypertrophic processes on Iba1 immunostained sections were defined as activated (but non-phagocytic) microglia, which can be transformed into phagocytotic cells (i.e., brain macrophages; Graeber and Moran, 2002). Immunocytochemical staining for ubiquitin was used to specifically label intranuclear inclusions in combination with cell-specific markers to identify the cell type. Whenever possible, immunocytochemical experiments using immunoperoxidase and immunofluorescence methods were carried out in parallel to verify staining patterns as well as to facilitate quantification.
4.7. Semiquantitative analysis of intranuclear inclusions in neurons
To evaluate the relative number and distribution of inclusions across brain regions, 6 female CGG KI mice 48–52 weeks of age with 70–100 CGG repeats, 6 female CGG KI mice 48–52 weeks of age with 140–200 CGG repeats, 6 female CGG KI mice 24–28 weeks of age with 70–100 CGG repeats, and 6 female CGG KI mice 24–28 weeks of age with 140–200 CGG repeats were processed for H&E staining. Standard histological sections in the parasagittal plane were selected for quantification. The following regions were evaluated: prelimbic cortex, motor cortex, parietal cortex, visual cortex, anterior cingulate cortex, piriform cortex, ventromedial and dorsolateral entorhinal cortex, the dentate gyrus, CA3, CA1, and subiculum in the hippocampus, the central, basolateral, and posterior nuclei of the amygdala, the medial and lateral septal nuclei, the lateral geniculate nucleus (LGN), the inferior and superior colliculi, medial vestibular nucleus, and lobe III in the inferior lobe of the cerebellum and the flocculus in the cerebellum. The semiquantitative analysis involved counting 200 cells at 1000 × magnification in each region of interest across mice at 20 × magnification. The 200 counted cells were selected randomly within the region of interest using the optical imaging system. A limited sample of female CGG KI mice with ≫200 CGG repeats and CGG KI mice at 3 months of age were evaluated in a similar manner, but the data from these mice were not directly compared to the other groups due to a limited number of mice evaluated. In all cases, adjacent sections were also immunoperoxidase stained for ubiquitin were compared with H&E stained sections to verify inclusion presence, but quantifications were performed using H&E stained tissues.
Differences among age groups were evaluated by computing Pearson’s correlation coefficients comparing CGG repeat length and inclusion number within each age group. Data across age groups were not statistically evaluated. Differences between the slope of the correlations the age group were evaluated using the standard error of the different slopes. These analyses were performed using the R 14.1 statistical environment (R Development Core Team, 2011).
Acknowledgments
The authors would like to acknowledge the contributions of Chou-Yu (James) Fan, Natalie N. Nguyen, Aimee Keyashian, and Kimberley C. D’Antonio for assistance with sectioning tissues used for this study, as well as Binh T. Ta and Dr. Dolores Garcia-Arocena, Ph.D. for assistance with mouse genotyping. The authors would also like to thank Dr. H. Jürgen Wenzel M.D., D.Sc. for helpful discussions concerning histological and immunohistological techniques.
Footnotes
Funding: This work was supported by National Institute of Health (NIH) Grants, NINDS RL1 NS062411 to RFB and RW. This work was also made possible by a Roadmap Initiative Grant (UL1 DE019583) from NIDCR in support of the NeuroTherapeutics Research Institute (NTRI) consortium; by a Grant (UL1 RR024146) from the NCRR to MRH; and an NSF Center for Biophotonics Science and Technology Internship to EWS. The contents of this manuscript are solely the responsibility of the authors and do not represent the official view of NCRR, NINDS, NIDCR, or NIH.
References
- Adams JS, Adams PE, Nguyen D, Brunberg JA, Tassone F, Zhang W, Koldewyn K, Rivera SM, Grigsby J, Zhang L, DeCarli C, Hagerman PJ, Hagerman RJ. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2007;69:851–859. doi: 10.1212/01.wnl.0000269781.10417.7b. [DOI] [PubMed] [Google Scholar]
- Al-Hinti JT, Nagan N, Harik SI. Fragile X premutation in a woman with cognitive impairment, tremor, and history of premature ovarian failure. Alzheimer Dis Assoc Disord. 2007;21:262–264. doi: 10.1097/WAD.0b013e31811ec130. [DOI] [PubMed] [Google Scholar]
- Allen Mouse Brain Atlas [Internet] Seattle (WA): Allen Institute for Brain Science ©; 2009. Available from: 〈 http://mouse.brain-map.org〉. [Google Scholar]
- Berman RF, Willemsen R. Mouse models of fragile X-associated tremor ataxia. J Invest Med. 2009;57 (8):837–841. doi: 10.231/JIM.0b013e3181af59d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Potanos K, Weinberg D, Zhou L, Goetz CG. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann Neurol. 2005;57:144–147. doi: 10.1002/ana.20360. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Huizer K, Severijnen LA, Hukema RK, Berman RF, Oostra BA, Willemsen R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107 (6):1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, Van der Linde HC, Nelson DL, Oostra BA, Willemsen R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated fragile X full mutation. Exp Cell Res. 2007;313 (2):244–253. doi: 10.1016/j.yexcr.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis E, Grigsby J, Leehey M, Tassone F, Brown WT, Greco C, Hagerman PJ. Fragile X premutation carriers: characteristic MR imaging findings in adult males with progressive cerebellar and cognitive dysfunction. Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Nguyen DV, Au J, Campos L, Berry-Kravis EM, Lohse K, Mu Y, Utari A, Hervey C, Wang L, Sorensen P, Cook K, Gane L, Tassone F, Hagerman RJ. Clinical involvement in daughters of men with fragile X-associated tremor ataxia syndrome. Clin Genet. 2010a;78:38–46. doi: 10.1111/j.1399-0004.2010.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Tassone F, Ashwood P, Hessl D, Schneider A, Campos L, Nguyen DV, Hagerman RJ. Autoimmune disease in mothers with the FMR1 premutation is associated with seizures in their children with fragile X syndrome. Hum Genet. 2010b;128:539–548. doi: 10.1007/s00439-010-0882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J, Doerflinger M, Hagerman PJ, RJH Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep AA, Hunsaker MR, Kwock R, Kim K, Willemsen R, Berman RF. Female CGG knock-in mice modeling the fragile X premutation are impaired on a skilled forelimb reaching task. Neurobiol Learn Mem. 2012;97 (2):229–234. doi: 10.1016/j.nlm.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene. 2007;395 (1–2):125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, La Spada AR. Intercellular (mis)communication in neurodegenerative disease. Neuron. 2012;73 (5):886–901. doi: 10.1016/j.neuron.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Srivastava S, Tassone F, Harvey D, Rivera SM, Simon TJ. Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments. Brain Cogn. 2011a;75 (3):255–260. doi: 10.1016/j.bandc.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Tassone F, Harvey D, Rivera SM, Simon TJ. Adult female fragile X premutation carriers exhibit age- and CGG repeat length-related impairments on an attentionally based enumeration task. Front Hum Neurosci. 2011b;5:63. doi: 10.3389/fnhum.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Tassone F, Harvey D, Rivera SM, Simon TJ. Enhanced manual and oral motor reaction time in young adult female fragile X premutation carriers. J Int Neuropsychol Soc. 2011c;21:1–5. doi: 10.1017/S1355617711000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Moran LB. Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol. 2002;12 (3):385–390. doi: 10.1111/j.1750-3639.2002.tb00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, Tassone F, Chudley A, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, Brunberg JA, Hagerman PJ, Hagerman RJ. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol. 2008;65:1114–1116. doi: 10.1001/archneur.65.8.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hagerman PJ. Testing for fragile X gene mutations throughout the life span. J Am Med Assoc. 2008;300:2419–2421. doi: 10.1001/jama.2008.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, Tassone F, Hessl D, Harris SW, Zhang L, Jardini T, Gane LW, Ferranti J, Ruiz L, Leehey MA, Grigsby J, Hagerman PJ. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin A, Liu Y, Nguyen DV, Tassone F, Zhang L, Hagerman RJ. Sleep apnea in fragile X premutation carriers with and without FXTA. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:923–928. doi: 10.1002/ajmg.b.31237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Goodrich-Hunsaker NJ, Willemsen R, Berman RF. Temporal ordering deficits in female CGG KI mice heterozygous for the fragile X premutation. Behav Brain Res. 2010;213 (2):263–268. doi: 10.1016/j.bbr.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, Kros JM, Severijnen LA, Berry-Kravis EM, Berman RF, Hagerman PJ, Willemsen R, Hagerman RJ, Hukema RK. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011a;122:467–479. doi: 10.1007/s00401-011-0860-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Tassone F, Berman RF, Willemsen R, Hagerman RJ, Hagerman PJ. Rare intranuclear inclusions in the brains of 3 older adult males with fragile X syndrome: implications for the spectrum of fragile X-associated disorders. J Neuropathol Exp Neurol. 2011b;70 (6):462–469. doi: 10.1097/NEN.0b013e31821d3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, von Leden RE, Ta BT, Goodrich-Hunsaker NJ, Arque G, Kim K, Willemsen R, Berman RF. Motor deficits on a ladder rung task in male and female adolescent and adult CGG knock-in mice. Behav Brain Res. 2011c;222 (1):117–121. doi: 10.1016/j.bbr.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Wenzel HJ, Willemsen R, Berman RF. Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav Neurosci. 2009;123 (6):1315–1324. doi: 10.1037/a0017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JE, Epstein MP, Tinker SW, Abramowitz A, Sherman SL. The FMR1 premutation and attention-deficit hyper-activity disorder (ADHD): evidence for a complex inheritance. Behav Genet. 2011;42 (3):415–422. doi: 10.1007/s10519-011-9520-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JE, Rohr JK, Sherman SL. Co-occurring diagnoses among FMR1 premutation allele carriers. Clin Genet. 2010;77 (4):374–381. doi: 10.1111/j.1399-0004.2009.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JE, Sherman S, Grigsby J, Kogan C, Cornish K. Capturing the fragile X premutation phenotypes: a collaborative effort across multiple cohorts. Neuropsychology. 2012;26 (2):156–164. doi: 10.1037/a0026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Farzin F, Hall D, Leehey M, Tassone F, Gane L, Zhang L, Grigsby J, Jardini T, Lewin F, Berry-Kravis E, Hagerman PJ, Hagerman RJ. Aging in individuals with the FMR1 mutation. Am J Ment Retard. 2004a;109:154–164. doi: 10.1352/0895-8017(2004)109<154:AIIWTF>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R, Berry-Kravis E, Brown WT, Schaeffer S, Kissel J, Tassone F, Hagerman PJ. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, Herman K, Grigsby J, Greco CM, Berry-Kravis E, Tassone F, Hagerman PJ. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. J Am Med Assoc. 2004b;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Orrico A, Galli L, Sahota PK, Brunberg JA, Anichini C, Leehey M, Schaeffer S, Hagerman RJ, Hagerman PJ, Tassone F. Spastic paraparesis, cerebellar ataxia, and intention tremor: a severe variant of FXTAS? J Med Genet. 2005;42:e14. doi: 10.1136/jmg.2004.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmon Y, Gadoth N. Fragile X associated tremor/ataxia syndrome (FXTAS) with dementia in a female harbouring FMR1 premutation. J Neurol Neurosurg Psychiatry. 2008;79:738–739. doi: 10.1136/jnnp.2007.139642. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Leehey MA, Legg W, Tassone F, Hagerman R. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology (Oxford) 2011;50:2233–2236. doi: 10.1093/rheumatology/ker273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Skewed X-chromosome inactivation is a common feature of x-linked mental retardation disorders. Am J Hum Genet. 2002;71:168–173. doi: 10.1086/341123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2011. URL: 〈 http://www.R-project.org/〉. [Google Scholar]
- Raske C, Hagerman PJ. Molecular pathogenesis of fragile X-associated tremor/ataxia syndrome. J Invest Med. 2009;57:825–829. doi: 10.231/JIM.0b013e3181be329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JE, Bailey DB, Jr, Mankowski J, Ford A, Sideris J, Weisenfeld LA, Heath TM, Golden RN. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet Part B, Neuropsychiatr Genet. 2009;150B:130–139. doi: 10.1002/ajmg.b.30786. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, Kulisevsky J, Gomez B, Mila M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17:1359–1362. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluto A, Brussino A, Tassone F, Arduino C, Cagnoli C, Pappi P, Hagerman P, Migone N, Brusco A. An enhanced polymerase chain reaction assay to detect pre-and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn. 2005;7 (5):605–612. doi: 10.1016/S1525-1578(10)60594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47 (6):719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Tassone F, Berry-Kravis EM. The Fragile X-Associated Tremor Ataxia Syndrome (FXTAS) Springer; 2010. [Google Scholar]
- Tassone F, Greco CM, Hunsaker MR, Berman RF, Seritan AL, Gane LW, Jacquemont S, Basuta K, Jin L-W, Hagerman PJ, Hagerman RJ. Neuropathological, clinical, and molecular pathology in female fragile X premutation carriers with and without FXTA. Genes Brain Behav. 2012 doi: 10.1111/j.1601-183X.2012.00779.x. http://dxdoi.org/10.1111/j.1601-183X.2012.00779.x. [DOI] [PMC free article] [PubMed]
- Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41:e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, van Unen L, Tassone F, Hoogeveen AT, Hagerman PJ, Mientjes EJ, Oostra BA. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12 (9):949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Hunsaker MR, Greco CM, Willemsen R, Berman RF. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010;1318:155–166. doi: 10.1016/j.brainres.2009.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachnis AT, Roth HL, Heilman KM. Fragile X dementia Parkinsonism syndrome (FXDPS) Cogn Behav Neurol. 2010;23:39–43. doi: 10.1097/WNN.0b013e3181b6e1b9. [DOI] [PubMed] [Google Scholar]
- Yonath H, Marek-Yagel D, Resnik-Wolf H, Abu-Horvitz A, Baris HN, Shohat M, Frydman M, Pras E. X-inactivation testing for identifying a non-syndromic X-linked mental retardation gene. J Appl Genet. 2011;52:437–441. doi: 10.1007/s13353-011-0052-2. [DOI] [PubMed] [Google Scholar]
- Zhang L, Coffey S, Lua LL, Greco CM, Schafer JA, Brunberg J, Borodyanskaya M, Agius MA, Apperson M, Leehey M, Tartaglia N, Tassone F, Hagerman PJ, Hagerman RJ. FMR1 premutation in females diagnosed with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2009;80:812–814. doi: 10.1136/jnnp.2008.160960. [DOI] [PubMed] [Google Scholar]