

Abstract
During our efforts to characterize regulatory properties of human liver pyruvate kinase (L-PYK), we have noted that the affinity of the protein for PEP becomes reduced after several days post cell lysis. A 1.8Å crystallographic structure of L-PYK with the S12D mimic of phosphorylation indicates that Cys436 is oxidized, the first potential insight into explaining the effect of “aging”. Interestingly, the oxidation is only to sulfenic acid despite the two-week time of crystal growth. Mutagenesis confirms that the 436 side-chain is energetically coupled to PEP binding. Mass spectrometry confirms that the oxidation is present in solution and is not an artifact due to X-ray exposure. Exposure to the L-PYK mutations to H2O2 also confirms that PEP affinity is sensitive to the nature of the side-chain at position 436. A 1.95Å structure of the C436M mutation of L-PYK, the only mutation at position 436 identified to strengthen PEP affinity, revealed that the methionine substitution results in the ordering of several N-terminal residues that have not been ordered in previous structures. This result allowed a speculation that oxidation of Cys436 and phosphorylation of the N-terminus at Ser12 may function through a similar mechanism, namely the interruption of an activating interaction between the non-phosphorylated N-terminus with the non-oxidized main body of the protein. Mutant cycles were used to provide evidence that mutations of Cys436 are energetically synergistic with N-terminal modifications, a result that is consistent with phosphorylation of the N-terminus and oxidation of Cys436 functioning through mechanisms with common features. Alanine-scanning mutagenesis was used to confirm that the newly ordered N-terminal residues were important to the regulation of enzyme function by the N-terminus of the enzyme (i.e. not an artifact due to the introduced methionine substitution) and to further define which residues in the N-terminus are energetically coupled to PEP affinity. Collectively, these studies indicate energetic coupling (and potentially mechanistic similarities) between the oxidation of Cys436 and phosphorylation of Ser12 in the N-terminus of L-PYK.
The primary focus of our laboratory is the characterization of the molecular mechanisms of allosteric and covalent regulation of human liver pyruvate kinase. However, while addressing these structure/function questions, we have noted that the apparent affinity of L-PYK for substrate, phosphoenolpyruvate (PEP), becomes weaker several days post cell lysis. Our previous solution for this problem was based on a rapid purification protocol (1), which facilitate data collection before the time-dependent changes occur. In fact, all activity-dependent data previously reported from our laboratory for the L-PYK protein (1–6) have been collected within two days after cell lysis. Our first insight into a potential mechanism for the time-dependent decrease in substrate affinity was through the observation of an oxidized cysteine (i.e. Cys436) in a protein structure determined by X-ray crystallography. We report here not only the nature of the time-dependent shift in PEP affinity and the structure that initiated insights into the mechanism of this shift, but also a series of experiments that indicate that there is energetic synergy in the outcomes elicited by mutation of the oxidized residue (Cys436) and by modification of the N-terminus. The latter is consistent with Cys436 oxidation and N-terminal modification functioning through a related mechanism. Therefore, a brief review of the mechanism by which phosphorylation modifies PEP affinity will become useful (5–7).
Of the four mammalian pyruvate kinase (PYK) isozymes, the one expressed in liver (L-PYK) provides key regulation to maintain the balance between gluconeogenesis and glycolysis. Like all PYK isozymes, L-PYK catalyzes the transfer of phosphate from phosphoenolpyruvate (PEP) to ADP to form pyruvate and ATP. Consistent with its regulatory role in metabolism, human L-PYK is inhibited by phosphorylation at Ser12, is allosterically inhibited by alanine, and is allosterically activated by fructose-1-6-bisphosphate (Fru-1,6-BP). All forms of covalent and allosteric regulation result in altered PEP affinity.
L-PYK and the isozyme expressed in erythrocytes (R-PYK) are products from the same gene due to the use of different start sites (the other two isozymes, M1-PYK and M2-PYK are products from a second gene). As a result of the altered start site for translation, R-PYK contains 31 additional amino acids at the N-terminus compared to L-PYK. However, little is known about the structure of the N-terminus of either isozyme. The previously reported 2.7Å structure of R-PYK contained a 49 N-terminal truncation (8) (equivalent to L-PYK without the initial 18 residues). Furthermore, additional residues were disordered such that the first ordered residue is equivalent to Gln26 of L-PYK. Therefore, the previous structure is not informative regarding how the N-terminus interacts with the main body of the protein or how phosphorylation modulates this interaction.
Previously we demonstrated that the S12D mutation mimics the effect of phosphorylation on L-PYK function (5). However, there were also indications in the literature that N-terminal truncation could mimic phosphorylation (9). Therefore, we completed a truncation series to provide additional experimental support to the hypothesis that phosphorylation of Ser12 interrupts an activating interaction between the N-terminus and the main body of the protein (5). As further evidence that the phosphorylated N-terminus does not interact with the main body of the protein, or does so with very low binding energy, we demonstrated that short peptide designed to mimic the non-phosphorylated N-terminus can cause the S12D protein to bind PEP with strengthened affinity (6). Therefore, the currently available data suggests that phosphorylation of Ser12 shifts a binding equilibrium to favor less of an activating interaction between the N-terminus and the main body of the L-PYK. As noted above, the summary of the studies reported here is consistent with the oxidation of Cys436 also functioning in an inhibitory fashion by shifting a binding equilibrium to favor less of the activating interaction between the N-terminus and the main body of L-PYK.
Materials and Methods
Mutagenesis and Protein Expression and Purification
Mutagenesis of the L-PYK gene to create desired mutations was with Quikchange (Stratagene). Wild type and mutant proteins were expressed in the FF50 strain of Escherichia coli (1). Cell lysis, ammonium sulfate fractionation and DEAE-cellulose column purification were carried out as previously reported (1). Mutant proteins for alanine-scanning mutagenesis were only partially purified using ammonium sulfate fractionation followed by protein dialysis (5).
Crystallization
Initial crystallization conditions for S12D-L-PYK were determined by submission of the protein to the high throughput crystallization facility at the Hauptman-Woodward Institute (Buffalo, NY) (10). Crystals of the S12D-L-PYK/Fru-1,6-BP/Mn/Na/Citrate complex were grown by the vapor diffusion hanging drop method. Briefly, S12D-L-PYK was dialyzed into 10 mM MES (pH 6.8), 5 mM MgCl2, 10 mM KCl and 2 mM DL-dithiothreitol (DTT) and then diluted in the same buffer to 5 mg/ml. Immediately before use, 25 μl of a Na-ATP/Na-Fru-1,6-BP solution was added to 100 μl protein to result in 4 mg/ml protein, 52 mM Na-ATP, and 1.3 mM Na-Fru-1,6-BP. The mother liquor solution consisted of 700 μl of 50 mM Na-citrate (pH 4.9), 26 mM MnCl2, and a range of 3 to 5% PEG 6000. The ratio of well solution to protein/Fru-1,6-BP/ATP solution was varied: (well:protein volumes in μl) 2:2, 4:2, 2:4, 4:4. Crystals were observed to grow over a period of 2 to 4 weeks at 4°C. Crystals were cryoprotected by sequential transfer of the crystals (at 4°C) into mother liquor solutions containing an increasing concentration of glycerol in 5% increments to a final concentration of 25%. During this transfer, the concentration of PEG 6000 was increased slightly and the concentration of other buffer components decreased slightly such that final concentrations were 25 mM Na-citrate (pH 4.9), 8.0% PEG 6000, 12 mM MnCl2, 25% glycerol, 0.5 mM MES, 0.5 mM KCl, 0.5 mM Fru-1,6-BP, and 20 mM ATP. The crystals were cryo-cooled prior to data collection by immersion directly into liquid nitrogen.
X-ray Diffraction Data Collection
Data on the cryo-cooled crystals at 100°K were collected at the Stanford Synchrotron Radiation Laboratory Beamline 11-1 (Menlo Park, CA). All data were integrated and scaled with HKL-2000 (11). Data statistics are presented in Table 1.
Table 1.
Crystallographic data and model statistics for the structure of S12D-L-PYK in complex with citrate, ATP, and Fru-1,6-BP.
| S12D-hLPYK Citrate/Mn/ATP/Fru-1,6-BP |
C436M-hLPYK Citrate/Mn/ATP/Fru-1,6-BP |
|
|---|---|---|
| Beamline | SSRL 11-1 | 19-ID, APS |
| Wavelength (Å) | 0.9 | 0.97921 |
| Space group | P21 | P21 |
| Unit cell | a = 78.1 Å | 82.7 |
| b = 205.1 Å | 204.7 | |
| C = 83.9 Å | 86.5 | |
| α = γ = 90.0° | 90.0 | |
| B = 92.2° | 96.7 | |
| Resolution Limits, Å | 50.0-1.8 | 50.0-1.95 |
| Total Reflections | 1019737 | 737037 |
| No. of unique reflections | 237180 | 200629 |
| Completeness, % (all data) | 97.8 (93.3) a | 97.7 (86.1) b |
| Redundancy | 4.3 (3.7) a | 3.7 (2.9) b |
| I/σ(I) | 17.7 (1.7) a | 14.9 (1.4) b |
| Rsym(%)* | 0.06 (0.51) a | 7.4 (51.8) b |
| No. of ASU molecules | 4 | 4 |
| No. reflections used | 224885 | 179645 |
| Rfree | 22.6 (35.1) a | 23.3 |
| Rwork | 18.9 (33.2) a | 19.8 |
| Bond length RMSD, Å | 0.02 | 0.007 |
| Bond angle RMSD, ° | 2.2 | 1.047 |
| Ramachandran: | ||
| Preferred, % | 96.2 | 97.07 |
| Allowed, % | 2.9 | 2.72 |
| Outliers, % | 0.9 | 0.21 |
| Average B Factor Protein: | ||
| Chain A | 37.7 | 40.0 |
| Chain B | 42.4 | 41.4 |
| Chain C | 43.1 | 41.8 |
| Chain D | 46.8 | 40.2 |
| Ligands: | ||
| Citrate | 32.0 | 35.8 |
| Adenosine | 34.6 | 43.2 |
| Fru-1,6-BP | 26.9 | 31.5 |
| Metals: | ||
| Mn2+ | 27.6 | 38.1 |
| Na+ | 39.8 | |
| Water | 38.4 | 42.4 |
Values in parentheses represent statistics for data in the highest resolution shell (1.86–1.80Å).
Values in parentheses represent statistics for data in the highest resolution shell (2.02–1.95Å).
Rsym = Σ|Iobs − Iavg|/ Σ Iavg; Rwork = Σ|| Fobs | − |Fcalc||/Σ Fobs
Rfree was calculated using 5% of data.
Structure Solution and Refinement
The initial phases for S12D-L-PYK were obtained by the method of molecular replacement using the program MOLREP (15) encoded in the CCP4 suite (16). X-ray coordinates of the R-PYK monomer (8), PDB entry 2VGB, were used as the search model after being stripped of all water molecules, ligands and ions. The molecular replacement solution resulted in an initial model containing a single tetramer in the asymmetric unit.
Refinement of the S12D-L-PYK model was carried out in Refmac5 (17) encoded in the CCP4 suite. After each round of refinement, Fo-Fc and 2Fo-Fc electron-density maps were produced and manual model building/optimization was carried out using the model-building/map-fitting program Coot (18). The addition of ligands, metals and water molecules was also carried out using Coot prior to a final round of restrained refinement in Refmac5. A final round of TLS refinement was carried out after the optimal number of TLS groups for each chain was determined using the TLSMD web server (http://skuld.bmsc.washington.edu/~tlsmd/). Each L-PYK chain contains 4 TLS groups for a total of 16 TLS groups. This resulted in a decrease in R/Rfree values from 19.2%/23.3% to 18.9%/22.6%. Structural validation was carried out using MolProbity (http://molprobity.biochem.duke.edu/) (12). The final model statistics are summarized in Table 1.
Kinetic Assays and data analysis
Activity measurements were carried out at 30°C using a lactate dehydrogenase coupled assay (4). For samples exposed to H2O2, the protein was added to a cocktail with all reagents necessary for activity, with the exception of PEP. H2O2 was added to this cocktail. After an 8 min. incubation, PEP was added and activity monitored as previously described (4). The lack of impact on Vmax activity confirms that activity reagents other than L-PYK were not sensitive to H2O2 during the brief incubation. C436A was the only mutation to largely prevent the H2O2 dependent-shift in Kapp-PEP. This was true whether proteins were used immediately after purification or if the four proteins were first diluted to similar activities and then dialyzed together against buffer with 2mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP). The latter was an experimental design to ensure that reducing agent added with protein was equivalent in all assays.
Data fitting was with the nonlinear least-squares analysis of Kaleidagraph (Synergy) software. Fits of PEP titrations of initial rates (ν) used to obtain Kapp-PEP are as previously described (1, 4). When evaluating allosteric regulation, Ka-PEP was determined by fitting a plot of the Kapp-PEP values as a function of effector concentration to equation 1 (13):
| equation 1 |
where Ka = Kapp-PEP when [Effector] = 0; Kix = the dissociation constant for effector (X) binding to the protein in the absence of substrate (A); and Qax is the allosteric coupling constant discussed in detail elsewhere (13). Due to the current focus of how different regions of the protein impact PEP affinity, only Kapp-PEP determined in the absence of effector or Ka-PEP values are included.
Results
Time-dependent shift in affinity for PEP
While studying the regulatory properties of purified L-PYK, we have noted that the affinity of the protein for PEP becomes weaker as the protein ages over days (Figure 1A). Although the data in Figure 1 shows a transition in Kapp-PEP after only one day, the exact time for the transition can vary considerably from 1 to 4 days (data not shown), but protein purified and stored in the presence of 2mM TCEP produces a constant Kapp-PEP for up to 7 days. (Recovery of “fresh” protein properties from long-term storage has now been accomplished by plunge freezing in thin walled PCR tubes followed by rapid thawing following a published method (14)). The change in PEP affinity due to “aging” appears to cause only slight changes in the magnitudes of the allosteric functions (Figure 1B).
Figure 1.

A time/aging dependent influence on the affinity of L-PYK for PEP. A) The initial velocity derived Kapp-PEP for wild type L-PYK as a function of storage time after purification (36 hrs. after cell lysis are required for purification). The line represents the data trend. B) The influence of “aging” on allosteric properties elicited by Fru-1,6-BP (squares) and by alanine (circles). The influence of ageing appears to primarily impact PEP affinity since the allosteric properties (the distance between the plateaus at low and high effector concentrations) is relatively unchanged for freshly purified (solid symbols) vs. “aged” (open symbols) enzyme. Lines represent the best fits to Equation 1.
Oxidation of cysteine 436 to sulfenic acid
We originally initiated crystallographic studies of L-PYK to characterize the structure of the N-terminus, which is not present and/or disordered in the previous structure (8) (an attempt to identify a previously proposed allosteric ATP binding site was a second goal, but the obtained structures provided little insight into the location of this ATP site; see Supportive Information). This effort resulted in a 1.8Å structure of the S12D enzyme (Table 1). Of particular interest, this structure presents evidence that Cys436 is partially oxidized to the sulfenic derivative (Figure 2). This partial oxidation is observed as density at an intermediate length between a hydrogen bonded water molecule and a covalently linked hydroxyl group, a result consistent with the averaging of oxidized and non-oxidized side-chains. Sulfenic acids are not typically stable. Instead, a sulfenic acid derivative is often rapidly further oxidized by reacting with additional oxygens to result in sulfinic and sulfonic derivatives (Supplemental Information). With this in mind, it is interesting that even though crystals used in this study required 2 to 3 weeks to grow, no evidence for oxidation beyond the sulfenic state was observed.
Figure 2.

A) Aberrant, unmodeled density adjacent to Cys436. 2Fo-Fc density rendered at 1.2σ and Fo-Fc density rendered at 2.5σ are illustrated as a blue and a green mesh, respectively. The dashed black line indicates a distance of 2.3Å from the center of the unmodeled density to the sulfur of C436. B) Spatial relationship of Cys436 (black spacefill) to other structural features. The four subunits of the hL-PYK homotetramer are in red, green, blue, and gray. The N-terminus of the red subunit is in cyan as determined by the C436M structure reported in this study. Citrate in the active site is in orange spacefill. Fru-1,6-BP is displayed in the top two subunits in magenta spacefill, but (although present in the structure) is not displayed in the two bottom subunits.
Confirmation of oxidation of Cys436
Mass spectrometry was used to confirm that Cys436 can be oxidized in purified protein in solution (Table 2). The observed mass is consistent with the sulfonic acid form of Cys436; sulfenic acids often further oxidize to sulfonic acid during mass spectrometry analysis (15). The sensitivity of Kapp-PEP to a brief incubation with low concentrations of H2O2 is also consistent with protein oxidation (Figure 3). These observations are also evidence that the oxidation in the structure is not solely an artifact due to exposure to X-ray, although exposure may have contributed to oxidation.
Table 2.
Cys436 containing peptides and their modifications detected by Sequest analysis of tandem mass spectraa
| Peptide sequence | MH+ | z | P (pep) | XC | ΔCn |
|---|---|---|---|---|---|
| AAPLSRDPTEVTAIGAVEAAFKC@CAAAIIVLTT | 3323.82 | 3 | 1.01E-05 | 3.72 | 0.36 |
| AAPLSRDPTEVTAIGAVEAAFKC#C#AAAIIVLTTTGR | 3704.27 | 3 | 2.81E-12 | 8.21 | 0.58 |
| C@C@AAAIIVLTTTGR | 1489.70 | 2 | 7.17E-05 | 2.68 | 0.35 |
| C#C#AAAIIVLTTTGR | 1507.80 | 2 | 1.24E-06 | 3.93 | 0.42 |
L-PYK was subjected to iodoamide alkylation previously to its overnight treatment with trypsin at 37°C. The resulting peptides were analyzed by HPLC-MS/MS on an LTQ-FT mass spectrometer. C# stands for carboxymethylcysteine; C@ stands for Cysteine sulfonic acid. Tandem mass spectra were searched against a human protein database using the Sequest algorithm and the following parameters for protein ID: peptide probability of random match <10-3, XC ≥ 1.9, 2.5 or 3.5 for z=1, 2 or 3 respectively and ΔCn ≥ 0.1. Peptides containing oxydized Cys are highlited in boldface. Since C# denoted cysteines were reactive modified by alkylation, they were not irreversibly oxidized.
Figure 3.

Sensitivity of Kapp-PEP for the wild type (black, filled circles) and C436A (red) L-PYK proteins to short (8 min.) incubation with H2O2. Exposure to concentrations up to 100 mM did not alter Vmax activity (data not shown). Wild type protein (black, open squares) in the presence of 2 mM DTT was included as a control. Data collection was replicated by multiple individuals using different protein preparations. Although general features were maintained in replicates (i.e. the wild type protein being much more sensitive to H2O2 as compared to C436A), there was considerable variability between protein preps in the Kapp-PEP determined for a given protein at a given H2O2 concentration. A representative comparison is included here.
The location of this regulatory oxidation at Cys436 is supported by the fact that much higher concentrations of H2O2 are required before the C436A mutant protein shows reduced PEP affinity (Figure 3); the impact of this mutation on apparent PEP affinity is presented below. To confirm the specificity, C338A was shown to maintain H2O2 sensitivity (Supplemental Material). C338A is a control protein with a mutant that also removes a cysteine residue but at a location distant from the N-terminus region in the structure. Therefore, the loss in sensitivity to oxidation in the C436A mutant protein is not a general effect due to a global modification of all cysteine residues in the protein (6 per subunit).
Several reports in the literature indicate roles for various PYK isozymes in oxidative stress (16–24). Characterization of cysteine oxidation in L-PYK in vivo was not our goal. However, we did find it reasonable to test the potential impact of oxidation of cysteine residues reported in the literature. Anastasiou et al. reported that M2-PYK is oxidized at the cysteine position equivalent to Cys370 in human L-PYK (17). This site of oxidation was determined by mutating three cysteine residues that reside in a subunit-subunit interface in the M2-PYK protein. In the current study to test the potential regulatory role of oxidation of Cys370 in L-PYK, this position was mutated to alanine. The mutant protein has a Kapp-PEP (0.223±0.006mM) similar to that of the wild type protein. Upon exposure to H2O2 (Supplemental Material), the C370A protein continues to show a response consistent with an oxidation event that modifies Kapp-PEP. Therefore, it appears that Cys370 is not the residue that responds to oxidation in L-PYK.
If the oxidation event responsible for weaker PEP affinity is truly the sulfenic acid derivitization of Cys436, then it seems reasonable to anticipate that this modification is reversible. However despite this prediction, we have not to-date successfully demonstrated reversibility using either DTT or TCEP additions.
Characterization of the C436M protein
Energetic coupling between the 436 position and the PEP binding site was confirmed by replacing cysteine at position 436 with a number of substitutions (Table 3). None of the mutant proteins retained wild type Ka-PEP. Of the mutant proteins, almost all substitutions caused weaker PEP affinity. C436A and C436H caused the smallest changes in Ka-PEP. C436S, C436D, C436N, and C436T caused larger reduction in PEP affinity. We are unaware if there is a single amino acid type that acts as a good mimic for oxidized cysteine in the same manner that glutamic acid and aspartic acid mimic phosphorylated serine. However based on the speculation from the summary of this study (i.e. that oxidation shifts the equilibrium of the binding interaction between the N-terminus and the main body of the protein), the only requirement for mimicry in this setting may be the ability to disrupt interactions that contribute positively to binding, hince the decreased PEP affinity in most mutants. The corollary is that C436M, the only mutant protein with Kapp-PEP less than that of the wild type protein, may stabilize the interaction between the N-terminus and the main body of the protein.
Table 3.
K a-PEP for L-PYK with mutations introduced at Cys436
| Protein | Ka-PEPa |
|---|---|
| Wild Type | 0.240±0.002 |
| C436A | 0.372±0.002 |
| C436S | 0.802±0.004 |
| C436D | 0.804±0.002 |
| C436N | 0.703±0.004 |
| C436M | 0.112±0.001 |
| C436T | 0.768±0.006 |
| C436H | 0.345±0.004 |
Fit parameter from Equation 1
The C436M protein was crystallized using the same crystallization conditions used earlier to obtain the structure of the S12D modified protein. Several N-terminal residues (residues 18–24) that were not ordered in previous structures are ordered in the structure of the C436M protein. Specifically, the side-chains of residues Leu20, Phe24, and Phe25 all make favorable van der Waals interactions with methionine at position 436 (Figure 4).
Figure 4.
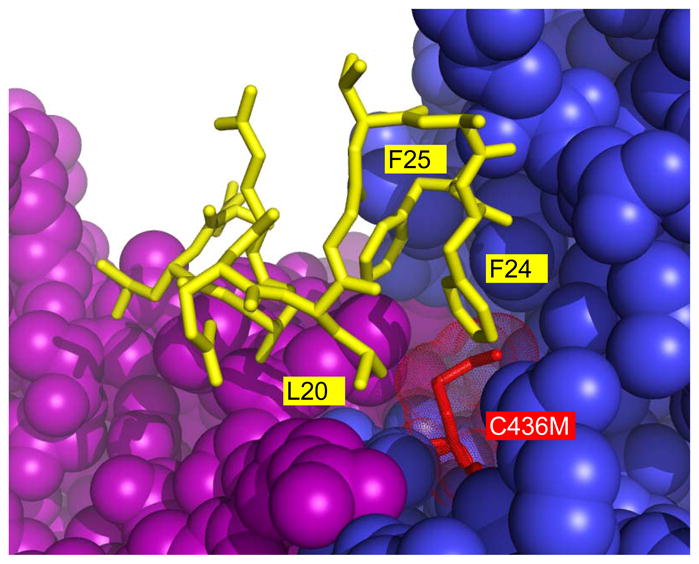
N-terminal residues (yellow stick) that become ordered as a result of the C436M (red stick) mutation. Specifically, residues 18–24 (QELGTAF) are ordered as a result of the mutation. The side-chains of residues Leu20, Phe24, and Phe25 from the N-terminus interact with methionine at position 436 of the same subunit (blue). The newly ordered residues cross a subunit interface to interact with a second subunit (purple).
N-terminus/Cys436 energetic synergy
Since the C436M mutation causes ordering of additional N-terminal residues, since these N-terminal residues directly interact with the 436 side-chain, and since oxidation of Cys436 and phosphorylation of the N-terminus(5) both result in weaker affinity for substrate, we considered that oxidation and phosphorylation may function through similar mechanisms. Such a shared mechanism would result in energetic synergy between outcomes resulting from modifications at the phosphorylation site and modifications at the oxidation site. The potential of this energyetic synergy was tested using mutant cycle analysis. This approach has previously been useful in testing if two different regions of a protein work synergistically in an allosteric mechanism (25). Mutant cycle analysis can be understood conceptually by considering our proposed mechanism for phosphorylation; i.e. that a modification at the N-terminus (i.e. phosphorylation, the S12D phosphorylation mimic, or N-terminal truncation (5)) disrupts an activating interaction between the N-terminal/main body of the protein (5). Oxidation of Cys436 on the main body of the protein may also interrupt this N-terminus/main body interaction. Therefore, introducing both a mutation that mimics Cys436 oxidation (i.e. C436S or C436N; see Table 3) and a modification at the N-terminus that mimics phosphorylation (S12D or an N-terminal truncation; (5)) will not introduce any further change compared to that introduced by adding only one of the two modifications. “Disrupts an interaction” is not intended to strictly imply removal of an all-or-none interaction and the current scenario may be better visualized as a shift in a binding equilibrium for N-terminal binding, in which the addition of phosphorylation or oxidation shifts the equilibrium more to the unbound state. Therefore, introducing both a mutation that mimics Cys436 oxidation and a modification at the N-terminus that mimics phosphorylation may minimally shift the binding equilibrium compared to that caused by adding only one of the two modifications. In both an all-or-none view of N-terminal binding or a shift in a binding equilibrium, this energetic synergy can be identified at the free energy level as non-additivity. In contrast, if the two modifications elicit their respective effects on PEP affinity through independent mechanisms, it should be expected that the sum of the change (relative to wild type) caused by each modification alone will be equivalent to the change observed in the doubly modified enzyme (Table 4).
Table 4.
Additivity of mutant cycles
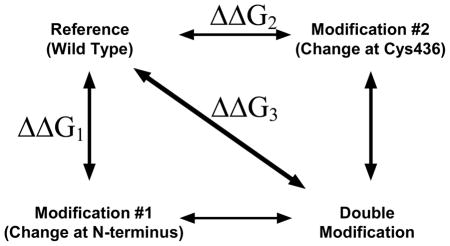
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Modification #1 | Modificaiton #2 | Double modification | |||||||
| Ka-PEP [mM] | Modification description | Ka-PEPb [mM] | ΔΔG1a (kcal/mol) | Modification description | Ka-PEP [mM] | ΔΔG2a (kcal/mol) | Calculated ΔΔG1 + ΔΔG2 (kcal/mol) | Ka-PEP [mM] | Δ ΔG3a (kcal/mol) | |
| Wild Type | 0.240±0.002 | Δ2–10 | 0.605±0.004 | 0.55±0.01 | C436S | 0.802±0.004 c | 0.726±0.008 | 1.28±0.02 | 1.139±0.003 | 0.938±0.007 |
| C436N | 0.703±0.004 c | 0.647±0.008 | 1.20±0.02 | 1.016±0.005 | 0.869±0.008 | |||||
| S12D | 0.552±0.007 | 0.50±0.01 | C436S | 0.802±0.004 c | 0.726±0.008 | 1.23±0.02 | 0.89±0.01 | 0.79±0.01 | ||
| C436N | 0.703±0.004 c | 0.647±0.008 | 1.15±0.02 | 0.95±0.01 | 0.83±0.01 | |||||
| C338A | 0.93±0.01 | 0.82±0.01 | 1.32±0.02 | 2.09±0.02 | 130±0.01. | |||||
ΔG = change in free energy associated with PEP binding. ΔΔG = the difference in ΔG of PEP binding of modified protein relative to that of the wild type enzyme. Conversion of Ka-PEP to ΔG is based on the generally accepted view that PYK isozymes function as rapid equilibrium systems (30). As such, the free energy associated with PEP binding was calculated by ΔG=−RTln Ka-PEP.
values from (12)
values from Table 2
Mutant cycle analysis was completed for all combinations of C436S or C436N vs. Δ2–10 (the protein without residues 2–10) or S12D. As a control for an example of additivity (i.e. no energetic coupling), a mutant cycle analysis was also completed between S12D and C338A, a mutation located at a site distant from the N-terminus region in the structure. By comparing the values in each row that are contained in the two shaded columns in Table 4, it is evident that the sum of the effects caused by S12D and C338A is equivalent to the effects caused when both mutants are added together. Therefore, these two mutations do not alter PEP affinity via perturbing the same energetic mechanism. However, for each combination of S12D or Δ2–10 with either C436S or C436N there is a difference of between 0.33 to 0.44 kcal/mol between the sum of changes caused by the individual modification (calculated Δ ΔG1 + Δ ΔG2) compared to the changes caused by the simultaneous addition of both mutations together (Δ ΔG3). As described above, this non-additivity indicates energetic synergy between modifications of the two regions of the protein. In turn, energetic synergy between these regions is consistent with the idea that introducing mutations at the Cys436 position modifies PEP affinity through a similar mechanism (or component of that mechanism) as that caused by introducing modifications at the N-terminus. Based on our previous work (5), this similar mechanism is likely a modification-dependent shift in the equilibrium of the interaction between the N-terminus and the main body of the protein. However, since the effect of a modification to the N-terminus in isolation is not completely equivalent in magnitude to the influence of a mutation of Cys436 (i.e. in Table 4, Ka-PEP=0.552/0.605 for modifications of the N-terminus vs. Ka-PEP-0.703/0.802 for modifications made at C436), we suggest similar, but not completely equivalent outcomes.
Alanine-scanning mutagenesis of the N-terminus for impact on PEP affinity
An alanine-scan across the N-terminus was initiated for two reasons: 1) to confirm that residues identified by structural studies to interact with the methionine at 436 contribute to N-terminal function (i.e. evidence that the observed interactions are not an artifact due to the introduced mutation); 2) to further detail individual residues that contribute to N-terminal functions. Of the unstructured N-terminal residues from the previous crystal structure (8),
MEGPAGYLRRASVAQLTQELGTAFF……,
non-alanine/non-glycine residues in the N-terminus were mutated to alanine as the best mimic of side-chain removal. The resulting series of mutant proteins each contain a single alanine substitution. Initially, the impact of these individual alanine substitutions on the apparent affinity of the enzyme for PEP was evaluated (Figure 5A). This apparent affinity was treated as a true Kd and converted to a free energy value. Using these free energy values, the mutant-dependent deviation from wild type was plotted as a function of the position of the mutated residue. For any residue at which an alanine or glycine is present in the wild type protein, the wild type value is used (i.e. no difference from wild type) in the graph. Interestingly, there is an apparent periodicity. In this periodicity, S12A and Q18A, and to a lesser extent T22A, cause strengthened PEP affinity. L20A and F24A, and to a lesser extent L16A, contribute to weakened PEP affinity. Considered in isolation, this periodicity could conceivably be consistent with a helical structure in which side-chain removal on one side of the helix removes critical interactions between the N-terminus and the main body of the protein (Supplemental Information). In contrast side-chain removal on the opposite side of the helix might remove hydrophilic residues that interact with water, favoring a shift in the propensity of the helix to bind to the protein. The residues ordered in the C436M structure are consistent with this periodicity, although the structure is not exactly helical in nature: Residues 24 and 20 are both interacting with the mutated 436 methionine and side-chains of residues 18 and 22 are orientated towards solvent.
Figure 5.

The impact of an N-terminus alanine scan on the ΔG associated with Ka-PEP. A) The impact of single residues substituted with alanine. Filled circles represent a comparison between measurements for mutant protein with that from wild type protein. Residues that are alanine or glycine in the wild type structure were not probed. Therefore, the wild type data used to represent Ka-PEP at these positions are represented by open circles to clearly distinguish data for mutant proteins. B) The impact of substituting two consecutive positions, both with alanine. C) The impact of substituting three consecutive positions with alanine. D) The overlay of the three data sets included in panels A, B, and C. In all panels, data are compared to that of the wild type protein. A positive deviation from zero on the y-axis indicates reduced PEP affinity; a negative value is improved PEP affinity. Panel D includes both the sum of effects caused by individual mutations (sum of Δ ΔG values for the two single alanine mutations; black) and the difference between the wild type value and that observed for the protein with both alanine substitutions of interest inserted simultaneously (Δ ΔG values for double alanine window mutations: red). As a means of simplifying panels, error bars are only included in panel A. However, error estimates are included for all data in the supplemental material. Green boxes are included in panels D and E to emphasize areas of interest.
The previously completed truncation series indicated the region including residues 7–10 as significant for binding of the non-phosphorylated N-terminus with the main body of the protein (5). Therefore, the lack of effects caused by removing side-chains of residues 2 or 4 is consistent with a lack of an effect due to truncation (residue 1 was not probed and residues 3, 5, and 6 are either Ala or Gly in the wild type protein and were not mutated). However, one surprise in the data in Figure 5A was the relatively small magnitude of the reduced affinity for PEP due to mutations in the region from 7–10. We considered that each residue in this region might contribute a fraction of the binding energy between the N-terminus and the main body of the protein. As such mutating a single residue (i.e. Figure 5A) would have only a slight effect on N-terminal binding and, thus, indirectly have only a slight effect on PEP affinity. In contrast, the previous deletion series would represent an additive effect: e.g. removal of residue 10 also removes residues 2–9. To test this possibility, a second alanine-scan was created in which two consecutive residues were replaced with alanine. Again, the impact on PEP affinity was determined (Figure 5B). A third alanine-scan was also repeated using a window of three consecutive alanine residues (Figure 5C). Outcomes of all three versions of the alanine-scans are overlaid in Figure 5D.
Consistent with the possibility that the binding energy for the non-phosphorylated N-terminus is spread over many interactions, combining consecutive alanine replacements highlights the region containing residues 7–10. To fully appreciate this additivity, consider that the data for mutations with three consecutive alanine replacements (blue points in Figure 5D) in the regions from 7–10 do not overlap with any of the data for single alanine replacements (black data). This is in contrast to examples like the data surrounding residue 20 in which any mutation combination that includes a modification of residue 20 is approximately equivalent (i.e. data in black overlaps data in blue). The comparison of data from various sizes of alanine “windows” is then fully consistent with the conclusion from the previous truncation series, and suggests that each residue in the 7–10 positions functions in an independent manner to add to the overall binding energy.
Driven by the data in-hand, we also questioned if there are regions in the N-terminus that function as a unit. In such interactions it is expected that modification of any one residue in the “unit” would alter the interaction of the entire unit. Therefore, the impact of introducing two mutations simultaneously into the unit should not be as drastic as the sum of changes caused by introducing the mutations individually; i.e. non-additivity. As exemplified above for mutations that modify Cys436 and the N-terminus, mutant cycle analysis can be used to identify non-additivity and therefore energetic synergy. However, this double mutant cycle analysis should be considered with some caution in this new application. We are unaware if mutant cycle analysis has previously been used with consecutive residue positions. Alanine, albeit our best mimic of side-chain removal, is known to change secondary structure propensity towards helix formation. Therefore, mutational analysis cannot consider an alanine substitution as merely the removal of the original side-chain (loss-of-function), but must keep in mind the potential of gain-of-function that could result from alanine driven helix formation. These considerations are also the basis of why we chose not to extend the non-additivity analysis to the three consecutive alanine data. Nonetheless, using the data from Figure 5A and 5B, Figure 5D considers when the combination of two consecutive alanine replacements results in a change in PEP affinity that is NOT equivalent to the sum of the effects caused by the two substitutions made independently in two different proteins. This non-additivity analysis identifies only one region with three or more consecutive positions, residues 15–20. In turn the energetic synergy indicated by non-additivity is consistent with residues 15–20 functioning as a unit.
Our exercise to provide data consistent with regions of the N-terminus that function as unit (i.e. non-additivity) and regions in which each residue contributing individually (i.e. additivity) should not overshadow the overall conclusion that many residues in the N-terminus contribute (both positively and negatively) to the overall PEP affinity. In particular, recall that the two goals of the alanine-scan were: 1) to confirm that residues identified by structural studies to interact with the methionine at 436 contribute to N-terminal function (i.e. evidence that the observed interactions are not an artifact due to the introduced mutation); 2) to further detail individual residues that contribute to N-terminal functions. Leu20 and Phe24, two residues identified to directly interact with methionine at 436, were included in the residues that impact N-terminal function. Therefore, both goals of the alanine-scan were accomplished.
Discussion
As introduced above, this study was initiated with a structural indication of a potentially oxidizable cysteine that could provide an explanation for an observed time-dependent shift in the substrate affinity displayed by a purified protein. However, the summation of the studies used to detail these initial observations has resulted in an advancement of our knowledge about the structure/function relationship of the N-terminus in regulation by oxidation and phosphorylation. In addition, clarification of the binding orientation of Fru-1,6-BP and the absence of bound ATP were secondary conclusions drawn from the new structure (Supplemental Information).
Phosphorylation
Despite a long standing understanding of the region of L-PYK that is phosphorylated in response to glucagon, mechanistic studies to understand how this phosphorylation results in weaker PEP affinity have only recently been initiated (5–7). It now seems likely that the Leu20 region of the N-terminus must interact with Cys436 before additional contacts involving residues 7–10 can be made. Therefore, modification of Cys436 may result in changes to a larger region of the N-terminal/main body interactions than modification of the Ser12 position. This would be consistent with the fact that modification of the N-terminus and Cys436 did not result in completely equivalent magnitudes in the changes each caused in PEP affinity and that the C436M mutation cause order of some, but not all N-terminal residues. All data continues to be consistent with the proposal that phosphorylation interrupts and activating interaction between the N-terminus and the main body of the protein.
Oxidation
We now have sufficient data to speculate that oxidation of Cys436 functions through shifting a binding equilibrium to favor less of an activating interaction between the N-terminus and the main body of the protein. However, this oxidation may perturb a slightly different set of interactions as compared to phosphorylation.
Opposite to phosphorylation, it is the potential physiological role (if any) of this modification that is uncharacterized and can only be speculated on at this time. PYK transcription is activated by the oxygen restriction in both tissues that express the L/R-PYK isozymes and those that express the M1/M2 isozymes (18–20). A potential metabolic rationale is that glycolysis is induced when oxygen is restricted, because glycolysis (as opposed to oxidative phosphorylation) is the primary source of cellular energy under these conditions. As additional support, up-regulation of M1-PYK increases tolerance to hypoxic stress in neuronal cells (21), whereas a mutation in R-PYK augments oxidative stress in erythroleukemia cells (22). Given this strong evidence for up-regulation of PYK protein concentration due to hypoxia, it may at first appear paradoxical that the enzymatic activity of a R-PYK protein is diminished upon exposure to oxygen radicals (23), a condition associated with hypoxia (24). However, this may be rationalized when considering that 1) much of the cellular injury during tissue hypoxia occurs during reoxygenation (i.e. reperfusion leads to oxygen-derived free radicals; (26, 27)) and 2) reversible oxidation can be a form of protein regulation (28). Therefore, although increased levels of PYK might be elicited in the absence of oxygen, rapid inhibition of the enzymatic activity of PYK may be necessary upon reoxygenation. This acute response might be possible through reversible protein oxidation. Not only would inhibition of PYK activity by oxidation be beneficial to prevent further pyruvate formation and overloading mitochondrial functions upon reoxygenation, but inhibition of lower glycolysis may force carbon flux through the pentose phosphate pathway to facilitate NADPH production. NADPH is used in the reduction of the glutathione redox control system that is, in turn, used to combat oxidative stress (such as that caused by overloaded mitochondria upon reoxygenation after hypoxyia) in the cell. This hypothesis has gained considerable support even during the time this report was being prepared (16, 17, 29).
Despite this potential scenario, we have provided no evidence that oxidation of Cys436 occurs in vivo. Therefore, at most we can review several observations that are consistent with regulatory oxidation. 1) The change in Kapp-PEP caused by oxidation is also the outcome of other forms of PYK regulation, including phosphorylation and allosteric effectors (1, 5). 2) There is a general growing consensus that reversible protein oxidation can be a physiologically important form of regulation (28, 30). Cysteine oxidation is reversible until a second oxygen is added to the sulfur group and reversibility is one of the criteria that supports a regulatory role of an oxidation event (28). Cys426 in the current structure is modified with a single oxygen, despite the weeks required for crystal growth and X-ray exposure. This brings up the speculation that some unique aspect of the protein prevents further oxidation, a property that could facilitate the reversibility of a physiologically important regulation. 3) Cys436 is the only oxidized cysteine in the structure, a specificity that is necessary to be consistent with regulation. 4) Although it is currently unclear how regulation is conserved among isozyme in a protein family (31, 32), Cys436 is conserved within mammalian liver PYK isozymes, as well as in other mammalian isozymes (Supportive Information). If regulation is conserved within a protein family, then oxidation of the equivalent of Cys436 in other isozymes may also contribute a regulatory response to oxidative stress to tissues other than the liver. Identification of oxidation of Cys436 using in vivo techniques will be required before further consideration of a physiological role of this oxidation will be warranted.
Implications for future studies
Although we have not yet elucidated the complete list of N-terminal/main body interactions, we have successfully identified a partial list. Furthermore, based on extensive data reported in this study, two strategies can be suggested for obtaining the remaining uncharacterized N-terminal/main body interactions. The additive effects of simultaneously introducing C436M with S12A and Q18A may aid in causing the N-terminus to be further ordered in structural studies. Alternatively, a computational approach may be used to search the surface of the protein for potential N-terminal (i.e. residues 7–10) binding sites, while considering the interactions identified in our C436M structure.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy Office of Basic Energy Sciences. The Stanford Synchrotron Radiation Laboratory Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We would like to thank Dr. Aileen Alontaga for her efforts to recover oxidized protein by addition of DTT and The Mass Spectrometry/Proteomics Core at The University of Kansas Medical Center (directed by Dr. Anotonio Artigues) for mass detection of oxidized cysteine.
Footnotes
This work was supported by NIH grant DK78076.
Supporting information includes 1) Other notable observations regarding the allosteric ATP binding site, 2) a helixal wheel representation for residues 11–24, 3) Other notable observations regarding citrate binding in the active site, 4) Other notable observations regarding the Fru-1,6-BP binding site, and 5) a table with all data used to generate Figure 5. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Fenton AW, Hutchinson M. The pH dependence of the allosteric response of human liver pyruvate kinase to fructose-1,6-bisphosphate, ATP, and alanine. Arch Biochem Biophys. 2009;484:16–23. doi: 10.1016/j.abb.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alontaga AY, Fenton AW. Effector analogues detect varied allosteric roles for conserved protein-effector interactions in pyruvate kinase isozymes. Biochemistry. 2011;50:1934–1939. doi: 10.1021/bi200052e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fenton AW. Identification of allosteric-activating drug leads for human liver pyruvate kinase. Methods Mol Biol. 2012;796:369–382. doi: 10.1007/978-1-61779-334-9_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fenton AW, Alontaga AY. The impact of ions on allosteric functions in human liver pyruvate kinase. Methods Enzymol. 2009;466:83–107. doi: 10.1016/S0076-6879(09)66005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fenton AW, Tang Q. An activating interaction between the unphosphorylated n-terminus of human liver pyruvate kinase and the main body of the protein is interrupted by phosphorylation. Biochemistry. 2009;48:3816–3818. doi: 10.1021/bi900421f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prasannan CB, Tang Q, Fenton AW. Allosteric Regulation of Human Liver Pyruvate Kinase by Peptides that Mimic the Phosphorylated/Dephosphorylated N-Terminus. Methods Mol Biol. 2012;796:335–349. doi: 10.1007/978-1-61779-334-9_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faustova I, Loog M, Jarv J. Probing L: -pyruvate kinase regulatory phosphorylation site by mutagenesis. The protein journal. 2012;31:592–597. doi: 10.1007/s10930-012-9438-1. [DOI] [PubMed] [Google Scholar]
- 8.Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, Wang C, Bianchi P, Zanella A, Mattevi A. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002;277:23807–23814. doi: 10.1074/jbc.M202107200. [DOI] [PubMed] [Google Scholar]
- 9.Bergstrom G, Ekman P, Humble E, Engstrom L. Proteolytic modification of pig and rat liver pyruvate kinase type L including phosphorylatable site. Biochim Biophys Acta. 1978;532:259–267. doi: 10.1016/0005-2795(78)90580-9. [DOI] [PubMed] [Google Scholar]
- 10.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 11.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 12.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- 14.Deng J, Davies DR, Wisedchaisri G, Wu M, Hol WG, Mehlin C. An improved protocol for rapid freezing of protein samples for long-term storage. Acta Crystallogr D Biol Crystallogr. 2004;60:203–204. doi: 10.1107/s0907444903024491. [DOI] [PubMed] [Google Scholar]
- 15.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 16.Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Antioxidant Responses. Science. 2011 doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krones A, Jungermann K, Kietzmann T. Cross-talk between the signals hypoxia and glucose at the glucose response element of the L-type pyruvate kinase gene. Endocrinology. 2001;142:2707–2718. doi: 10.1210/endo.142.6.8200. [DOI] [PubMed] [Google Scholar]
- 19.Zierz S, Katz N, Jungermann K. Distribution of pyruvate kinase type L and M2 in microdissected periportal and perivenous rat liver tissue with different dietary states. Hoppe Seylers Z Physiol Chem. 1983;364:1447–1453. doi: 10.1515/bchm2.1983.364.2.1447. [DOI] [PubMed] [Google Scholar]
- 20.Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA. Hypoxia regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J Biol Chem. 1998;273:26087–26093. doi: 10.1074/jbc.273.40.26087. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu T, Uehara T, Nomura Y. Possible involvement of pyruvate kinase in acquisition of tolerance to hypoxic stress in glial cells. J Neurochem. 2004;91:167–175. doi: 10.1111/j.1471-4159.2004.02702.x. [DOI] [PubMed] [Google Scholar]
- 22.Aisaki K, Aizawa S, Fujii H, Kanno J, Kanno H. Glycolytic inhibition by mutation of pyruvate kinase gene increases oxidative stress and causes apoptosis of a pyruvate kinase deficient cell line. Exp Hematol. 2007;35:1190–1200. doi: 10.1016/j.exphem.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Narli N, Satar M, Kayrin L, Yapicioglu H, Ozlu F, Budgayci R. The activity and kinetics of pyruvate kinase in hypoxic newborns. Pediatr Hematol Oncol. 2005;22:567–573. doi: 10.1080/08880010500198616. [DOI] [PubMed] [Google Scholar]
- 24.Ogasawara Y, Funakoshi M, Ishii K. Pyruvate kinase is protected by glutathione-dependent redox balance in human red blood cells exposed to reactive oxygen species. Biol Pharm Bull. 2008;31:1875–1881. doi: 10.1248/bpb.31.1875. [DOI] [PubMed] [Google Scholar]
- 25.Alexiev U, Mollaaghababa R, Khorana HG, Heyn MP. Evidence for long range allosteric interactions between the extracellular and cytoplasmic parts of bacteriorhodopsin from the mutant R82A and its second site revertant R82A/G231C. J Biol Chem. 2000;275:13431–13440. doi: 10.1074/jbc.275.18.13431. [DOI] [PubMed] [Google Scholar]
- 26.Bulkley GB. Reactive oxygen metabolites and reperfusion injury: aberrant triggering of reticuloendothelial function. Lancet. 1994;344:934–936. doi: 10.1016/s0140-6736(94)92276-4. [DOI] [PubMed] [Google Scholar]
- 27.Grace PA. Ischaemia-reperfusion injury. The British journal of surgery. 1994;81:637–647. doi: 10.1002/bjs.1800810504. [DOI] [PubMed] [Google Scholar]
- 28.Jacob C, Holme AL, Fry FH. The sulfinic acid switch in proteins. Organic & biomolecular chemistry. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
- 29.Gruning NM, Rinnerthaler M, Bluemlein K, Mulleder M, Wamelink MM, Lehrach H, Jakobs C, Breitenbach M, Ralser M. Pyruvate kinase triggers a metabolic feedback loop that controls redox metabolism in respiring cells. Cell metabolism. 2011;14:415–427. doi: 10.1016/j.cmet.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiley PJ, Storz G. Exploiting thiol modifications. PLoS biology. 2004;2:e400. doi: 10.1371/journal.pbio.0020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pendergrass DC, Williams R, Blair JB, Fenton AW. Mining for allosteric information: Natural mutations and positional sequence conservation in pyruvate kinase. IUBMB Life. 2006;58:31–38. doi: 10.1080/15216540500531705. [DOI] [PubMed] [Google Scholar]
- 32.Livesay DR, Kreth KE, Fodor AA. A critical evaluation of correlated mutation algorithms and coevolution within allosteric mechanisms. Methods Mol Biol. 2012;796:385–398. doi: 10.1007/978-1-61779-334-9_21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.