

Abstract
In several types of thalassemia (including β039-thalassemia), stop codon mutations lead to premature translation termination and to mRNA destabilization through nonsense-mediated decay. Drugs (for instance aminoglycosides) can be designed to suppress premature termination, inducing a ribosomal readthrough. These findings have introduced new hopes for the development of a pharmacologic approach to the cure of this disease. However, the effects of aminoglycosides on globin mRNA carrying β-thalassemia stop mutations have not yet been investigated. In this study, we have used a lentiviral construct containing the β039- thalassemia globin gene under control of the β-globin promoter and a LCR cassette. We demonstrated by fluorescence-activated cell sorting (FACS) analysis the production of β-globin by K562 cell clones expressing the β039-thalassemia globin gene and treated with G418. More importantly, after FACS and high-performance liquid chromatography (HPLC) analyses, erythroid precursor cells from β039-thalassemia patients were demonstrated to be able to produce β-globin and adult hemoglobin after treatment with G418. This study strongly suggests that ribosomal readthrough should be considered a strategy for developing experimental strategies for the treatment of β0-thalassemia caused by stop codon mutations.
Introduction
Nonsense mutations, giving rise to UAA, UGA, and UAG premature translation termination codons (PTTCs) within the coding region of mRNAs, account for ~10–30% of all described gene lesions causing human inherited diseases [1–5]. As recently reviewed by Mort et al. [6], pathological nonsense mutations resulting in TGA (38.5%), TAG (40.4%), and TAA (21.1%) occur in different proportions to naturally occurring stop codons. Of the 23 different nucleotide substitutions that cause nonsense mutations, the most frequent are CGA → TGA (21%; resulting from methylation-mediated deamination) and CAG → TAG (19%) [6].
There are numerous examples of inherited diseases caused by nonsense mutations, such as cystic fibrosis [7,8], lysosomal storage disorders [9], Duchenne muscular dystrophy [10,11], and thalassemia [12,13]. There are also noninherited diseases associated to de novo formation of stop codons. For instance, in cancers many tumor suppressor genes exhibit a disproportionate number of somatic nonsense mutations [14], many of which were found to occur recurrently in the hypermutable CpG dinucleotide, as expected [14].
The major molecular consequences of stop mutations are the promotion of premature translational termination and the nonsense-mediated RNA decay (NMD) [15–18]. These two features are strictly associated. NMD, in fact, recognizes and degrades transcripts harboring PTTCs, thereby preventing the production of truncated and faulty proteins. NMD is considered as a very important pathway in an mRNA surveillance system that typically degrades transcripts containing PTTCs to prevent unnecessary processing of RNA precursors and unnecessary translation of aberrant transcripts [15–18]. Failure to eliminate these mRNAs with PTTCs may result in the synthesis of abnormal proteins that can be toxic to cells through dominant-negative or gain-of-function effects.
As far as thalassemia syndromes, in the β039-thalassemia, the CAG (Gln) codon is mutated to an UAG stop codon [12,13], leading to premature translation termination and to mRNA destabilization through NMD [19,20]. The β039-thalassemia mutation is very frequent in Italy (about 70% of the total β-thalassemia mutations) [21] and, in general, in the whole Mediterranean area. Other examples of stop mutation of the β-globin mRNA occur at position 15, 37, 59, and 127 of the mRNA sequence [22–27].
In the last few years, it has been demonstrated that drugs can be designed and produced to suppress premature termination, inducing a ribosomal readthrough of premature, but not normal termination codons [28–30]. The molecular basis of this phenomenon is related to the sequence “context” surrounding normal termination codons, which makes the normal termination codons refractory to the drug-mediated readthrough [28]. Therefore, this approach has been considered very promising for the treatment of all the pathologies caused by nonsense mutations [29–31].
Among drugs able to induce mammalian ribosomes to readthrough premature stop codon mutations, aminoglycosides are the most studied and they have been recently proposed for the development of novel therapeutic approaches for the treatment of human diseases caused by PTTCs [32,33]. As recently reviewed by Kellermayer [31], this new and challenging task has opened new research avenues in the field of aminoglycoside applications.
In the case of cystic fibrosis, in vitro studies in cell lines expressing stop mutations [34,35] and in mice [36,37] have shown that aminoglycosides caused a dose-dependent increase in CFTR expression and restored functional CFTR to the apical membrane. Clinical studies also provided evidence that the aminoglycoside gentamicin can suppress these CFTR premature stop mutations in affected patients [38]. A recent double-blind, placebo-controlled, crossover study has demonstrated restoration of CFTR function by topical application of gentamicin to the nasal epithelium of cystic fibrosis patients carrying stop mutations. In 21% of the patients, there was a complete normalization of all the electrophysiologic abnormalities caused by the CFTR defect, and in 68% there was restoration of either chloride or sodium transport. Despite the fact that it is still unknown how much corrected mutant CFTR must reach the apical membrane to induce a clinically relevant beneficial effect [39], the data strongly support the concept that this is a suitable approach and new compounds should be developed. Safe compounds could then be administered to small children from the time of diagnosis. The use of aminoglycosides to correct PTTCs occurring in muscular Duchenne dystrophy has also been reported both in vitro and in vivo [40–42].
Because of the importance of PTTCs in β-thalassemia, such as for β039-thalassemia, these findings have introduced new hopes for the development of a pharmacologic approach to cure this disease. However, the effects of aminoglycosides on the possible correction of β-thalassemia stop mutations have not yet been investigated.
In a recent article, we have described the development of a novel experimental system suitable to screen potential modifiers of biological consequences of stop mutations [43]. We have generated two lentiviral constructs, one containing the human normal β-globin gene and the other containing the β039-thalassemia globin gene, both under the control of the β-globin promoter and a LCR cassette (see Fig. 1A for the map of the construct). These vectors were transfected to K562 cells and several K562 cell clones isolated, expressing either the normal β-globin or the β039-thalassemia globin genes at different levels. This system was proved to be suitable to detect readthrough activity [43].
Figure 1.

A: Map of the vector pCCL.βwt. PGW used to generate the K562 cellular clones carrying the wild-type and the β039-thal mutated globin mRNA. βp, beta-globin promoter. The three exons, the two introns and the genomic region including the 3′ enhancer are indicated. B: Effects of 400 μg/ml G418 on the production of β-globin in K562-wt3 (D–G) and K562-m5 (H–M). As a reference control, the immunohistochemistry analysis of original wild-type K562 cells (not expressing β-globin mRNA) is shown in panels B and C; analysis performed on untreated (D, E, H, I) versus G418-treated (F, G, L, M) K562-wt3 and K562-m5 cells is shown. Staining of the cells with the β-globin-PE (PE, phycoerythrin) (Santa Cruz Biotechnology, Santa Cruz, CA) is shown in panels C, E, G, I, and M. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We report in this article the treatment and characterization with G418 of one K562 clone carrying the β039-thalassemia globin gene. Characterization was performed by immunostaining, FACS, and proteomic assays. In addition, we treated erythroid precursor cells from six homozygous β039-thalassemia patients with G418 and analyzed the hemoglobin (Hb) production by HPLC, to confirm the ability to cause β-globin synthesis and allow production of HbA in primary erythroid cells from β039-thalassemia patients.
Results
Effects of geneticin (G418) on β-globin production in wt3 and m5 clones, expressing normal β-globin genes (wt3) and β-globin genes carrying the β039-thalassemia mutation (m5)
To test the effects of G418 on erythroid cells mimicking β039-thalassemia, K562 cell clones carrying multiple copies of the normal and β039-globin gene were used. The production of the lentiviral vectors used for the generation of the K562 cells clones carrying either βwt or β039-globin genes has been reported elsewhere [43]. Briefly, the original 13,824 bp construct (pCCL.βwt.PGW) displays two LTR sequences, the SV40 origin of replication, a GFP gene under the control of the PGK promoter, the β-globin gene under the control of the β-globin gene promoter, and a minimal LCR of the human β-like globin gene cluster (see the map shown in Fig. 1A). The presence of GFP allows a high-throughput screening of transduced cells, giving at the same time some preliminary information about the number of integration events [43]. We have developed a second construct by substituting the wild-type β-globin gene with a β039-globin gene produced by site-directed mutagenesis [43].
Among the different clones produced [43], clones wt3 and m5 were chosen because they display similar levels of accumulation of β-globin mRNA, facilitating, therefore, a correct interpretation of the results obtained after treatment with G418. Despite the fact that clones wt3 and m5 exhibited hybridization efficiency compatible with at least 5 and 7–8 integrated copies/genome, respectively [43], they express similar levels of GFP. As far as expression of the β-globin gene, clone m5 presumably produces, with respect to K562-wt3, higher amounts of β-globin mRNA primary transcripts, which undergo NMD, leading to accumulation amounts of mature β-globin mRNA sequences similar to those of clone K562-wt3 [43].
The effects of G418 on the β-globin production by K562-wt3 and K562-m5 clones were analyzed following two complementary approaches, immunohistochemistry and FACS analysis. Figure 1B, C shows that, as expected, no β-globin is produced by control wild-type K562 cells. It is well known, indeed, that K562 cells are committed to embryo-fetal globin gene expression and produce only very low levels of β-globin mRNA. RT-PCR analyses demonstrate that β-globin mRNA is transcribed in both K562-wt3 and K562- m5, probably due to the fact that the integrated β-globin genes lack in these clones the chromatinic context inhibiting, in original K562 cells, the transcription of adult β-globin genes. As far as protein production is concerned, β-globin protein is synthesized in K562-wt3 cells, and addition of G418 does not have any effect on β-globin production (panels D–G of Fig. 1). The results obtained using K562- m5 cells are shown in panels H–M of Fig. 1. In this clone, despite the high levels of β039-globin mRNA produced (data not shown), no accumulation of β-globin is detectable (Fig. 1H, I). However, when K562-m5 cells are treated with G418 (400 μg/ml), production of β-globin is detected, after staining the cells with the PE-conjugated β-globin antibody (Fig. 1L, M).
To better quantify the β-globin production in G418-treated K562-m5 cells, FACS analysis was performed (Fig. 2). K562-wt3 and K562-m5 cells were either untreated or treated with increasing (100, 200, and 400 μg/ml) concentrations of G418. At the end of the treatment, cells were recovered and labeled. This labeling allows discrimination by FACS analysis of the green fluorescence of GFP from the red fluorescence of β-globin-PE antibody. The two different fluorochromes, one associated with the beta-globin chains, PE, and the other expressed directly by the cells transduced with the lentiviral construct (GFP), are easily distinguished by flow cytometry, because of their different absorbance properties.
Figure 2.

A–N: Effects of G418 on the production of β-globin by K562-wt3 and K562-m5 cells. The FACS analysis is shown of untreated K562-wt3 (A, B) and K562-m5 (C, D) cells versus cells treated with 200 μg/ml (E–H) and 400 μg/ml (I–N) G418. A, B, E, F, I, L = K562-wt3 cells; C, D, G, H, M, N = K562-m5 cells. The arrows in panels D, H, and N are positioned on the intensity of the β-globin-PE peak of untreated cells (D), to help the reader to follow the shift of the right in G418-treated cells (H and N). O, P: Quantitative analysis of the FACS obtained in three independent experiments. GFP (closed symbols) and β-globin-PE (open symbols) fluorescence in K562-wt3 (O) and K562-m5 (P) cells treated with 100–400 μg/ml of G418 is reported. Data represent the average ± SD of fluorescence intensity. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 2A, B, E, F, I, L clearly shows that G418 treatment of K562-wt3 cells does not alter GFP production (panels A, E, and I) and reactivity to the anti-β-globin monoclonal anti-body (panels B, F, and L). On the contrary, Figure Figure 1. Figure 2. 2C, D, G, H, M, N clearly shows that, although G418 treatment of K562-m5 cells does not induce major changes in GFP production (panels C, G, and M), it induces a concentration- dependent increase of red fluorescence, indicating significant increase of the β-globin chain production (P > 0.01 when panel D of Fig. 2 is compared with panels H and N). G418 did not affect cellular morphology when administered to both K562-wt3 and K562-m5 clones (data not shown). Figure 2 (panels O and P) shows the quantification of the data of three independent experiments, including the results of the representative experiment shown in panels A–N of Fig. 2. Despite the fact that it is hard to use GFP expression as an internal control for comparing effects on K562-wt3 and K562-m5 clones, because the integration sites and overall transcriptional efficiency are expected to be different, it is interesting to note that the level of β-globin production/cell in the G418-treated K562-m5 clone approaches that of K562-wt3 clones. Similar effects of G418 were found using other cellular clones carrying the β039-globin gene.
In conclusion, the results shown in Figs. 1 and 2 consistently suggest that synthesis of β-globin in a context of a β039-thalassemia phenotype can be obtained after treatment of K562-m5 cells with G418. This is not associated with major damages of the cellular shape and block of cell growth (data not shown). However, to determine whether G418 has effects of protein production and overall control of gene expression, proteomic studies were undertaken.
The effects of G418 on β-globin production by K562 cells are not associated with major changes in protein expression
K562 cells were cultured in the presence or in the absence of the highest dose of G418 used for the studies on the K562 cell clones (400 μg/ml) for 3 days and protein extracts prepared. Proteomic studies were performed by bidimensional gel electrophoresis. Gels were performed in quadruplicate. Figure 3 shows representative results obtained. The same amounts of protein extract were loaded on the gels. To obtain better resolution at the high molecular weight, allowing comparative analysis of the highest number of protein spots, a small proportion of low-molecular- weight proteins were allowed to run out the gels. The two-dimensional gel electrophoresis (2DE) gels were scanned using the Quantity One (1D Analysis Software), version 4.6.1 (Bio-Rad), to acquire images. The spot analysis was performed by the PDQuest™ Basic (2D Analysis Software), version 8.0 (Bio-Rad), creating two analysis sets from the protein patterns, each referring to a specific sample (control K562 cells, G418-treated K562 cells). After normalizing spot amounts to remove nonexpression-related variations, the results were evaluated in terms of spot intensities. Statistical analysis allowed the identification of the spots which were constantly reproduced, as well as those which showed a twofold differential intensity.
Figure 3.
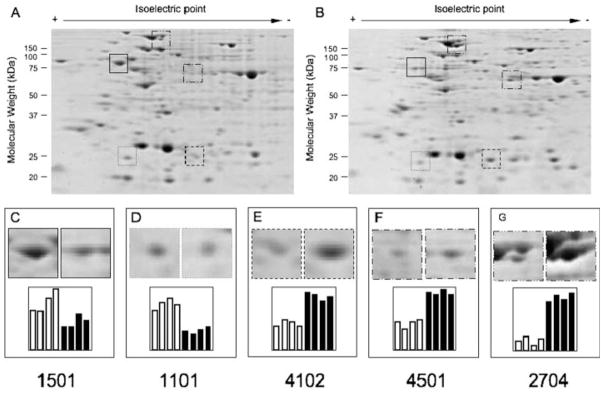
Proteomic analysis of untreated (A) and G418-treated (B) K562-m5 cells. In panels C–G, examples are reported, relative to two downmodulated spots (panels C and D) and three upmodulated spots (panels E–G). The quantitative data of four independent proteomic analysis are shown in the bottom of panels C–G. To obtain these data, the 2DE gels were scanned by a GS-800 Calibrated Densitometer (Bio-Rad, Hercules, CA), using the Quantity One (1D Analysis Software), version 4.6.1
The data obtained firmly demonstrate that no major changes occur in the protein profile after G418 treatment. Out of more than 300 protein spots analyzable, only five (1501, 1101, 4102, 4501, and 2704) displayed quantitative twofold changes (three were upregulated and two were downregulated) and no extra spots were detectable. These results were further confirmed by performing nuclear protein analysis (data not shown and Breveglieri et al., manuscript in preparation) and allow to conclude that, up to the concentration of 400 μg/ml, G418 does not change the proteomic profile of treated cells. Despite the fact that further analyses are required (a) to identify the proteins whose expression is altered by G418 and (b) to rule out read-through effects on low-copy-number cellular mRNAs, these data suggest that the correction of mutated β039-globin mRNA occurs with high efficiency in respect to the read-through of the other potential cellular mRNA targets. In the experimental conditions used, the globins migrate outside the gel.
Effects of G418 on HbA production by erythroid precursor cells isolated from homozygous β039-thalassemia patients
This set of experiments was performed to determine whether β-globin production is achieved by treatment of primary erythroid cells from β039-thalassemia patients with G418. To this aim, erythroid precursors from the peripheral blood of six homozygous β039-thalassemia patients were isolated and cultured following the two-phase procedure described by Fibach et al. [44,45]. During the second phase, the cells were cultured with erythropoietin (EPO) with or without G418. A representative FACS analysis is shown in Fig. 4A and clearly indicates that the majority of the G418-treated cell population increase its positivity to the PE-anti-β-globin monoclonal antibody, suggesting high level of readthrough and translation of the β039-globin mRNA in these primary erythroid cells. HPLC analysis (a representative experiment is shown in panel B of Fig. 4) demonstrates production of HbA by homozygous β039-thalassemia erythroid precursor cells treated with G418. The HPLC data, therefore, confirm the FACS results (Fig. 4A), demonstrating that the ex novo production of β-globin after G418 treatment leads to HbA accumulation. The summary of 10 independent experiments conducted on cells from the six homozygous β039-thalassemia patients is depicted in Fig. 4C, indicating a consistent increase in the proportion of HbA in erythroid precursor cells from homozygous β039- thalassemia patients after G418 treatment (P < 0.01). In addition to the increase of HbA, it is observed a sharp decrease of a peak, close to HbF, which we demonstrated to be constituted only of α-globin chains and which we consider as an internal marker of the reachment of clinically relevant results (Breda et al., manuscript in preparation). The excess of α-globin chains is in fact a major factor causing the pathophysiological alterations of thalassemic cells [46,47].
Figure 4.

A, B: Effect of 400 μg/ml G418 on the production of β-globin and HbA in erythroid precursor cells isolated from the peripheral blood of homozygous β039-thalassemia patients. (A) FACS analysis; (B) HPLC analysis of lysates from untreated (upper panel) and G418-treated (lower panel) cells. C: Summary of the data on the increase of the percentage of HbA accumulation in erythroid precursor cells from β039-thalassemic patients after treatment with G418; the data represent the mean ± SD from 10 different independent experiments using erythroid precursor cells from six homozygous β039-thalassemic patients. which is available at www.interscience.wiley.com.]
Effects of G418 on β-globin mRNA content in erythroid precursor cells from homozygous β039-thalassemia patients
This set of experiments were undertaken to understand whether G418 treatment might lead to changes in globin mRNA accumulation. Figure 5A clearly indicates that no changes in β-globin mRNA content occur in K562-wt3 cells treated with G418. These data were reproducibly obtained in several experiments and strongly suggest that G418 has no major effects on the transcription, processing, and stability of the wt-β-globin mRNA. On the contrary, when the same experiment was performed on K562-m5 cells, a net increase in β039-globin mRNA content was demonstrated, together with the induction of β-globin production documented in Figs. 1 and 2. The same phenomenon is evident in erythroid precursor cells from β039-thalassemia subjects, as shown in Fig. 5 (panels B and C). The data on erythroid precursor cells allow us to make the following statements: (a) in untreated cells from β039 homozygous patients, the β0-39 globin mRNA is very low (observed mRNA is about 7% than that expected in the absence of this mutations, P < 0.01) (Fig. 5B); (b) after G418 treatment, the β039-globin mRNA content sharply increases (Fig. 5C), reaching about 35% when comparison is done with the levels of β-globin mRNA produced by cells isolated from normal donors and exposed to the same experimental conditions (P < 0.01). Taken together, these data are compatible with a G418-mediated stabilization of the β039-globin mRNA transcript. When data presented in Fig. 4B are presented together with those of Fig. 5B, C, it appears clear that the β039-globin mRNA is translated at high levels in the presence of G418.
Figure 5.

A: RT-PCR quantitative analysis performed on RNA isolated from K562-wt3 (A, black symbols), K562-m5 (A, open symbols) and from erythroid cells from β039-thalassemia patients (B, C), using primers amplifying β-globin mRNA sequences. In panels A and C, cells were treated with the indicated amounts of G418. In panels A and C, results are presented as fold induction of β-globin (panel A) and β0-globin mRNA (panel C) of G418-treated cells with respect to untreated controls (mean ± SD from three different determinations).
Discussion
The first result of this article is that the aminoglycoside geneticin (G418) is able to induce production of β-globin in cells carrying β-globin genes with the β 039-thalassemia mutation, by the readthrough mechanism leading to translation of β039-globin mRNA and ultimate production of HbA. This was reproducibly obtained using K562 cell clones carrying β039-globin genes, generated by stable transduction with a lentiviral vector carrying the β039-globin gene under the control of a minimal LCR region. This effect was demonstrated not associated with alteration in proteomic profile (see Fig. 3), major alteration of cellular morphology, and block of cellular proliferation (data not shown).
The major result of our article is that efficient production of β-globin by β039-globin mRNA occurs in erythroid precursor cells isolated from β039-thalassemia patients. To verify this interesting possibility, we recruited six homozygous β039-thalassemia patients. The erythroid progenitor cells of these patients were isolated from peripheral blood, and Hb production was stimulated after treatment with EPO with or without G418. Using G418, we consistently obtained the conversion of an high proportion of these cells from being negative for β-globin chain synthesis to β-globin producing cells. This was firmly established by both FACS (Fig. 4A) and HPLC analyses (Fig. 4B). These findings were reproducibly obtained in erythroid precursor cells from different β039-thalassemia patients (Fig. 4C) and support the notion that this strategy might be considered a therapeutic approach for treating β039-thalassemia. The effects observed are associated with an increase of β-globin mRNA content, presumably due to stabilization of the unstable β039-globin mRNA in the presence of G418. In agreement with this hypothesis, no changes in β-globin mRNA are detectable when K562-wt3 cells, expressing the wild-type β-globin mRNA, are treated with G418 (see Fig. 5).
The data presented in this article should be considered as a “proof of principle” that drug-induced ribosomal read-through might lead to β-globin production by β039-globin mRNA. The first effect of this β-globin production in homozygous β039-thalassemic cells leads to a decrease of the excess of α-globin, indicating the achievement of a first therapeutic relevant objective. Despite we are far away to the reachment by this strategy of a full restoration of HbA content in homozygous β039-thalassemic cells, due to the fact that the β039-globin mRNA is present in very low amounts, we like to underline that even a partial increase of HbA might be beneficial in patients carrying selected genotypes (for instance β0-39/β+IVSI-110) or when this approach is carried on in combination with other treatments (for instance those using hydroxyurea as inducers of HbF production) [48,49]. Further experiments are necessary for clarifying these very important points and to verify the effects of increased concentrations of G418, even if this would lead to alterations of cell growth. Furthermore, in the future, further issues might be approached, i.e., the combinations of NMD inhibitors and readthrough inducers using two different compounds within the same target cell. For instance, silencing RNAs against SMG-1 and Upf-1 strongly inhibit NMD with a mechanism of action clearly different from G418-mediated effects [50,51]. In addition, inhibition of NMD can be reached under hypoxic conditions, as suggested by Gardner [52]. Finally, the functionality of the HbA produced should be clearly investigated, because in our article, we have not characterized the aminoacid substition(s) following G418- mediated readthrough.
In any case, the readthrough strategy to overcome, even partially, stop mutations occurring in β-globin genes of β-thalassemic patients might turn to be a novel alternative approach to cure β-thalassemia in a subset of β-thalassemic patients (carrying pathological stop codons in homozygous or heterozygous state).
In addition to the data presented in this article, several considerations available in the literature support this hypothesis. First of all, it has been firmly demonstrated that aminoglycosides-mediated readthrough is dependent on the “sequence context,” in which the stop codons are located, introducing the possibility of a lower effects of G418 on normal stop codons [53,54]. Second, even if G418-mediated readthrough is occurring to some extent in nonglobin mRNAs, this is expected to cause production of very low amounts of altered proteins [51], which are expected to be degraded by the proteasome machinery.
Recent literature, in agreement with a possible read-through strategy to cure the diseases caused by nonsense mutations, has demonstrated the application of this strategy to cystic fibrosis [34,36], DMD [40,41], hemophilia [55,56], ataxia-telangiectasia [57], and Hurler syndrome [58].
Accordingly, the importance of projects aimed at identifying aminoglycoside analogs is reinforced by the results described here, and the identification of novel molecules exhibiting better parameters of administration to the patients, availability, and in vivo toxicity was reported with great interest from the research community. As far as the use of aminoglycosides of possible therapeutic applications, gentamycin should be carefully analyzed, despite the facts that it is expected to be less efficient of G418 in our cellular systems [43] and erythroid precursor cells from homozygous β039-thalassemic patients.
In this respect, we like to outline recent reports describing a molecule (PTC124) able to suppress stop mutations by a readthrough activity. Interestingly, this molecule is administered orally and is expected to be very promising in therapy. PCT 124 is a 284.24 Da, achiral, 1,2,4,-oxadiazole linked to fluorobenzene and benzoic acid rings (3-[5-(2-fluorophenyl)-[1,2,4]oxadiazol-3-yl]-benzoic acid; C15H9FN2O3) with no structural similarity to aminoglycosides or other clinically developed drugs [28–30].
At last, we would like to underline that thalassemia and sickle cell anemia are among the major health problems in developing countries, where affected patients and healthy carriers are numerous, mainly because of the absence of genetic counseling and prenatal diagnosis [59,60]. It should be pointed out that pharmacological therapy of β-thalassemia is expected to be crucial for several developing countries, unable to efficiently sustain the high-cost clinical management of β-thalassemia patients requiring regular transfusion regimen, chelation therapy, and advanced hospital facilities. It is well known that, in addition of “direct costs,” blood transfusions requires accurate monitoring of the blood safety, using expensive technologies, some of which are based on multiple PCR covering all the possible hematological infectious diseases [59].
As far as alternative therapeutic approaches are concerned, gene therapy [61,62] and bone-marrow transplantation [63,64] are very promising strategies, but they are expected to be useful only for a minority of patients, selected on the basis of biological/genetic parameters and the economic possibility to afford these therapies.
On the other hand, large investments by pharmaceutical companies finalized to the design, production, and testing of novel drugs for the treatment of β-thalassemia is discouraged by the fact that this pathology is a rare disease in developed countries, because of the recurrent campaigns for prevention, genetic counseling, and prenatal diagnosis [59]. Therefore, the search of molecules exhibiting the property of inducing β-globin is of great interest.
We believe that this field will be exciting from the scientific point of view, but also represent a hope for several patients, whose survival will depend on the possible use of drugs rendering not necessary blood transfusion and chelation therapy.
Materials and Methods
Human K562 cell cultures and K562 cells clones carrying the bwt and the β039-globin genes
The human leukemia K562 cells [43,65] were cultured in humified atmosphere of 5% CO2/air in RPMI 1640 medium (SIGMA, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Analitical de Mori, Milan, Italy), 50 U/ml penicillin, and 50 mg/ml streptomycin. Cell growth was studied by determining the cell number per ml with a ZF Coulter Counter (Coulter Electronics, Hialeah, FL) [66–68]. Two lentiviral constructs were used to generate stable K562 clones integrating human normal (pCCL.β. PGW) and β039-globin (pCCL.β039. PGW) genes. Transduction was carried out by plating 105 K562 cells in 9.5-cm2 dishes with 45% RPMI and 45% I-MDM (Iscove’s Modified Dulbecco’s Medium, CAMBREX–Biowhittaker Europe), 10% FBS (Biowest, Nuaillé, France), 2 mM l-glutamine (CAMBREX–Biowhittaker Europe, Milan, Italy), 100 U/ml penicillin, and 100 mg/ml streptomycin (Pen-Strep, CAMBREX – Biowhittaker) in humified atmosphere of 5% CO2/air and adding the decided volume of the viral supernatant. To facilitate the cell infection, 10 μl of the 800 μg/μl transduction agent polybrene (Chemicon International, Millipore, Billerica, MA) was added to the K562 cells plated, which were subsequently cultured in a 5% CO2 incubator. After 7 days, K562 cells were cloned by limiting dilutions and GFP-producing clones identified under a fluorescence microscope and further characterized. Treatment with G418 (GIBCO—Invitrogen-Life Technologies, Carlsbad, CA) was carried out by adding the appropriate drug concentrations at the beginning of the experiment (cells were usually seeded at 30,000 cells/ml). The medium was not changed during the induction period. Details of the production of these clones have been included in a previous article [43].
Human erythroid cell cultures
Blood samples were obtained after receiving informed consent. The two-phase liquid culture procedure was used as previously described [44,45,67]. Mononuclear cells were isolated from peripheral blood samples by Ficoll-Hypaque density gradient centrifugation and seeded in α-minimal essential medium (α-MEM, SIGMA) supplemented with 10% FBS (Celbio, Milano, Italy), 1 μg/ml cyclosporine A (Sandoz, Basel, Switzerland), and 10% conditioned medium from the 5637 bladder carcinoma cell line. The cultures were incubated at 37°C, under an atmosphere of 5% CO2 in air, with extra humidity. After 7 days incubation in this phase I culture, the nonadherent cells were harvested, washed, and then cultured in fresh medium composed of α-MEM (SIGMA), 30% FBS (Celbio), 1% deionized bovine serum albumin (BSA, SIGMA), 10−5 M β-mercaptoethanol (SIGMA), 2 mM l-glutamine (SIGMA), 10−6 M dexamethasone (SIGMA), and 1 U/ml human recombinant EPO (Tebu-bio, Magenta, Milano, Italy) and stem cell factor (BioSource International, Camarillo, CA). This part of the culture is referred to as phase II [46]. Erythroid differentiation was determined by counting benzidine-positive cells after suspending the cells in a solution containing 0.2% benzidine in 0.5 M glacial acetic acid, 10% H2O2, as elsewhere described [45,67]. Treatment with G418 was carried out by adding the appropriate drug concentrations at the beginning of the experiment (cells were usually seeded at 106 cells/ml). The medium was not changed during the induction period. For analysis of haemoglobins, cells were harvested, washed once with phosphate-buffered saline (PBS), and the pellets were lysed in lysis buffer (sodium dodecyl sulfate, SDS, 0.01%). After spinning for 1 min in a microcentrifuge, the supernatant was collected and stored at 4°C.
RNA isolation and RT-PCR analysis
K562 clones and erythroid precursor cells were collected by centrifugation at 1,200 rpm for 5 min at 4°C, washed in PBS, lysed in 1 ml of TRIZOL® Reagent (GIBCO—Invitrogen- Life Technologies), according to the manufacturer’s instructions. The isolated RNA was washed once with cold 75% ethanol, dried, and dissolved in diethylpyrocarbonate-treated water before use. For gene expression analysis, 1 μg of total RNA was reverse transcribed by using random hexamers. Quantitative real-time PCR assay was carried out using gene-specific double fluorescently labeled probes in a 7700 Sequence Detection System version 1.7 (Applied Biosystems, Warrington Cheshire, UK) as described elsewhere [43,67]. The nucleotide sequences used for real-time PCR analysis of the K562 clones β-globin mRNA are as follows: primer forward, 5′-CAG GCT GCT GGT GGT CTA C-3′; primer reverse, 5′-AGT GGA CAG ATC CCC AAA GGA-3′; probe βwt, 5′-VIC-AAA GAA CCT CTG GGT CCA-TAMRA; probe β039, 5′-FAM-CAA AGA ACC TCT AGG TCC A-TAMRA-3′. The probes βwt and β039 were fluorescently labeled with VIC and FAM (Applied Biosystems), respectively, as to quantify the βwt and β039-globin mRNA in a single reaction. While, the primers and probe sequences for the quantitative PCR analysis of the human erythroid cells β-globin mRNA are as follows: β-globin forward primer, 5′-CAA GAA AGT GCT CGG TGC CT-3′, β-globin reverse primer, 5′-GCA AAG GTG CCC TTG AGG T-3′, and β-globin probe, 5′-FAM-TAG TGA TGG CCT GGC TCA CCT GGA C-TAMRA-3′. For real-time PCR analysis, we used as reference gene the endogenous control human GAPDH kit (Applied Biosystems). The fluorescent reporter and the quencher of the GAPDH probe were as follows: VIC and 6-carboxy-N,N,N′,N′-tetramethylrhodamine (TAMRA), respectively.
High-performance liquid chromatography
Human erythroid precursor cells were harvested, washed once with PBS, and the pellets were lysed in lysis buffer (SDS 0.01%). After incubation on ice for 15 min and spinning for 5 min at 14,000 rpm in a microcentrifuge, the supernatant was collected and injected. Hb proteins present in the lysates were separated by cation-exchange HPLC [69], using a Beckman Coulter instrument System Gold 126 Solvent Module-166 Detector. Hbs were separated using a Syncropak CCM 103/25 (250 mm × 4.6 mm) column, samples were eluted in a solvent gradient using aqueous sodium acetate-BisTris-KCN buffers, and detection was performed at 415 nm. The standard controls were the purified HbA (SIGMA, St. Louis, MO) and HbF (Alpha Wassermann, Milano, Italy) [69].
Immunocytochemistry and FACS
K562 cells treated with G418 were permeabilized and marked with the antibody against β-globin using the Cytofix/Citoperm™ Kit (BD Biosciences Pharmingen, Franklin Lakes, NJ). A total of 1.5 × 106 cells were first washed with 500 μl of PBS 1× (CAMBREX—Biowhittaker Europe) and then incubated with 500 μl of BD Cytofyx-Citoperm solution for 20 min at 4°C, to permit the cellular permeabilization. After incubation, the cells were washed twice and incubated with 300 μl of PBS 1×–BSA 1% (SIGMA) solution for 1 hr at room temperature in darkness. The BSA has the capacity to block the aspecific binding sites. The cells were then collected by centrifugation and incubated with 30 μl of β-globin-PE (PE-phycoerythrin) (Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:10 in PBS 1×–BSA 1%, for about 20 hr at 4°C in darkness. After incubation, the cells were washed with 500 μl of PBS 1× and resuspended with 30 μl of PBS 1×. A total of 1/3 of the cellular suspension was placed on a chamber slide (CultureSlide, FALCON, Becton-Dickinson), previously treated with 0.01% poly-l-lysine (SIGMA), drained, fixed with 4% formalin (SIGMA), and mounted for examination. The slides were analyzed with the Olympus BX60 fluorescence microscope and the imagines acquired with a Nikon DS-2Mv digital camera. The left 2/3 of the cellular suspension was transferred to a FACS tube and 500 μl of staining buffer (PBS 1× plus 1% FBS) was added. The analysis of these cells was performed with FACScan (flow-activated cell sorting, Becton-Dickinson), using the software Cell Quest Pro (Becton-Dickinson).
Extract preparation
Cytoplasmic extracts from treated or untreated K562 cells were prepared by the technique reported by Andrews and Faller [70]. Briefly, K562 cells (2 × 107 cells) were collected and washed three times with cold PBS (Lonza-Biowhittaker, Basel, Switzerland). Cellular pellets were then resuspended in cold buffer A (10 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 0.2 mM PMSF) (400 μl for 9 × 106 cells), allowed to swell on ice for 10 min, and vortexed for 10 sec. Samples were finally centrifuged at 13,200g for 10 sec, and the supernatant cytoplasmic fractions were collected and immediately frozen at −80°C. Protein concentration was determined according to the Bradford method [71].
Two-dimensional gel electrophoresis
Approximately 300 μg of each sample protein extract was treated with ReadyPrep™ 2D Cleanup Kit (Bio-Rad) to eliminate high levels of salts and other interfering compounds. Pellets were resuspended in 600 μl rehydration buffer (8 M urea, 2% w/v CHAPS, 50 mM DTT, 0.2% w/v Bio-Lyte 3/10 ampholyte, 0.002% w/v Bromophenol Blue) for isoelectric focusing (IEF). After determining the concentration of purified proteins according to the Bradford method [71], about 100 μg of sample was used to rehydrate 7 cm long, pH 3–10 immobilized linear pH gradient strips (Ready-Strip™ IPG Strip, Bio-Rad), allowing a passive rehydration at room temperature for about 16–18 hr. IEF was then performed at 20°C using a Protean IEF Cell (Bio-Rad): after a first step at 250 V for 20 min, a gradient of 250–4,000 V was applied to the strips, followed by constant 4,000 V, with focusing complete after 10,000 Vh; a last maintenance step at 500 V was performed. After IEF, IPG strips were equilibrated for 10 min with equilibration buffer I (0.375 M Tris-HCl pH 8.8, 6 M urea, 20% v/v glycerol, 2% w/v SDS, 2% w/v DTT). The procedure was then repeated with equilibration buffer II, containing 2.5% w/v iodoacetamide instead of DTT. The second dimension run was performed using a MiniProtean® 3 (Bio-Rad) electrophoresis system, gel size 8.3 cm × 7.3 cm, 4% acrylamide stacking gel and 12% acrylamide running gel: equilibrated strips were inserted into the vertical slab gel and sealed with 0.5% low-melting point agarose, then SDS-PAGE was performed at 200 V for 50 min at room temperature. Precision Plus Protein Standard Plugs Unstained (Bio-Rad) was used as molecular weight marker. Gels were stained overnight with Bio-Safe Coomassie Stain (Bio-Rad), whereas destaining was performed with distilled water, until a clear background was achieved. Four replicas for each condition (control and G418-treated) were made and the same experiments were repeated twice.
Image acquisition and analysis
The 2DE gels were scanned by a GS-800 Calibrated Densitometer (Bio-Rad, Hercules, CA), using the Quantity One (1D Analysis Software), version 4.6.1 (Bio-Rad), to acquire images. The spot analysis was performed by the PDQuest™ Basic (2D Analysis Software), version 8.0 (Bio-Rad), creating two analysis sets from the protein patterns, each referring to a specific sample (control K562 cells, G418-treated K562 cells). After normalizing spot amounts to remove nonexpression-related variations, the results were evaluated in terms of spot intensities. Statistical analysis allowed the identification of the spots which were constantly reproduced, as well as those which showed a twofold differential intensity.
Statistical analysis
The statistical significance of difference in between different treatments was analyzed using one-way analysis of variance and the Student-Newman Keuls test. P values lower than 0.01 were considered statistically significant.
Acknowledgments
Contract grant sponsor: Telethon; Contract grant number: GGP07257; Contract grant sponsors: Fondazione CARIPARO (Cassa di Risparmio di Padova e Rovigo), AIRC, Cofin-2005, STAMINA Project (University of Ferrara), UE ITHANET Project (eInfrastructure for the Thalassaemia Research Network), Regione Emilia-Romagna (Spinner Project), Associazione Veneta per la Lotta alla Talassemia (AVLT), Rovigo.
Footnotes
Conflict of interest: Nothing to report.
References
- 1.Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson J, Martin R. Mutations to nonsense codons in human genetic disease: Implications for gene therapy by nonsense suppressor tRNAs. Nucleic Acids Res. 1994;22:1327–1334. doi: 10.1093/nar/22.8.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urlaub G, Mitchell PJ, Ciudad CJ, Chasin LA. Nonsense mutations in the dihydrofolate reductase gene affect RNA processing. Mol Cell Biol. 1989;9:2868–2880. doi: 10.1128/mcb.9.7.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inácio A, Silva AL, Pinto J, et al. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem. 2004;279:32170–32180. doi: 10.1074/jbc.M405024200. [DOI] [PubMed] [Google Scholar]
- 5.Mashima Y, Murakami A, Weleber RG, et al. Nonsense-codon mutations of the ornithine aminotransferase gene with decreased levels of mutant mRNA in gyrate atrophy. Am J Hum Genet. 1992;51:81–91. [PMC free article] [PubMed] [Google Scholar]
- 6.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 7.Feldmann D, Laroze F, Troadec C, et al. A novel nonsense mutation (Q1291X) in exon 20 of CFTR (ABCC7) gene. Hum Mutat. 2001;17:356. doi: 10.1002/humu.50. [DOI] [PubMed] [Google Scholar]
- 8.Pagani F, Buratti E, Stuani C, Baralle FE. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J Biol Chem. 2003;278:26580–26588. doi: 10.1074/jbc.M212813200. [DOI] [PubMed] [Google Scholar]
- 9.Brooks DA, Muller VJ, Hopwood JJ. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol Med. 2006;12:367–373. doi: 10.1016/j.molmed.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Tran VK, Takeshima Y, Zhang Z, et al. A nonsense mutation-created intraexonic splice site is active in the lymphocytes, but not in the skeletal muscle of a DMD patient. Hum Genet. 2007;120:737–742. doi: 10.1007/s00439-006-0241-y. [DOI] [PubMed] [Google Scholar]
- 11.Ito T, Takeshima Y, Yagi M, et al. Analysis of dystrophin mRNA from skeletal muscle but not from lymphocytes led to identification of a novel nonsense mutation in a carrier of Duchenne muscular dystrophy. J Neurol. 2003;250:581–587. doi: 10.1007/s00415-003-1040-1. [DOI] [PubMed] [Google Scholar]
- 12.Trecartin R, Liebhaber SA, Chang JC, et al. β0 thalassemia in Sardinia is caused by a nonsense mutation. J Clin Invest. 1981;68:1012–1017. doi: 10.1172/JCI110323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piras I, Vona G, Falchi A, et al. Beta-globin cluster haplotypes in normal individuals and beta(0)39-thalassemia carriers from Sardinia, Italy. Am J Hum Biol. 2005;17:765–772. doi: 10.1002/ajhb.20442. [DOI] [PubMed] [Google Scholar]
- 14.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med. 2002;80:367–376. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- 15.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 16.Stalder L, Mühlemann O. The meaning of nonsense. Trends Cell Biol. 2008;18:315–321. doi: 10.1016/j.tcb.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Isken O, Maquat LE. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 18.Behm-Ansmant I, Kashima I, Rehwinkel J, Saulière J. mRNA quality control: An ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett. 2007;581:2845–2853. doi: 10.1016/j.febslet.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 19.Takeshita K, Forget BG, Scarpa A, Benz EJ., Jr Intracellular defect in beta-globin mRNA accumulation due to a premature translation termination codon. Blood. 1984;64:13–22. [PubMed] [Google Scholar]
- 20.Lim SK, Sigmund CD, Gross KW, Maquat LE. Nonsense codons in human beta-globin mRNA result in the production of mRNA degradation products. Mol Cell Biol. 1992;12:1149–1161. doi: 10.1128/mcb.12.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huisman THJ, Carver MFH, Baysal E. A Syllabus of Thalassemia Mutations. Augusta, GA: The Sickle Cell Anemia Foundation; 1997. [Google Scholar]
- 22.Ahmed M, Stuhrmann M, Bashawri L, et al. The beta-globin genotype E121Q/W15X (cd121GAA-->CAA/cd15TGG-->TGA) underlines Hb d/beta-(0) thalassaemia marked by domination of haemoglobin D. Ann Hematol. 2001;80:629–633. doi: 10.1007/s002770100376. [DOI] [PubMed] [Google Scholar]
- 23.Kornblit B, Taaning P, Birgens H. Beta-thalassemia due to a novel nonsense mutation at codon 37 (TGG-->TAG) found in an Afghanistani family. Hemoglobin. 2005;29:209–213. doi: 10.1081/hem-200066319. [DOI] [PubMed] [Google Scholar]
- 24.Li D, Liao C, Li J, Tang X. The codon 37 (TGG-->TAG) beta(0)-thalassemia mutation found in a Chinese family. Hemoglobin. 2006;30:171–173. doi: 10.1080/03630260600642385. [DOI] [PubMed] [Google Scholar]
- 25.Préhu C, Pissard S, Al-Sheikh M, et al. Two French Caucasian families with dominant thalassemia-like phenotypes due to hyper unstable hemoglobin variants: Hb Sainte Seve [codon 118 (−T)] and codon 127 [CAG-->TAG (Gln-->stop]) Hemoglobin. 2005;29:229–233. doi: 10.1081/hem-200066335. [DOI] [PubMed] [Google Scholar]
- 26.Patterson M, Walker L, Chui DH, et al. Identification of a new beta-thalassemia nonsense mutation [codon 59 (AAG-->TAG)] Hemoglobin. 2003;27:201–203. doi: 10.1081/hem-120023385. [DOI] [PubMed] [Google Scholar]
- 27.Amato A, Pia Cappabianca M, Ponzini D, et al. Detection of a rare beta-globin nonsense mutation [codon 59 (AAG-->TAG)] in an Italian family. Hemoglobin. 2006;30:405–407. doi: 10.1080/03630260600755948. [DOI] [PubMed] [Google Scholar]
- 28.Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 29.Du M, Liu X, Welch EM, et al. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirawat S, Welch EM, Elfring GL, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 31.Kellermayer R. Translational readthrough induction of pathogenic nonsense mutations. Eur J Med Genet. 2006;49:445–450. doi: 10.1016/j.ejmg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Zingman LV, Park S, Olson TM, et al. Aminoglycoside-induced translational read-through in disease: Overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther. 2007;81:99–103. doi: 10.1038/sj.clpt.6100012. [DOI] [PubMed] [Google Scholar]
- 33.Hainrichson M, Nudelman I, Baasov T. Designer aminoglycosides: The race to develop improved antibiotics and compounds for the treatment of human genetic diseases. Org Biomol Chem. 2008;6:227–239. doi: 10.1039/b712690p. [DOI] [PubMed] [Google Scholar]
- 34.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 35.Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 36.Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. doi: 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 37.Du M, Keeling KM, Fan L, et al. Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J Mol Med. 2006;84:573–582. doi: 10.1007/s00109-006-0045-5. [DOI] [PubMed] [Google Scholar]
- 38.Wilchanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 39.Kerem E. Pharmacologic therapy for stop mutations: How much CFTR activity is enough? Curr Opin Pulm Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 40.Bidou L, Hatin I, Perez N, et al. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11:619–627. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- 41.Howard MT, Anderson CB, Fass U, et al. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 42.Howard MT, Shirts BH, Petros LM, et al. Sequence specificity of aminoglycoside- induced stop condon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–169. [PubMed] [Google Scholar]
- 43.Salvatori F, Cantale V, Breveglieri G, et al. Development of K562 cell clones expressing beta-globin mRNA carrying the beta039 thalassaemia mutation for the screening of correctors of stop codon mutations. Biotechnol Appl Biochem. 2009;54:41–52. doi: 10.1042/BA20080266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fibach E, Manor D, Oppenheim A, Rachmilewitz EA. Proliferation and maturation of human erythroid progenitors in liquid culture. Blood. 1989;73:100–103. [PubMed] [Google Scholar]
- 45.Pope SH, Fibach E, Sun J, et al. Two-phase liquid culture system models normal human adult erythropoiesis at the molecular level. Eur J Haematol. 2000;64:292–303. doi: 10.1034/j.1600-0609.2000.90032.x. [DOI] [PubMed] [Google Scholar]
- 46.Weatherall DJ. Pathophysiology of thalassaemia. Baillieres Clin Haematol. 1998;11:127–146. doi: 10.1016/s0950-3536(98)80072-3. [DOI] [PubMed] [Google Scholar]
- 47.Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol. 2002;9:123–126. doi: 10.1097/00062752-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 48.Fibach E, Burke LP, Schechter AN, et al. Hydroxyurea increases fetal haemoglobin in cultured erythroid cells derived from normal individual and patients with sickle cell anemia or beta-thalassemia. Blood. 1993;81:1630–1635. [PubMed] [Google Scholar]
- 49.Koren A, Levin C, Dgany O, et al. Response to hydroxyurea therapy in betathalassemia. Am J Hematol. 2008;83:366–370. doi: 10.1002/ajh.21120. [DOI] [PubMed] [Google Scholar]
- 50.Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsensemediated mRNA decay. PLoS Biol. 2008 Apr;29:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Usuki F, Yamashita A, Kashima I, et al. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol Ther. 2006;14:351–360. doi: 10.1016/j.ymthe.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–3741. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol. 1995;251:334–345. doi: 10.1006/jmbi.1995.0438. [DOI] [PubMed] [Google Scholar]
- 54.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.James PD, Raut S, Rivard GE, et al. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood. 2005;106:3043–3048. doi: 10.1182/blood-2005-03-1307. [DOI] [PubMed] [Google Scholar]
- 56.Pinotti M, Rizzotto L, Pinton P, et al. Intracellular readthrough of nonsense mutations by aminoglycosides in coagulation factor VII. J Thromb Haemost. 2006;4:1–7. doi: 10.1111/j.1538-7836.2006.01915.x. [DOI] [PubMed] [Google Scholar]
- 57.Lai CH, Chun HH, Nahas SA, et al. Correction of ATM gene function by aminoglycoside- induced read-through of premature termination codons. Proc Natl Acad Sci USA. 2004;101:15676–15681. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keeling KM, Brooks DA, Hopwood JJ, et al. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of α-L-iduronidase activity and reduces lysosomal-glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 59.Gambari R, Fibach E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr Med Chem. 2007;14:199–212. doi: 10.2174/092986707779313318. [DOI] [PubMed] [Google Scholar]
- 60.Bianchi N, Zuccato C, Lampronti I, et al. Fetal hemoglobin inducers from the natural world: A novel approach for identification of drugs for the treatment of beta-thalassemia and sickle-cell anemia. Evid Based Complement Altern Med. 2009;6:141–151. doi: 10.1093/ecam/nem139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sadelain M. Globin gene transfer as a potential treatment for the β-thalassemias and sickle cells disease. Vox Sang. 2004;87 (Suppl 2):S235–S242. doi: 10.1111/j.1741-6892.2004.00495.x. [DOI] [PubMed] [Google Scholar]
- 62.Puthenveetil G, Scholes J, Carbonell D, et al. Successful correction of the human β-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- 63.Lucarelli G, Clift RA, Galimberti M, et al. Bone marrow transplantation in adult thalassemic patients. Blood. 1999;93:1164–1167. [PubMed] [Google Scholar]
- 64.Lawson SE, Roberts IAG, Amrolia P, et al. Bone marrow transplantation for β-thalassemia major: The UK experience in two pediatric centres. Br J Haematol. 2003;120:289–295. doi: 10.1046/j.1365-2141.2003.04065.x. [DOI] [PubMed] [Google Scholar]
- 65.Lozzio CB, Lozzio BB. Human chronic myelogenous leukaemia cell-line with positive Philadelphia chromosome. Blood. 1975;45:321–334. [PubMed] [Google Scholar]
- 66.Lampronti I, Bianchi N, Borgatti M, et al. Accumulation of gamma-globin mRNA in human erythroid cells treated with angelicin. Eur J Haematol. 2003;71:189–195. doi: 10.1034/j.1600-0609.2003.00113.x. [DOI] [PubMed] [Google Scholar]
- 67.Fibach E, Bianchi N, Borgatti M, et al. Effects of rapamycin on accumulation of α-, β- and γ-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur J Haematol. 2006;77:437–441. doi: 10.1111/j.1600-0609.2006.00731.x. [DOI] [PubMed] [Google Scholar]
- 68.Zuccato C, Bianchi N, Borgatti M, et al. Everolimus is a potent inducer of erythroid differentiation and gamma-globin gene expression in human erythroid cells. Acta Haematol. 2006;117:168–176. doi: 10.1159/000097465. [DOI] [PubMed] [Google Scholar]
- 69.Huisman THJ. Separation of Hemoglobins and hemoglobin chains by HPLC. J Chromatogr. 1987;418:277–304. doi: 10.1016/0378-4347(87)80012-9. [DOI] [PubMed] [Google Scholar]
- 70.Andrews NC, Faller DV. A rapid micropreparetion technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]