

Abstract
Enhanced susceptibility to inflammatory and autoimmune disease can be related to impairments in HPA axis activity and associated hypocortisolism, or to glucocorticoid resistance resulting from impairments in local factors affecting glucocorticoid availability and function, including the glucocorticoid receptor (GR). The enhanced inflammation and hypercortisolism that typically characterize stress-related illnesses, such as depression, metabolic syndrome, cardiovascular disease or osteoporosis, may also be related to increased glucocorticoid resistance. This review focuses on impaired GR function as a molecular mechanism of glucocorticoid resistance. Both genetic and environmental factors can contribute to impaired GR function. The evidence that glucocorticoid resistance can be environmentally induced has important implications for management of stress-related inflammatory illnesses and underscores the importance of prevention and management of chronic stress. The simultaneous assessment of neural, endocrine, and immune biomarkers through various noninvasive methods will also be discussed.
Keywords: stress, cortisol, cytokines, autoimmune, depression, psychoneuroimmunology
Glucocorticoid resistance: from HPA axis to glucocorticoid receptor
Endogenous glucocorticoids play an important role in regulating homeostatic processes under basal and challenge conditions, including metabolism, immune function and behavior.1 The essential role of glucocorticoids in protecting the host from the detrimental consequences of an overactive inflammatory immune response has been well established.2-5 In 1989, we first showed that an impaired hypothalamic–pituitary–adrenal (HPA) axis was an important risk factor for susceptibility to and severity of inflammatory arthritis in autoimmune disease prone rats.6 Since that time, impaired HPA responsiveness has been shown in numerous animal models and human inflammatory and autoimmune diseases, including rheumatoid arthritis;7-10 Crohn’s disease, colitis, or inflammatory bowel disease;11-12 multiple sclerosis and its animal equivalent autoimmune encephalomyelitis (EAE);13 and the allergic conditions, asthma and dermatitis.14-15 HPA axis disturbances have also been demonstrated in somatic fatigue and pain disorders, such as chronic fatigue syndrome and fibromyalgia,16-18 and psychiatric disorders, such as depression and PTSD,19-23 which are also associated with an enhanced inflammatory state.18,24-32 This enhanced inflammatory susceptibility can be related to impairments at any level in the HPA axis, whether at the level of hypothalamic corticotrophin releasing hormone (CRH),33-35 pituitary ACTH6,36,37 or adrenal glucocorticoid secretion,6 leading to overall hypocortisolism (Fig. 1); or impairments in local factors affecting glucocorticoid availability and function, including the glucocorticoid receptor (GR) (see below), which can render a state of glucocorticoid resistance by preventing cells and tissues in the body from responding adequately to glucocorticoids.
Figure 1.
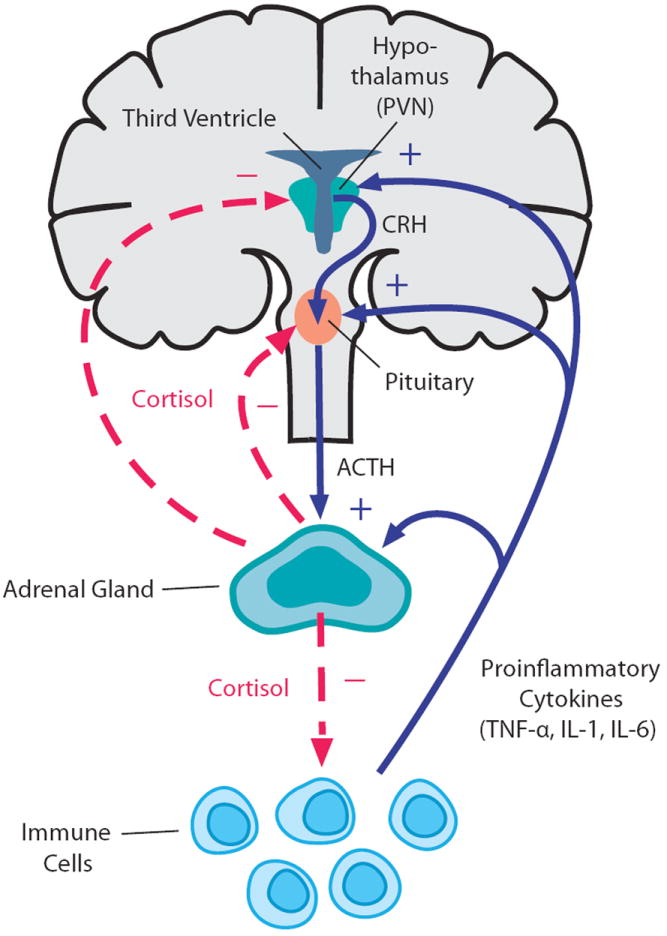
Bidirectional communication between the immune system and the HPA axis. Proinflammatory cytokines, such as TNF, IL-1, and IL-6, stimulate glucocorticoid release (cortisol in humans; corticosterone in rodents) by acting at all three levels of the HPA axis (solid blue lines). In turn, glucocorticoids negatively feedback on the immune system to suppress the further synthesis and release of proinflammatory cytokines (dashed red line). In addition, glucocorticoids regulate their own production through negative feedback on the upper levels of the HPA axis, including corticotropin-releasing hormone (CRH) in the paraventricular nucleus (PVN) of the hypothalamus and adrenocorticotropin (ACTH) in the anterior pituitary (dashed red lines). Reprinted with modifications.98
Once glucocorticoids are released into circulation, glucocorticoid availability at the cellular level is influenced by various local factors, including corticosteroid binding globulin (CBG, binding over 90% of circulating glucocorticoids), the multidrug resistance (MDR) P-glycoprotein transporter (an efflux pump that decreases intracellular concentrations of potentially toxic drugs or hormones) and 11β-hydroxysteroid dehydrogenase (11β-HSD, an enzyme with two isoforms: #1 converts inactive glucocorticoids into their active form (e.g., corticosterone in rodents; cortisol in humans); #2 breaks down active glucocorticoids into inactive metabolites). Enhanced CBG levels and MDR expression, as well as a decrease in 11β-HSD-1 or increase in 11β-HSD-2, reduce the levels of free/active glucocorticoids in the cell, contributing to a state of glucocorticoid resistance.10 Glucocorticoid effects are ultimately determined at the level of the GR. An impaired GR, whether as a consequence of reduced expression, binding affinity to its ligand, nuclear translocation, DNA binding or interaction with other transcription factors (i.e., NFκB, AP-1), could also lead to a state of glucocorticoid resistance,38,39 increasing an individual’s vulnerability to exaggerated inflammatory responses (Fig. 2). Taken together, even when circulating glucocorticoid concentrations are normal or elevated, impaired counter-regulatory control of immune responses can still occur at the cellular and molecular level.
Figure 2.
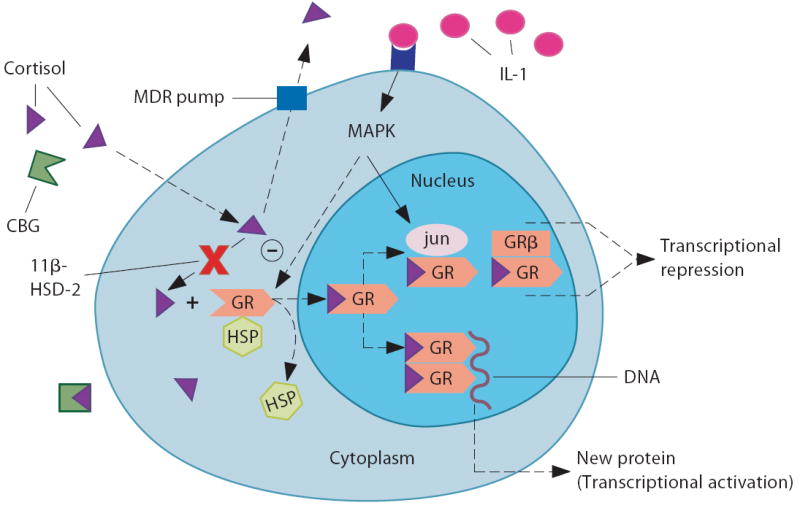
Glucocorticoid resistance can be attributed to changes in local/cellular factors regulating glucocorticoid bioavailability and action. (1) increased corticosterone binding globulin (CBG); (2) increased expression of the multidrug resistance transporter (MDR pump); (3) decreased expression of 11β-hydroxy steroid dehydrogenase (HSD)-1 or increased expression of 11β-HSD-2; (4) reduced glucocorticoid binding to the glucocortoid receptor (GRα); (5) reduced GR translocation from the cytoplasm to the nucleus, which could be affected by its phosphorylation state (via mitogen-activated protein kinase (MAPK) pathways); (6) increased GR interaction with inflammatory-related transcription factors, such as NFκB or AP-1 (jun/fos); and 7) increased GRα interaction with GRβ. HSP = heat shock protein. Reprinted with modifications.98
Since glucocorticoid resistance occurs through many different molecular mechanisms, conditions that appear phenotypically similar may have different molecular bases in different individuals or populations. When combined with the many molecular pathways that could potentially disrupt the HPA axis, at the level of CRH, ACTH, or glucocorticoid production, secretion, availability, or receptor function, the number of potential molecular mechanisms that might result in impaired HPA function may seem daunting. This review will focus on impaired GR function as a molecular mechanism of glucocorticoid resistance, which may play a role in the final common pathway of many inflammatory-related conditions.
Genetic and environmental factors can impair GR function
Both genetic and environmental factors can contribute to impaired GR function. Numerous polymorphisms and mutations of the GR have been linked with glucocorticoid resistance and associated with various disease states.40-42 Alternative splicing of the human GR primary transcript produces multiple isoforms.43 Increased expression of the GRβ isoform—an inactive form of the GR that competes with the active GRα form—can cause relative glucocorticoid resistance (Fig. 2).44 Increased expression of GRβ is stimulated by proinflammatory cytokines,45 and has been found in immune cells of patients with various inflammatory conditions.10,45 In addition, alternative translation initiation of GR mRNA and various posttranslational protein modifications can lead to the expression of GR isoforms with different transcriptional activity.43 Interestingly, GR polymorphisms that confer reduced glucocorticoid sensitivity have been found in both the human GRα and GRβ gene. The A3669G variant of GRβ increases its mRNA stability and dominant negative function,46 and the ER22/23EK polymorphism in GRα produces higher expression of the GR translational variant GR-A, leading to decreased expression of the more transcriptionally active GR-B protein.47 (See Fig. 1 in Ref. 41 and Fig. 3 in Ref. 43.) Epigenetic changes in the GR gene have also been shown as a result of early life behavioral programming in both rats and humans.48,49
Other environmental factors that can induce glucocorticoid resistance include chronic inflammation, exposure to infectious agents, chronic exposure to exogenous glucocorticoids and chronic stress. Cytokines and their downstream signaling molecules can influence the expression and activity of various factors that regulate local glucocorticoid bioavailability and GR function, which are altered in various chronic inflammatory disease states, usually favoring glucocorticoid resistance.10,50,51 We have recently shown that exposure to bacterial toxins, including Bacillus anthracis lethal toxin and Clostridia sordelli lethal toxin, can induce a state of glucocorticoid resistance, with impaired GR function, reduced GR transactivation, and enhanced mortality in mice.52-54 Chronic exposure to high-dose glucocorticoid therapy also induces glucocorticoid resistance, as in steroid resistant asthma.15 Chronic psychosocial stress has also been shown to induce glucocorticoid resistance in immune cells in humans and mice.55-58 Moreover, both psychogenic and immune stressors can induce similar neuroendocrine (HPA-activating) and neurotransmitter changes in the brain, therefore, sensitizing the brain to subsequent stressors (of either type) and, hence, inducing a state of increased stress vulnerability as seen in various psychiatric and somatic disorders.59 By synergizing, neuroendocrine-immune disturbances present in chronic exposure to either psychogenic or immune stressors can further contribute to the development of glucocorticoid resistance via impaired GR function,60,61 and therefore, produce a vicious feed-forward cycle perpetuating reduced inhibitory feedback on the HPA axis and inflammatory responses.
This suggests a mechanism for the long observed phenomenon of severe or chronic stress triggering the onset or exacerbations of inflammatory/autoimmune diseases, psychiatric and somatic disorders,18,50,62 and even metabolic disorders, such as metabolic syndrome.63,64 It would appear counterintuitive to predict that stress-related illnesses such as depression, metabolic syndrome, cardiovascular disease, or osteoporosis have inflammation as a common final pathway, since they are commonly characterized by a hyperactive HPA axis,65 and the presence of chronically elevated levels of glucocorticoids should suppress inflammation. However, the induction of glucocorticoid resistance and enhanced inflammation in these states provides a potential mechanism for this effect21 and may explain their high comorbidity.
Impaired GR function as a mechanism for exaggerated cytokine-induced behavioral and metabolic alterations: from sickness behavior to depression and metabolic syndrome
Depression is a case in point. Unrestrained inflammation and HPA axis activity, nonresponsiveness to suppression by the synthetic glucocorticoid dexamethasone, as well as metabolic alterations, are commonly observed in certain subsets of depressed patients and may be due to glucocorticoid resistance via impaired GR function.19,21,61,66 This phenomenon may be of particular relevance to depression in the medically ill, which is more consistently characterized by an enhanced inflammatory state.24,26,27 Innate immune cytokines can influence virtually every pathophysiologic domain relevant to depression, including neuroendocrine function, neurotransmitter metabolism, regional brain activity, and ultimately, behavior.28,67 Cytokines have been shown to induce a constellation of symptoms referred to as “sickness behavior,” in which an animal’s motivational state is shifted towards a behavioral depression (lethargy, reduced locomotor activity and food intake, increased sleep) in the effort to conserve energy for fever production and immune activation.68 Many features of sickness behavior overlap with symptoms of depression, particularly neurovegetative symptoms (i.e., psychomotor slowing, fatigue, anorexia, weight loss, hypersomnia). Sickness behavior is usually acute, considered adaptive for the recovery from infection and usually subsides along with the resolution of inflammation.68 However, sustained levels of low-level inflammation (as seen in many chronic inflammatory disorders) can contribute to prolonged expression of these behaviors, and in some conditions, culminate in the maladaptive development of depression.24,26-28 Indeed, greater expression of sickness behaviors/neurovegetative symptoms is a prominent feature of immunotherapy (e.g., IFNα)-induced depression (in patients without pretreatment depression), compared with depression in medically healthy individuals.69 The importance of intact glucocorticoid responses in keeping inflammatory responses and their behavioral sequelae in check is supported by the observation that glucocorticoid resistance in animal models given an immune challenge, either due to hypocortisolemia or impaired GR signaling, is associated with exaggerated peripheral and central inflammatory responses, as well as enhanced sickness and depressive-like behaviors.70-74
Similar to the metabolic effects of glucocorticoids (promoting muscle proteolysis, adipose lipolysis and hepatic gluconeogenesis),65 proinflammatory cytokines promote a catabolic state to provide energy sources for increased glucose availability (required for thermogenesis/fever and sustained immune activation).75-76 Therefore, sustained metabolic effects of an enhanced inflammatory state may contribute to an “inflammatory” metabolic syndrome.63,64 Moreover, metabolic syndrome has been associated with depressive symptomatology characterized primarily by neurovegetative features.77 Although enhanced inflammation appears to represent a link between depressive-like behavior and metabolic alterations, the molecular mechanisms by which an impaired GR may be a vulnerability factor for inflammation-induced behavioral and metabolic dysfunction requires further elucidation.
The GR is a ligand-dependent transcription factor that belongs to the nuclear hormone receptor super family. The two main molecular pathways by which GR controls transcription are by: 1) dimerizing and directly binding to positive or negative glucocorticoid response elements (GREs) in the promoter regions of genes leading to transcriptional increases (i.e., anti-inflammatory and metabolic molecules) or decreases (i.e, negative feedback on the HPA axis), and 2) GR monomers indirectly influencing the transcription of genes through protein-protein interactions with other transcription factors (i.e., inhibiting the transcriptional activity of NFκB and AP-1 on proinflammatory molecules) (Fig. 2).38,39 Whereas the metabolic actions of glucocorticoids are believed to be mediated by GR-DNA binding mechanisms, the extent to which GR-DNA binding mechanisms contribute to anti-inflammatory effects of GR remains an issue of debate.78,79 The predominant role of GR protein–protein interactions with other transcription factors (e.g., NFκB, AP-1) in mediating GR’s anti-inflammatory effects is mainly supported by in vitro studies.39,80,81 Under in vivo conditions, the dependence of GR’s anti-inflammatory effects on GR-DNA binding–dependent mechanisms has been shown to differ according to the type of immune challenge.81-84
To dissect the relative importance of GR-DNA binding–dependent mechanisms versus protein–protein interactions in GR regulation of the sickness syndrome (HPA, inflammatory, behavioral, and metabolic responses) to a systemic inflammatory challenge, we administered low-dose lipopolysaccharide (LPS) to GRdim mice. GRdim mice contain a point mutation that renders them deficient in GR dimerization, and hence, GR-DNA binding–dependent mechanisms of transcriptional regulation are impaired, but GR protein–protein interactions with other transcription factors remain intact.80 We have shown that GRdim mice administered low-dose LPS exhibit exaggerated peripheral and central inflammatory responses in a cytokine-, time-, and tissue specific manner. In addition, these mice exhibit sustained LPS-induced plasma corticosterone levels and sickness behaviors, as well as enhanced metabolic alterations and impaired thermoregulation (Silverman, unpublished data). Taken together, these data show that GR dimerization, and hence GR-DNA binding–dependent mechanisms, are essential for the proper regulation and recovery of HPA axis, inflammatory, behavioral, and metabolic responses to a systemic immune challenge, such as LPS.
Our data also support the concept that GR protein–protein interactions are not sufficient for glucocorticoids to exert their full anti-inflammatory effects, and suggest that endogenous glucocorticoid responses or exogenous glucocorticoid therapy limited to GR protein–protein interactions could predispose individuals to prolonged behavioral and metabolic sequelae of an enhanced inflammatory state. These include a continued state of sickness/depressive behaviors, such as lethargy/fatigue, reduced locomotor activity and anorexia, and metabolic alterations favoring a sustained catabolic state (especially increased proteolysis in skeletal muscle required for increased gluconeogenesis in the liver), which can lead to muscle wasting, fat gain (cachectic obesity), insulin resistance (selective inhibition of fuel storage in liver, adipose tissue, and muscle), osteopenia (reduced calcium absorption in bone) and anemia (reduced iron in blood) to provide sustained fuel to the activated immune system.63 Therefore, many features traditionally attributed to elevated glucocorticoids could actually result from impaired GR function, and hence glucocorticoid resistance and ensuing enhanced inflammation.21
This suggests a general principle for a protective role of intact glucocorticoid negative feedback and a fully functioning GR in a range of illnesses in which inflammation plays a central role. And, hence, at the molecular level, the functional status of the GR may play a role in the final common pathway in determining relative vulnerability versus resilience to many inflammatory-related conditions. Further elucidation of the molecular mechanisms involved in GR regulation of inflammation will help inform the pathophysiology of and potential risk factors for clinical conditions associated with HPA axis dysfunction and prolonged inflammation (e.g., autoimmune disease, depression, chronic fatigue, fibromyalgia, PTSD, cardiovascular disease, type 2 diabetes, metabolic syndrome, osteoporosis)18,21,63,64,67 and may also explain their high comorbidity. Moreover, these insights can better inform the development of glucocorticoid-related therapeutic agents for inflammatory disorders and their downstream correlates.79,85,86
Neural-immune biomarker assessment and therapeutic implications
Given the many levels of the HPA axis that can be potentially interrupted, and the combination of environmental and genetic factors that can induce glucocorticoid resistance, it is not surprising that stress-induced inflammatory diseases vary greatly in their presentation in different individuals and populations, and may include features that are inflammatory, behavioral and/or metabolic related. The evidence that glucocorticoid resistance can be environmentally induced has important implications for management of stress-related inflammatory illnesses and underscores the importance of prevention and management of chronic stress. If the initial goals of research in the fields encompassed by the terms “psychoneuroimmunology,” “neuroendocrine immunology,” or “neuroimmunomodulation” were to prove and elucidate the connections between the immune and central nervous systems, the goals for the next generation of research in these fields should be to bring this knowledge into the clinic to help prevent and treat stress-related diseases. In this regard, a thorough understanding of the role of glucocorticoid resistance in illness and of environmental factors that modify or prevent it will be essential in taking neuroimmunomodulation to the translational level to prevent and treat human illness.
To do so, new tools will be required to fully characterize the functioning of the central nervous and immune systems at functional, physiological, hormonal, and molecular levels. Ideally, such tools should be noninvasive or minimally invasive and should provide profiles of multiple biomarkers to obtain signature patterns of analytes indicative of the status of these systems. We are currently developing a method to measure neural and immune biomarkers in sweat patch eluates extracted from cutaneous patches worn on the abdomen. Our previous proof-of-principle studies showed that sweat patch analytes (i.e., proinflammatory cytokines and neuropeptides) correlated with plasma levels in healthy controls87 and with both plasma levels and symptom severity in a population of women with a history of major depressive disorder in remission.88 Salivary cortisol has long been used to noninvasively assess the circadian rhythm of cortisol.89 More recently, attention has been paid to the salivary cortisol awakening response (CAR) as a sensitive measure to detect HPA axis dysregulation.90 Cortisol measurements in hair provide another promising new method on the horizon to measure the cumulative burden of chronic stress over a longer time period (months).91 Although we have not discussed the role of the autonomic nervous system (ANS) in neuroimmune interactions92-95 or HPA–ANS interactions21 in this review, heart rate variability (HRV) is another minimally-invasive method, which measures the relative activity of the parasympathetic and sympathetic nervous systems. Decreased HRV, indicative of reduced parasympathetic-vagal tone, is an independent risk factor for morbidity and mortality.96 In addition, salivary alpha-amylase measurements have been used as a non-invasive biomarker for the sympathetic nervous system.97
These methods coupled with other more invasive methods requiring blood draws will provide a snapshot of an individual’s health status and will allow more comprehensive approaches to predicting those at risk and monitoring effectiveness of therapeutic interventions and coping strategies in stress-related disorders. Ultimately, prediction and management of stress-related illnesses will require a comprehensive work-up, screening for both genetic predisposition factors, such as polymorphisms of the GR and related proteins, and functional status of the neuroendocrine stress response, the autonomic nervous system and immune/inflammatory responses. Simultaneous assessment of neural, endocrine, and immune biomarkers may help inform the design of more effective therapeutic interventions, whether pharmacological or behavioral, to optimize the reversal of chronic stress’ averse effects on the body, normalize stress-induced neuroendocrine-immune disturbances, and restore proper GR function.
Acknowledgments
This work was supported by the NIH, NIMH Division of Intramural Research.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 2.Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 3.McEwen BS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- 4.Webster JI, Sternberg EM. Role of the hypothalamic-pituitary-adrenal axis, glucocorticoids and glucocorticoid receptors in toxic sequelae of exposure to bacterial and viral products. J Endocrinol. 2004;181:207–221. doi: 10.1677/joe.0.1810207. [DOI] [PubMed] [Google Scholar]
- 5.Glezer I, Rivest S. Glucocorticoids: protectors of the brain during innate immune responses. Neuroscientist. 2004;10:538–552. doi: 10.1177/1073858404263494. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg EM, et al. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci. 1989a;86:2374–2378. doi: 10.1073/pnas.86.7.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crofford LJ. The hypothalamic-pituitary-adrenal axis in the pathogenesis of rheumatic diseases. Endocrinol Metab Clin North Am. 2002;31:1–13. doi: 10.1016/s0889-8529(01)00004-4. [DOI] [PubMed] [Google Scholar]
- 8.Chikanza IC, Kuis W, Heijnen CJ. The influence of the hormonal system on pediatric rheumatic diseases. Rheum Dis Clin North Am. 2000;26:911–925. doi: 10.1016/s0889-857x(05)70176-9. [DOI] [PubMed] [Google Scholar]
- 9.Straub RH, et al. How psychological stress via hormones and nerve fibers may exacerbate rheumatoid arthritis. Arthritis Rheum. 2005;52:16–26. doi: 10.1002/art.20747. [DOI] [PubMed] [Google Scholar]
- 10.Silverman MN, Sternberg EM. Neuroendocrine-immune interactions in rheumatoid arthritis: mechanisms of glucocorticoid resistance. Neuroimmunomodulation. 2008;15:19–28. doi: 10.1159/000135620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stasi C, Orlandelli E. Role of the brain-gut axis in the pathophysiology of Crohn’s disease. Dig Dis. 2008;26:156–166. doi: 10.1159/000116774. [DOI] [PubMed] [Google Scholar]
- 12.Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut. 2005;54:1481–1491. doi: 10.1136/gut.2005.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gold SM, et al. The role of stress-response systems for the pathogenesis and progression of MS. Trends Immunol. 2005;26:644–652. doi: 10.1016/j.it.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Buske-Kirschbaum A. Cortisol responses to stress in allergic children: interaction with the immune response. Neuroimmunomodulation. 2009;16:325–332. doi: 10.1159/000216190. [DOI] [PubMed] [Google Scholar]
- 15.Adcock IM, et al. Steroid resistance in asthma: mechanisms and treatment options. Curr Allergy Asthma Rep. 2008;8:171–178. doi: 10.1007/s11882-008-0028-4. [DOI] [PubMed] [Google Scholar]
- 16.Parker AJ, Wessely S, Cleare AJ. The neuroendocrinology of chronic fatigue syndrome and fibromyalgia. Psychol Med. 2001;31:1331–1345. doi: 10.1017/s0033291701004664. [DOI] [PubMed] [Google Scholar]
- 17.Neeck G, Crofford LJ. Neuroendocrine perturbations in fibromyalgia and chronic fatigue syndrome. Rheum Dis Clin North Am. 2000;26:989–1002. doi: 10.1016/s0889-857x(05)70180-0. [DOI] [PubMed] [Google Scholar]
- 18.Silverman MN, et al. Neuroendocrine and immune contributors to fatigue. Physical Med Rehab. 2010;2:338–346. doi: 10.1016/j.pmrj.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 20.Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25:1–35. doi: 10.1016/s0306-4530(99)00035-9. [DOI] [PubMed] [Google Scholar]
- 21.Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 22.Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann NY Acad Sci. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- 23.Anacker C, et al. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology. 2011;36:415–425. doi: 10.1016/j.psyneuen.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollak Y, Yirmiya R. Cytokine-induced changes in mood and behaviour: implications for ‘depression due to a general medical condition’, immunotherapy and antidepressive treatment. Int J Neuropsychopharmacol. 2002;5:389–399. doi: 10.1017/S1461145702003152. [DOI] [PubMed] [Google Scholar]
- 25.Maes M. Inflammatory and oxidative and nitrostative stress pathways underpinning chronic fatigue, somatization and psychosomatic symptoms. Curr Opin Psychiatry. 2008;22:75–83. doi: 10.1097/yco.0b013e32831a4728. [DOI] [PubMed] [Google Scholar]
- 26.Maes M, et al. The inflammatory and neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009;24:27–53. doi: 10.1007/s11011-008-9118-1. [DOI] [PubMed] [Google Scholar]
- 27.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dantzer R, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–57. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loftis JM, Huckans M, Morasco MJ. Neuroimmne mechanisms of cytokine-induced depression: current theories and novel treatment strategies. Neurobiology of Disease. 2010;37:519–533. doi: 10.1016/j.nbd.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pace TW, Heim CM. A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav Immun. 2011;25:6–13. doi: 10.1016/j.bbi.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Gill JM, et al. PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr Care. 2009;45:262–277. doi: 10.1111/j.1744-6163.2009.00229.x. [DOI] [PubMed] [Google Scholar]
- 32.Gur A, Oktayoglu P. Status of immune mediators in fibromyalgia. Curr Pain Headache Rep. 2008;12:175–181. doi: 10.1007/s11916-008-0031-4. [DOI] [PubMed] [Google Scholar]
- 33.Sternberg EM, et al. A central nervous system defect in biosynthesis of corticotropin-releasing hormone is associated with susceptibility to streptococcal cell wall-induced arthritis in Lewis rats. Proc Natl Acad Sci. 1989b;86:4771–4775. doi: 10.1073/pnas.86.12.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zelazowski P, et al. Release of hypothalamic corticotropin-releasing hormone and arginine-vasopressin by interleukin 1 beta and alpha MSH: studies in rats with different susceptibility to inflammatory disease. Brain Res. 1993;631:22–26. doi: 10.1016/0006-8993(93)91181-q. [DOI] [PubMed] [Google Scholar]
- 35.Calogero AE, et al. Neurotransmitter-induced hypothalamic-pituitary-adrenal axis responsiveness is defective in inflammatory disease-susceptible Lewis rats: in vivo and in vitro studies suggesting globally defective hypothalamic secretion of corticotropin-releasing hormone. Neuroendocrinology. 1992;55:600–608. doi: 10.1159/000126173. [DOI] [PubMed] [Google Scholar]
- 36.Zelazowski P, et al. In vitro regulation of pituitary ACTH secretion in inflammatory disease susceptible Lewis (LEW/N) and inflammatory disease resistant Fischer (F344/N) rats. Neuroendocrinology. 1992;56:474–482. doi: 10.1159/000126264. [DOI] [PubMed] [Google Scholar]
- 37.Bernardini R, et al. Adenylate-cyclase-dependent pituitary adrenocorticotropin secretion is defective in the inflammatory-disease-susceptible Lewis rat. Neuroendocrinology. 1996;63:468–474. doi: 10.1159/000127073. [DOI] [PubMed] [Google Scholar]
- 38.Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J. 2006;27:413–426. doi: 10.1183/09031936.06.00125404. [DOI] [PubMed] [Google Scholar]
- 39.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25:6868–6886. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 40.van Rossum EF, van den Akker EL. Glucocorticoid resistance. Endocr Dev. 2010;20:127–136. doi: 10.1159/000321234. [DOI] [PubMed] [Google Scholar]
- 41.Manenschijn L, et al. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci. 2009;1179:179–198. doi: 10.1111/j.1749-6632.2009.05013.x. [DOI] [PubMed] [Google Scholar]
- 42.DeRijk R, de Kloet ER. Corticosteroid receptor genetic polymorphisms and stress responsivity. Endocrine. 2005;28:263–270. doi: 10.1385/ENDO:28:3:263. [DOI] [PubMed] [Google Scholar]
- 43.Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70:407–417. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9. doi: 10.1196/annals.1351.001. [DOI] [PubMed] [Google Scholar]
- 45.Webster JC, et al. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci. 2001;98:6865–6870. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derijk RH, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed] [Google Scholar]
- 47.Russcher H, et al. Increased expression of the glucocorticoid receptor-A translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19:1687–1696. doi: 10.1210/me.2004-0467. [DOI] [PubMed] [Google Scholar]
- 48.Weaver IC, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 49.McGowan PO, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–38. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marques AH, Silverman MN, Sternberg EM. Glucocorticoid dysregulations and their clinical correlates. From receptors to therapeutics. Ann N Y Acad Sci. 2009;1179:1–18. doi: 10.1111/j.1749-6632.2009.04987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tait AS, et al. The large clostridial toxins from Clostridium sordellii and C. difficile repress glucocorticoid receptor activity. Infect Immun. 2007;75:3935–3940. doi: 10.1128/IAI.00291-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webster JI, Sternberg EM. Anthrax lethal toxin represses glucocorticoid receptor (GR) transactivation by inhibiting GR-DNA binding in vivo. Mol Cell Endocrinol. 2005;241:21–31. doi: 10.1016/j.mce.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 54.Webster JI, et al. Anthrax lethal factor represses glucocorticoid and progesterone receptor activity. Proc Natl Acad Sci. 2003;100:5706–5711. doi: 10.1073/pnas.1036973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohleder N. Acute and chronic stress induced changes in sensitivity of peripheral inflammatory pathways to the signals of multiple stress systems - 2011 Curt Richter Award Winner. Psychoneuroendocrinology. 2012 doi: 10.1016/j.psyneuen.2011.12.015. Jan 4, 2012 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 56.Miller GE, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of proinflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. 2002;21:531–541. doi: 10.1037//0278-6133.21.6.531. [DOI] [PubMed] [Google Scholar]
- 58.Avitsur R, et al. Social interactions, stress, and immunity. Immunol Allergy Clin North Am. 2009;29:285–293. doi: 10.1016/j.iac.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Hayley S, Merali Z, Anisman H. Stress and cytokine-elicited neuroendocrine and neurotransmitter sensitization: implications for depressive illness. Stress. 2003;6:19–32. doi: 10.1080/1025389031000091167. [DOI] [PubMed] [Google Scholar]
- 60.Barden N. Implication of the hypothalamic-pituitary-adrenal axis in the physiopathology of depression. J Psychiatry Neurosci. 2004;29:185–193. [PMC free article] [PubMed] [Google Scholar]
- 61.Zunszain PA, et al. Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:722–9. doi: 10.1016/j.pnpbp.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nature Rev Immunol. 2006;6:318–328. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Straub RH, et al. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 2010;267:543–560. doi: 10.1111/j.1365-2796.2010.02218.x. [DOI] [PubMed] [Google Scholar]
- 64.Black PH. The inflammatory response is an integral part of the stress response: Implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav Immun. 2003;17:350–364. doi: 10.1016/s0889-1591(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 65.Chrousos GP, Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress. 2007;10:213–219. doi: 10.1080/10253890701292119. [DOI] [PubMed] [Google Scholar]
- 66.McIntyre RS, et al. Should Depressive Syndromes Be Reclassified as “Metabolic Syndrome Type II”? Ann Clin Psychiatry. 2007;19:257–264. doi: 10.1080/10401230701653377. [DOI] [PubMed] [Google Scholar]
- 67.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 69.Capuron L, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119:181–185. doi: 10.1016/j.jad.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goujon E, et al. Regulation of cytokine gene expression in the central nervous system by glucocorticoids: mechanisms and functional consequences. Psychoneuroendocrinology. 1997;22:S75–80. doi: 10.1016/s0306-4530(97)00009-7. [DOI] [PubMed] [Google Scholar]
- 71.Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23:5536–5544. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson RW, Propes MJ, Shavit Y. Corticosterone modulates behavioral and metabolic effects of lipopolysaccharide. Am J Physiol. 1996;270:R192–198. doi: 10.1152/ajpregu.1996.270.1.R192. [DOI] [PubMed] [Google Scholar]
- 73.Pezeshki G, Pohl T, Schöbitz B. Corticosterone controls interleukin-1 beta expression and sickness behavior in the rat. J Neuroendocrinol. 1996;8:129–135. doi: 10.1111/j.1365-2826.1996.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 74.Wang D, et al. Chronic blockade of glucocorticoid receptors by RU486 enhances lipopolysaccharide-induced depressive-like behaviour and cytokine production in rats. Brain Behav Immun. 2011;25:706–714. doi: 10.1016/j.bbi.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 75.Beisel WR. Metabolic response to infection. Annu Rev Med. 1975;26:9–20. doi: 10.1146/annurev.me.26.020175.000301. [DOI] [PubMed] [Google Scholar]
- 76.Chang HR, Bistrian B. The role of cytokines in the catabolic consequences of infection and injury. JPEN J Parenter Enteral Nutr. 1998;22:156–166. doi: 10.1177/0148607198022003156. [DOI] [PubMed] [Google Scholar]
- 77.Capuron L, et al. Depressive symptoms and metabolic syndrome: is inflammation the underlying link? Biol Psychiatry. 2008;64:896–900. doi: 10.1016/j.biopsych.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 79.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 80.Reichardt HM, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 81.Reichardt HM, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grose R, et al. A role for endogenous glucocorticoids in wound repair. EMBO Rep. 2002;3:575–582. doi: 10.1093/embo-reports/kvf119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tuckermann JP, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. 2007;117:1381–1390. doi: 10.1172/JCI28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kleiman A, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2012 doi: 10.1096/fj.11-192112. Oct 31, 2011 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 85.De Bosscher K, et al. Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur J Pharmacol. 2008;583:290–302. doi: 10.1016/j.ejphar.2007.11.076. [DOI] [PubMed] [Google Scholar]
- 86.Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol. 2007;275:98–108. doi: 10.1016/j.mce.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 87.Marques-Deak A, et al. Measurement of cytokines in sweat patches and plasma in healthy women: validation in a controlled study. J Immunol Methods. 2006;315:99–109. doi: 10.1016/j.jim.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 88.Cizza G, et al. Elevated neuroimmune biomarkers in sweat patches and plasma of premenopausal women with major depressive disorder in remission: the POWER study. Biol Psychiatry. 2008;64:907–911. doi: 10.1016/j.biopsych.2008.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirschbaum C, Hellhammer DH. Salivary cortisol in psychoneuroendocrine research: recent developments and applications. Psychoneuroendocrinology. 1994;19:313–333. doi: 10.1016/0306-4530(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 90.Clow A, Hucklebridge F, Thorn L. The cortisol awakening response in context. Int Rev Neurobiol. 2010;93:153–175. doi: 10.1016/S0074-7742(10)93007-9. [DOI] [PubMed] [Google Scholar]
- 91.Dettenborn L, et al. Introducing a novel method to assess cumulative steroid concentrations: Increased hair cortisol concentrations over 6 months in medicated patients with depression. Stress. 2012 doi: 10.3109/10253890.2011.619239. Nov 1, 2011 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 92.Elenkov IJ, et al. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 93.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system. Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tracey KJ. Physiology and immunology of the cholinergic anti-inflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thayer JF. Vagal tone and the inflammatory reflex. Cleve Clin J Med. 2009;76:S23–26. doi: 10.3949/ccjm.76.s2.05. [DOI] [PubMed] [Google Scholar]
- 96.Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361–372. doi: 10.1196/annals.1366.014. [DOI] [PubMed] [Google Scholar]
- 97.Nater UM, Rohleder N. Salivary alpha-amylase as a non-invasive biomarker for the sympathetic nervous system: current state of research. Psychoneuroendocrinology. 2009;34:486–496. doi: 10.1016/j.psyneuen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 98.Silverman MN, Pearce BD, Miller AH. Cytokines and HPA axis regulation. In: Kronfol Z, editor. Cytokines and Mental Health. Kluwer Adademic Publishers; Norwell, MA: 2003. pp. 85–122. [Google Scholar]