

Abstract
Traumatic brain injury (TBI) causes chronic microglial activation that contributes to subsequent neurodegeneration, with clinical outcomes declining as a function of aging. Microglia/macrophages (MG/MΦ) have multiple phenotypes, including a classically activated, pro-inflammatory (M1) state that may contribute to neurotoxicity, and an alternatively activated (M2) state that may promote repair. In this study we used gene expression, immunohistochemical and stereological analyses to show that TBI in aged versus young mice caused larger lesions associated with an M1/M2 balance switch and increased numbers of reactive (bushy and hypertrophic) MG/MΦ in the cortex, hippocampus and thalamus. Ym1, an M2 phenotype marker, displayed heterogeneous expression after TBI with amoeboid-like Ym1-positive MG/MΦ at the contusion site and ramified Ym1-positive MG/MΦ at distant sites; this distribution was age related. Aged injured mice also showed increased MG/MΦ expression of MHC II and NADPH oxidase, and reduced antioxidant enzyme expression- which was associated with lesion size and neurodegeneration. Thus, altered relative M1/M2 activation and an NADPH oxidase-mediated shift in redox state may contribute to worse outcomes observed in older TBI animals by creating a more pro-inflammatory M1 MG/MΦ activation state.
Keywords: microglia/macrophage, alternative activation, traumatic brain injury, aging, NADPH oxidase, neurodegeneration, Ym1
1. Introduction
Elderly individuals are particularly vulnerable to traumatic brain injury (TBI) (Faul et al., 2010); starting at age 65 years, TBI incidence doubles each additional 10 years of age (Coronado et al., 2005). Elderly patients have clinically worse outcomes after TBI, including increased morbidity and mortality (Galbraith, 1987; Pennings et al., 1993; Stocchetti et al., 2012), and reduced functional recovery (Livingston et al., 2005; Rapoport et al., 2006; Testa et al., 2005). Despite these clinical observations, research is limited with regard to the underlying mechanisms involved.
Secondary injury mechanisms after TBI have been well studied in experimental models. However, whereas age-related changes in secondary injury mechanisms have been suggested to contribute to poorer neurological outcomes in aged animals (Hamm et al., 1991; Hamm et al., 1992; Onyszchuk et al., 2008), the effects of aging on secondary injury cascades and how it contributes to worsened neurologic function after injury are not well understood. Mitochondrial dysfunction is increased in the aged TBI brain, with enhanced oxidative damage of synaptic mitochondria contributing to significant impairments in bioenergetic function (Gilmer et al., 2010). Following TBI, aged animals show increased evidence of oxidative stress (e.g. 4-hydroxynonenal, acrolein) and reduced antioxidant capacity (e.g. ascorbate) when compared to younger animals (Anderson et al., 2009; Moor et al., 2006; Shao et al., 2006). Post-traumatic neuroinflammation is also significantly impacted by aging, with more pronounced and prolonged glial cell activation in the hippocampus (Sandhir et al., 2008) that is associated with altered expression of C/EBP transcription factors (Sandhir and Berman, 2010).
Microglia (MG) are the primary innate immune cells in the central nervous system (CNS). MG, like peripheral macrophages (MΦ), have multiple activation phenotypes (Gordon, 2003; Mantovani et al., 2004), and depending on the stimuli in their local microenvironment they can be polarized to have distinct molecular phenotypes and effector functions (Colton, 2009). For example, lipopolysaccharide (LPS) or the pro-inflammatory cytokine interferon-γ (IFNγ) promote a ‘classical’ (M1) phenotype, which produces high levels of pro-inflammatory cytokines and oxidative metabolites that are essential for host defense and phagocytic activity, but that also can cause damage to healthy cells and tissue (Lynch, 2009). Conversely, activating MG in the presence of anti-inflammatory cytokines such as interleukin (IL)-4 (IL-4) or IL-10 promote ‘alternative’ (M2a) or ‘acquired deactivated’ (M2c) phenotypes respectively (Colton et al., 2006; Ponomarev et al., 2007), and both M2a and M2c phenotypes reduce M1 cytokines and other pro-inflammatory mediators (Gordon, 2003; Mantovani et al., 2004). It is thought that much like M2-polarized MΦ, M2 MG can promote repair processes such as angiogenesis and extracellular matrix (ECM) remodeling while also suppressing destructive immunity (Colton, 2009). In vitro studies have demonstrated that IL-4-polarized M2 MΦ promote extensive neurite elongation and outgrowth across inhibitory surfaces (Kigerl et al., 2009), indicating a role for M2 MG/MΦ in repair. MG possess an M2 phenotype in the intact CNS (Ponomarev et al., 2007); however, following an acute insult or injury to the brain it is likely that M1 and M2 MG/MΦ exist in a state of dynamic equilibrium within the lesion microenvironment. Whether these cells differentiate into an M1 phenotype that exacerbates tissue injury or into an M2 phenotype that promotes CNS repair likely depends on the local signals in the lesion microenvironment.
NADPH oxidase is implicated in the ongoing pathology of several age-associated neurodegenerative diseases such as Alzheimer’s disease (Bruce-Keller et al., 2010; Shimohama et al., 2000) and Parkinson’s disease (Wu et al., 2003), and it has been shown to play a major role in oxidative stress and neuroinflammation following ischemic brain injury (Chen et al., 2011; Chen et al., 2009), and TBI (Dohi et al., 2010; Zhang et al., 2012). NADPH oxidase is a multi-subunit enzyme complex responsible for the production of both extracellular and intracellular reactive oxygen species (ROS) by phagocytic cells including MG. NADPH oxidase has been implicated as a common and essential mechanism of MG-mediated neurotoxicity (Lull and Block, 2010), and NADPH oxidase plays a critical role in the modulation of MG/MΦ activation phenotype and subsequent inflammatory response (Choi et al., 2012). Recently we demonstrated that NADPH oxidase plays a key role in MG/MΦ activation after TBI, and that its membrane bound catalytic subunit gp91phox (also known as NOX2) is highly expressed in chronically activated MG/MΦ up to 4 months after TBI and is associated with chronic neurodegeneration (Byrnes et al., 2012).
In the present studies we hypothesize that exaggerated MG/MΦ activation in the aged brain after TBI is associated with an imbalance in the M1 and M2 MG/MΦ activation phenotypes and a reduced potential of MG/MΦ to participate in repair and resolution processes after injury. These changes may be associated with an age-related and NADPH oxidase-dependent shift in the redox state towards an oxidized microenvironment which may contribute to acquiring and retaining MG/MΦ in a pro-inflammatory M1 activation state, thereby enhancing neurodegeneration and tissue loss in the aged brain after TBI.
2. Materials and methods
2.1 Animals
Studies were performed using young adult (3 month old, 22–26g) and aged (24 ± 2 month old, 28–34g) male C57Bl/6 mice which were housed in the AAALAC-accredited Georgetown University Division of Comparative Medicine under a 12 hour light-dark cycle, with ad libitum access to food and water. All surgical procedures were carried out in accordance with protocols approved by Georgetown University Medical Center Institutional Animal Care and Use Committee (IACUC).
2.2 Controlled cortical impact injury
Our custom-designed Controlled Cortical Impact (CCI) injury device (Fox et al., 1998) consists of a microprocessor-controlled pneumatic impactor with a 3.5 mm diameter tip. Young (3 month old) and aged (24 month old) male C57Bl/6 mice were anesthetized with isoflurane evaporated in a gas mixture containing 70% N2O and 30% O2 and administered through a nose mask (induction at 4% and maintenance at 2%). Depth of anesthesia was assessed by monitoring respiration rate and pedal withdrawal reflexes. Mice were placed on a heated pad, and core body temperature was maintained at 37°C. The head was mounted in a stereotaxic frame, and the surgical site was clipped and cleaned with Nolvasan and ethanol scrubs. A 10-mm midline incision was made over the skull, the skin and fascia were reflected, and a 4-mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was then extended to its full stroke distance (44 mm), positioned to the surface of the exposed dura, and reset to impact the cortical surface. Moderate-level injury was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm as previously described (Loane et al., 2009b). After injury, the incision was closed with interrupted 6-0 silk sutures, anesthesia was terminated, and the animal was placed into a heated cage to maintain normal core temperature for 45 minutes post-injury. All animals were monitored carefully for at least 4 hours after surgery and then daily. Sham animals underwent the same procedure as injured mice except for the impact.
Study 1
3 month old sham-injured (n=9) and TBI (n=5) and 24 month old sham-injured (n=7) and TBI (n=4) mice were anesthetized (100 mg/kg sodium pentobarbital, I.P.) at 24 h post-injury and transcardially perfused with ice-cold 0.9% saline (100ml). Ipsilateral cortical tissue was rapidly dissected and snap-frozen on liquid nitrogen for RNA extraction.
Study 2
3 month old sham-injured (n=3) and TBI (n=5) and 24 month old sham-injured (n=3) and TBI (n=4) mice were anesthetized at 7 d post-injury and transcardially perfused with ice-cold 0.9% saline (100ml), followed by 4% paraformaldehyde (10% buffered formalin solution, 300 ml; Fisher Scientific). The sham and injured brains were removed and post-fixed in 4% paraformaldehyde overnight and cryoprotected in 30% sucrose for histological analysis.
2.3 Real-time PCR analysis
Total RNA was extracted from snap-frozen sham and TBI (24 h) cortical tissue using a miRNeasy isolation kit (Qiagen, Valencia, CA) with on-column DNase treatment (Qiagen), and cDNA synthesis was performed on 1 μg of total RNA using a Verso cDNA RT kit (Thermo Scientific, Pittsburg, PA); the protocols used were according to the manufacturer’s instructions. Real-time PCR was performed using TaqMan gene expression assays (Applied Biosystems, Carlsbad, CA) on an ABI 7900 HT FAST Real Time PCR (Applied Biosystems). Gene expression was calculated relative to the endogenous control samples (GAPDH) to give a relative quantity value (2−ΔΔCt, where Ct is the threshold cycle).
2.4 Immunohistochemistry
Immunohistochemistry was performed on 20 or 60μm μm coronal sections and standard immunostaining techniques were employed. Sections were incubated with anti-Iba-1 (1:1000, #019-19741; Wako Chemicals, Richmond, VA), anti-Ym1 (1:600, #01404; Stem Cell Technologies, Vancouver, Canada) or anti-MHC II (1:800, #M0775, Clone CR3/43, Dako, Carpinteria, CA) overnight, washed in 1 x PBS (3 times) and incubated with biotinylated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) for 2 h at room temperature. Sections were incubated in avidin–biotin–horseradish peroxidase solution (Vectastain elite ABC kit, Vector Laboratories) for 1 h and then reacted with 3,3′-diaminobenzidine (Vector Laboratories) for color development and mounted for immunohistochemical analysis using a Leica DM4000B microscope.
2.5 Unbiased stereologicial assessment of microglia/macrophage (MG/MΦ) activation
Classification of MG/MΦ activation phenotypes was performed on sham and 7 d TBI tissue as previously described (Byrnes et al., 2012). Stereoinvestigator software (MBF Biosciences, Williston, VT) was used to count the number of MG/MΦ in each of the three MG/MΦ morphological phenotypes (ramified, hypertrophic and bushy) in the injured cortex, hippocampus and thalamus using the optical fractionator method of unbiased stereology. The sampled region was the ipsilateral cortex, hippocampus and thalamus between −1.22mm and −2.54mm from bregma. Every fourth 60μm section was analyzed beginning from a random start point. The optical dissector had a size of 50μm by 50μm in the x and y-axis with a height of 10μm and guard zone of 4μm from the top of the section. For cortical and hippocampal analysis a grid spacing of 150μm in the x and y-axis was used, resulting in an area fraction of one-ninth. For thalamic analysis a grid spacing of 250μm in x-axis and 150μm in the y-axis was used, resulting in an area fraction of one-fifteenth. MG/MΦ phenotypic classification was based on the length and thickness of the projections, the number of branches and the size of the cell body as previously described (Byrnes et al., 2012; Soltys et al., 2001). Briefly, ramified MG/MΦ possessed long thin processes (>650μm in length), had a small cell body volume (<10μm3) and many branches (20–30). Hypertrophic MG/MΦ possessed medium length processes (300–550μm in length), had larger cell body volumes (50–75μm3) and many branches (20–30). Bushy MG/MΦ possessed short thick processes (<200μm in length), had a larger cell body volume (80–100μm3) and very few branches (<10). The volume of the region of interest was measured using a Cavalieri estimator method with a grid spacing of 100μm. The estimated number of MG/MΦ in each phenotypic class was divided by the volume of the region of interest to obtain the cellular density expressed in cells/mm3.
2.6 Immunofluorescence analysis
Triple immunofluorescence staining was performed on 20 μm coronal sections and standard immunostaining techniques were employed. Sections were washed with 1 x PBS (3 times), blocked for 1 h in goat serum containing 0.4% Triton X-100, and incubated overnight at 4°C with the appropriate primary antibodies. Primary antibodies were as follows: mouse anti-CD11b [OX-42] (1:100, #MCA275GA; AbD Serotec, Raleigh, NC), rabbit anti-Ym1 (1:50, #01404; Stem Cell Technologies), rat anti-ED1 [CD68] (1:200, AbD Serotec), mouse anti-GFAP (1:200, #MAB360; Millipore, Billerica, MA), rabbit anti-Iba-1 (1:200; Wako Chemicals), mouse anti-gp91phox (1:200, #611415, BD Transduction, Franklin Lakes, NJ), mouse anti-MHC II (1:200, #M0775, Clone CR3/43, Dako). Sections were washed with 1x PBS (3 times) and incubated with appropriate Alexa Fluor conjugated secondary antibodies (Life Technologies, Grand Island, NY). Sections were washed with 1x PBS (3 times), and counterstaining was performed with 4′, 6-diamidino-2-phenylindole (DAPI) (1 μg/ml; Sigma), followed by mounting with glass coverslips with Hydomount (National diagnostics, Atlanta, GA). Fluorescence microscopy was performed using a LEICA (TCS SP5 II) confocal microscope system (Leica Microsystems, Exton, PA).
2.7 Neuronal survival and lesion volume assessment
Stereoinvestigator software (MBF Biosciences) was used to count the total number of surviving neurons in the Cornu Ammonis (CA) 1, CA3 and dentate gyrus (DG) sub-regions of the hippocampus and the thalamus using the optical fractionator method of unbiased stereology as previously described (Byrnes et al., 2012). Lesion volume was quantified based on the Cavalieri method of unbiased stereology using Stereologer 2000 program software (Systems Planning and Analysis, Alexandria, VA) as previously described (Kabadi et al., 2012).
2.8 Statistical Analysis
Lesion volume and unbiased stereological analysis were performed by an investigator blinded to groups. Quantitative data were expressed as mean ± standard errors of the mean (s.e.m.). Gene expression and MG/MΦ phenotype classification were analyzed by analyzed by two-way ANOVA, followed by post-hoc adjustments using Student-Newman-Keuls test. Remaining data were analyzed using Student’s t test or one-way ANOVA followed by Student-Newman-Keuls multiple comparison test, where appropriate. Statistical tests were performed using either SigmaPlot 12 (Systat Software, San Jose, CA) or GraphPad Prism Program, Version 3.02 for Windows (GraphPad Software, San Diego, CA). A p<0.05 was considered statistically significant.
3. Results
3.1 MG/MΦ M1 (classical) and M2 (alternative) activation markers differ in the young and aged TBI brain
Exacerbated glial cell activation has have been reported in the aged TBI brain and it is associated with poorer neurological recovery and increased neurodegeneration (Hamm et al., 1991; Hamm et al., 1992; Onyszchuk et al., 2008; Sandhir et al., 2008). We hypothesize that exaggerated MG/MΦ activation in the aged brain following TBI is related to an imbalance in the M1 and M2 MG/MΦ activation phenotypes and reduced potential of MG/MΦ to participate in repair and resolution processes after injury. To test this hypothesis, we anesthetized young adult (3 month old) or aged (24 month old) C57Bl/6 mice and subjected them to moderate-level controlled cortical impact TBI, and collected cortical tissue after 24 h to determine the expression of MG/MΦ genes associated with classical (M1), alternative activation (M2a), and acquired deactivation (M2c), based on identifying antigenic markers for each MG/MΦ state (Colton, 2009). Figure 1A shows the relative MG/MΦ mRNA expression of M1 genes: interleukin-1β (IL-1β), tumor necrosis factor-α (TNFα), CD86, inducible nitric oxide synthase (iNOS), chemokine (C-C motif) ligand 2 (CCL2) and chemokine (C-C motif) ligand 3 (CCL3). TBI significantly increased the mRNA levels of IL-1β, TNFα, iNOS, CCL2, and CCL3 in young mice when compared to age-matched sham-injured controls (Figure 1A; p<0.05 [iNOS], p<0.001 [all other M1 genes]). Similarly, TBI in the aged mice resulted in increased mRNA levels for each of the M1 genes when compared to age-matched sham-injured controls (p<0.001). Moreover, there were significant increases in M1 gene expression levels in the aged TBI mice when compared to the young TBI mice (p<0.05 [CCL3], p<0.01 [IL-1β, iNOS, CD86], p<0.001 [TNFα, CCL2]).
Figure 1. Aging modulates M1 and M2 MG/MΦ activation genes following TBI.

Quantitative real-time PCR was used to assess the expression levels of classical (M1; A), alternative (M2a; B), acquired deactivation (M2c; C) MG/MΦ activation genes and IL-4 signal transduction pathway (D) genes in cortical tissue from young (3 month old) and aged (24 month old) mice at 24 h post-injury. (A) M1 gene expression levels (IL-1β, TNFα, CD86, iNOS, CCL2 and CCL3) in the aged TBI mice were significantly increased when compared to the young TBI mice (p<0.05 [CCL3], p<0.01 [IL-1β, iNOS, CD86], p<0.001 [TNFα, CCL2]). (B) M2a gene expression levels (Arg-1, Ym1, Mrc) in the aged TBI mice were significantly increased when compared to young TBI mice (p<0.05 [Mrc], p<0.001 [Arg-1, Ym1]). The M2a gene, Fizz-1, was significantly decreased in aged sham mice when compared to young sham mice (p<0.001), and TBI resulted in significantly reduced Fizz-1 expression in young mice (p<0.01). (C) M2c gene expression levels (IL-4Rα, SOCS3, TGFβ) were significantly increased in young TBI mice when compared to young sham mice (p<0.001), and expression levels were significantly lower in aged TBI mice when compared with young TBI mice (p<0.01 [IL-4Rα], p<0.001 [SOCS3, TGFβ]). (D) STAT 6 expression was significantly increased in the young TBI mice when compared to young sham mice (p<0.01), and STAT6 expression was significantly lower in aged TBI mice when compared to young TBI mice (p<0.05). Statistical analysis was by two-way ANOVA (injury x aging), followed by post-hoc adjustments using Student-Newman-Keuls Multiple Comparison test; *p<0.05, +p<0.05, ##p<0.01, ++p<0.01, ^^p<0.01, +++p<0.001, ###p<0.001 and ***p<0.001, where + = TBI 3 month vs. sham 3 month, * = TBI 24 month vs. sham 24 month, # = TBI 3 month vs. TBI 24 month, ^ = sham 3 month vs. sham 24 month. Bars represent mean ± s.e.m.
The MG/MΦ M2a gene expression changes in young and aged TBI mice were more complex with some M2a genes increasing and others decreasing with injury and age (Figure 1B). For example, arginase-1 (Arg-1) and chitinase3-like 3 (Ym1) expression levels were significantly increased in young TBI mice (p<0.01 and p<0.05 respectively versus young sham), with further significant increases detected in aged TBI mice (p<0.001 versus young TBI). Similarly, there was a significant increase in the expression levels of mannose receptor (Mrc) in the aged TBI mice when compared to the young TBI mice (p<0.05). In contrast, aging alone resulted in significantly lower expression levels of the M2a gene, found in inflammatory zone 1 (Fizz-1; p<0.001 versus young sham), and TBI in young and aged mice resulted in significantly lower Fizz-1 mRNA expression levels (p<0.01 versus young sham). The injury-induced changes in MG/MΦ M2c gene expression were also modulated by aging. The M2c genes IL-4 receptor-α (IL-4Rα), suppressor of cytokine signaling 3 (SOCS3) and transforming growth factor-β (TGFβ) were increased in the young TBI mice when compared to age-matched sham-injured controls (Figure 1C; p<0.001 versus young sham). Although M2c genes in the aged TBI brain were increased compared to age-matched sham-injured controls (p<0.05 for TGFβ only), the gene expression levels of IL-4Rα, SOCS3 and TGFβ were significantly lower in aged TBI mice when compared to young TBI mice (p<0.01 [IL-4Rα], p<0.001 [SOCS3, TGFβ]). In addition, we assessed STAT6 expression levels for signal transduction pathways downstream of IL-4Rα (Figure 1D). STAT 6 mRNA expression was significantly increased in the young TBI mice compared to age-matched sham-injured controls (p<0.01), however, STAT6 expression was impaired in aged TBI mice and expression levels were significantly lower when compared to young TBI mice (0<0.05). These results indicate that there is an imbalance of M1 and M2 MG/MΦ expression patterns in the aged brain following TBI, with significantly increased expression of M1 and a subset of M2a (Arg-1, Ym1 and Mrc) genes, and lower expression of M2c genes in the injured cortex of aged mice when compared to young mice. In addition, the expression levels of another M2a gene (Fizz-1) were significantly lowered by aging and injury.
3.2 Highly reactive MG/MΦ activation phenotypes predominate the aged TBI brain
Brain injury due to stroke or trauma results in a switch in MG/MΦ phenotype from a resting form displaying ramified cellular morphologies to more activated forms displaying hypertrophic or bushy morphologies (Byrnes et al., 2012; Soltys et al., 2001). Based on cell morphological features, we classified MG/MΦ into three cellular phenotypes corresponding to increasing activation status: ramified, hypertrophic, and bushy (Byrnes et al., 2012). As shown in Figure 2B ramified MG/MΦ have small cell bodies, and thin, long and highly branched processes. In contrast, hypertrophic MG/MΦ have larger cell bodies, with thicker, shorter and highly branched processes, whereas bushy MG/MΦ have multiple short processes that form thick bundles around an enlarged cell body (Figure 2B). We performed an unbiased stereological assessment of MG/MΦ cell number and activation phenotype in the injured cortex, hippocampus and thalamus of young (3 month old) and aged (24 month old) TBI mice by analyzing Iba-1 stained coronal sections at 7 d post-injury.
Figure 2. Aging increases the number of highly activated MG/MΦ (hypertrophic and bushy cellular phenotype) in the cortex, hippocampus and thalamus of TBI mice.

(A) Representative Iba-1 immunohistochemistry images of MG/MΦ activation in the cortex of young and aged TBI mice. Bar = 100μm. (B) Representative images of ramified, hypertrophic and bushy MG/MΦ activation phenotypes. There was a significant difference between MG/MΦ activation phenotypes in the cortex of young and aged TBI mice (F(2,18)=4.268, p=0.03), with significantly reduced number of ramified activation phenotypes (p<0.05), and trends to increased numbers of hypertrophic and bushy activation phenotypes in the aged TBI mice. (C) There were significant differences between MG/MΦ activation phenotypes in the hippocampus and thalamus of young and aged TBI mice (CA1 subregion (F(2.18)=7.489, p=004); CA3 subregion (F(2,18)=5.060, p=0.018); thalamus (F(2.18)=5.006, p=0.019)). There were increased numbers of hypertrophic and bushy MG/MΦ in the aged TBI mice in the CA1 and CA3 subregions (p<0.001 for hypertrophic, and p<0.05 for bushy), and significantly reduced numbers of ramified MG/MΦ (p<0.01) in the thalamus when compared to young TBI mice. Bars = 100μm (dentate gyrus) or 50μm (thalamus). Statistical analysis by two-way ANOVA (injury x activation phenotype), followed by post-hoc adjustments using Student-Newman-Keuls Multiple Comparison test. ***p<0.001, +p<0.05, ##p<0.01, where * = 3 month hypertrophic vs. 24 month hypertrophic, + = 3 month bushy vs. 24 month bushy, # = 3 month ramified vs. 24 month ramified. Bars represent mean ± s.e.m.
There were increased numbers of MG/MΦ displaying the highly activated (hypertrophic and bushy) phenotype and reduced ramified phenotypes in the cortex of aged TBI mice when compared to young TBI mice (Figure 2A). The interaction of ‘age x activation phenotype’ was statistically significant (F(2,18)=4.268, p=0.03) in the injured cortex, and there were significantly reduced numbers of the ramified MG/MΦ (p<0.05), and trends to increased numbers of highly activated phenotypes in the aged TBI mice (Figure 2B). Similarly, there were significantly increased highly activated MG/MΦ in the hippocampus of aged TBI mice when compared with young TBI mice (CA1 sub-region (F(2.18)=7.489, p=004); CA3 sub-region (F(2,18)=5.060, p=0.018), and there were significantly increased numbers of highly activated MG/MΦ in the aged TBI mice in the CA1 and CA3 sub-regions (Figure 2C; p<0.001 for hypertrophic, and p<0.05 for bushy). Trends to increased numbers of highly activated MG/MΦ in the dentate gyrus of aged TBI mice were observed but did not reach statistical significance. Furthermore, there were significantly increased numbers of highly activated MG/MΦ in the thalamus of aged TBI mice when compared with young TBI mice (F(2.18)=5.006, p=0.019), and there were significantly reduced numbers of ramified MG/MΦ (p<0.01), and trends to increased numbers of highly activated MG/MΦ in the aged TBI mice (Figure 2C). Taken together, our data suggest that aging modulates the MG/MΦ activation phenotype after TBI, with increases in the most highly activated MG/MΦ cellular phenotypes (hypertrophic and bushy) in the cortex, hippocampus and thalamus of moderate-level injured aged mice when compared with equal level injury in young mice.
3.3 MHC II is highly expressed on MG/MΦ in the aged TBI brain
Expression of major histocompatibility complex (MHC) II is a hallmark of antigen presenting cells, and its expression on activated MG/MΦ enables their interaction and signaling with other cells such as T cells (Sedgwick et al., 1998). We measured the expression levels of MHC II mRNA in the cortex from young (3 month old) and aged (24 month old) TBI mice at 24 h post-injury. Compared to age-matched sham-injured controls TBI resulted in a modest but non-significant increase in MHC II mRNA expression in young mice and a significant increase in MHC II mRNA in aged mice (Figure 3A, p<0.05). Notably, the expression levels of MHC II were significantly higher in the aged TBI group when compared to the young TBI group (p<0.05). We also evaluated MHC II expression in 7 d TBI tissue from young (3 month old) and aged (24 month old) TBI mice, and representative immunohistochemical images from the injured thalamus are shown in Figure 3B. MHC II immunoreactivity was detected in cells that displayed a ramified/hypertrophic-like MG/MΦ cellular morphology in ventrolateral thalamic subregions of the young TBI brain (Figure 3B, i and iii). There was more intense MHC II immunoreactivity detected in the ventrolateral thalamus of aged TBI brain, and MHC II staining was observed in cells that displayed hypertrophic/bushy-like MG/MΦ cellular morphologies (Figure 3B, ii and iv). Similar MHC II immunostaining patterns were observed in the cortex and hippocampus of young and aged TBI mice (data not shown). We then performed triple immunofluorescence staining for reactive MG/MΦ (Iba-1/ED1-positive) that expressed MHC II in 7 d TBI tissue from young (3 month old) and aged (24 month old) TBI mice (Figure 3C). There was minimal expression of MHC II in young TBI brain that co-localized with Iba-1/ED1-positive MG/MΦ in the injured thalamus. In contrast, MHC II was highly expressed in the aged TBI brain and co-localized with Iba-1/ED1-positive MG/MΦ that displayed a highly activated cell morphology.
Figure 3. MHC II is expressed on MG/MΦ in the aged TBI brain.

(A) Quantitative real-time PCR was used to assess the expression levels of MHC II mRNA in cortical tissue from young (3 month) and aged (24 month) mice at 24 h post-injury. MHC II mRNA was significantly increased in the cortex of aged TBI brain when compared to the young TBI brain (p<0.05). Statistical analysis was by two-way ANOVA (injury x aging), followed by post-hoc adjustments using Student-Newman-Keuls Multiple Comparison test; *p<0.05, #p<0.05, where * = TBI 24 month vs. sham 24 month, # = TBI 3 month vs. TBI 24 month. Bars represent mean ± s.e.m. (B) Immunohistochemistry for MHC II in the thalamus of young and aged TBI mice at 7 d post-injury. MHC II immunoreactivity was detected in cells that displayed a ramified/hypertrophic-like MG/MΦ cellular morphology in ventrolateral thalamic subregions of the young TBI brain (i and iii). In contrast, intense MHC II immunoreactivity was detected in cells that displayed hypertrophic/bushy-like MG/MΦ cellular morphologies in the ventrolateral thalamus of aged TBI brain (ii and iv). Bar = 200μm, inset ii bar = 50μm, inset iv bar = 10μm. (C) Triple immunofluorescence staining for Iba-1 (green), MHC II (red) and ED1 (magenta) in the thalamus of young and aged TBI mice at 7 d post injury. In contrast to the young TBI brain MHC II was highly expressed in Iba-1/ED1-positive MG/MΦ in the aged TBI brain. Bar = 20μm.
3.4 Heterogeneous expression of Ym1-positive M2 MG/MΦ after TBI
In order to assess the localization and expression of Ym1-positive M2 MG/MΦ following TBI we performed triple immunfluorescence staining for Ym1, CD11b (MG/MΦ marker) and ED1 (marker of phagocytic MG/MΦ) in 7 d TBI tissue (Figure 4A). Our results revealed that Ym1 was expressed in heterogeneous cell populations displaying distinct cellular morphologies that were spatial separated in the injured cortical tissue. For example, Ym1 was highly expressed in the perilesional regions of the injured cortex and these Ym1-expressing cells assumed the typical morphology of the highly activated and amoeboid MG/MΦ. In contrast, in subcortical regions distant from the lesion core Ym1 was expressed in cells displaying a ramified morphology with numerous highly branched processes. High magnification images of these Ym1-positive cell populations demonstrated that both cell populations were positive for the MG/MΦ marker, CD11b (Figure 3Ai, ii). XZ- and YZ-orthogonal projection analysis of individual cells demonstrated that the majority (>90%) of Ym1-positive amoeboid MG/MΦ were negative for ED1, but were localized adjacent to ED1-positive cells (Figure 3Ai, panel a and b). Conversely, ED1-positive amoeboid MG/MΦ were Ym1-negative (panel c). However, this analysis also revealed a small number of true double-positive cells which indicated that following TBI some amoeboid MG/MΦ co-expressed Ym1 and ED1 (panel d; yellow cell); but this cellular phenotype was in the minority. Taken together, these data suggest that Ym1-positive MG/MΦ may have altered functional capacity that is not involved in phagocytosis. In addition, the Ym1-positive ramified MG/MΦ were also ED1-negative, and there were no ED1-positive MG/MΦ located in the surrounding subcortical regions (Figure 3Aii), thereby suggesting that M2 MG/MΦ that displayed a ramified cellular phenotype may have alternative functional roles after TBI. Ym1 expression was not detected in astrocytes surrounding the lesion area as there was non-overlapping expression of Ym1-positive cells with GFAP-positive cells in perilesional areas (Figure 3B).
Figure 4. Heterogeneous expression of Ym1-positive M2 MG/MΦ after TBI.

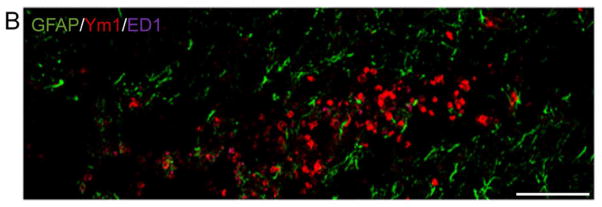
(A) Triple immunofluorescence staining for Ym1 (red), CD11b (green) and ED1 (magenta) in the cortex of young and aged TBI mice at 7 d post injury. Ym1-positive MG/MΦ displaying a highly activated (bushy) and amoeboid cellular phenotype (i) were localized in the perilesional cortex surrounding the lesion site. In addition, Ym1-positive MG/MΦ displaying a ramified cellular phenotype (ii) were observed in subcortical areas distant from the lesion site. Bar = 100μm. For the purpose of xz- and yz-orthogonal projection analysis the ED1 fluorescent signal was changed to pseudocolor green to facilitate differentiation between single positive, Ym1 (red) or ED1 (green) and double-positive, Ym1/ED1 cells (yellow). Xz- and yz-views demonstrate that individual Ym1-positive cells (red) were ED1-negative, although ED1-positive cells (green) were located in close proximity to the Ym1-positive cells (panels i, a and b), and ED1-positive cells (green) were Ym1-negative (panel i, c). Within the field of view a single true double-positive cell (yellow) was detected, which indicated that this amoeboid MG/MΦ expressed Ym1 and ED1 (panel i, d). Inset bar = 20μm. (B) Ym1 immunoreactivity (red) did not colocalize with GFAP (green), thereby demonstrating that Ym1-positive cells were not astrocytes. Bar = 100μm.
We evaluated Ym1 expression in 7 d TBI tissue from young (3 month old) and aged (24 month old) TBI mice, and representative immunohistochemical images from the injured cortex, corpus callosum and hippocampus are shown in Figure 5. In the aged TBI mice Ym1-positive amoeboid MG/MΦ surrounded the lesion site and were dispersed throughout the injured cortex (Figure 5Aii). In contrast, there were fewer Ym1-positive MG/MΦ in the cortex of young TBI mice and some of these cells displayed a more ramified/hypertrophic cellular morphology (Figure 5Ai). Further analysis revealed that there were numerous Ym1-positive ramified/hypertrophic MG/MΦ displayed throughout the corpus callosum of young TBI mice (Figure 5Biii, iv), whereas Ym1-positive MG/MΦ in the corpus callosum of aged TBI mice displayed heterogeneous cellular morphologies as both Ym1-positive bushy (amoeboid; Figure 5Bv) and ramified (Figure 5Bvi) cellular morphologies were observed in the aged TBI mice. Notably, the highly branched Ym1-positive ramified MG/MΦ were dispersed throughout the hippocampus of young TBI mice (Figure 5Cvii), and fewer of these cells were detected in the hippocampus of aged TBI mice. These results indicate that Ym1 is highly expressed in less activated ramified MG/MΦ in the young TBI brain whereas Ym1 is expressed in both the highly activated bushy (amoeboid) and ramified MG/MΦ in the aged TBI brain, with increased amoeboid cells surrounding the lesion site and fewer ramified cells at more distant sites from the injury (e.g. hippocampus).
Figure 5. Ym1-positive M2 MG/MΦ phenotypic heterogeneity in the young and aged TBI mice.

Immunohistochemistry for Ym1-positive MG/MΦ in the cortex, corpus callosum and hippocampus of young and aged TBI mice. (A) Ym1-positive amoeboid MG/MΦ surrounded the lesion site and were dispersed throughout the cortex of aged TBI mice (ii), whereas there were fewer Ym1-positive MG/MΦ in the cortex of young TBI mice and some cells displayed a ramified/hypertrophic cellular morphology (i). Bar = 100μm. (B) In the corpus callosum there were numerous Ym1-positive ramified/hypertrophic MG/MΦ in young TBI mice (iii, iv), whereas Ym1-positive MG/MΦ in the aged TBI mice displayed heterogeneous cellular morphologies as both Ym1-positive amoeboid (v) and ramified/hypertrophic (vi) cellular morphologies were observed. Bar = 50μm. (C) In the hippocampus of young TBI mice there were numerous highly branched Ym1-positive ramified/hypertrophic MG/MΦ (vii). In contrast, there were fewer of these cells observed in the hippocampus of aged TBI mice. Bar = 50μm.
3.4 NADPH oxidase is up-regulated in reactive MG/MΦ in the aged TBI brain and is associated with reduced antioxidant gene expression
We measured the expression levels of superoxide dismutase 1 (SOD1) and glutathione peroxidase 1 (GPX1), two key enzymes involved in antioxidant defense mechanisms, in the cortex from young (3 month old) and aged (24 month old) TBI mice at 24 h post-injury. The SOD1 mRNA expression levels were significantly lower in aged sham-injured controls when compared with young sham-injured controls (Figure 6A; p<0.01), and TBI in young mice resulted in a non-significant increase in SOD1 expression when compared to the young sham-injured controls. TBI in aged mice did not induce SOD1 expression and its expression levels were significantly lower when compared with the young TBI group (p<0.01). TBI in young mice resulted in a significant increase GPX1 expression when compared to the young sham-injured controls (Figure 6B; p<0.05), while TBI in aged mice did not induce GPX1 expression. However, there were significantly lower GPX1 expression levels in aged TBI mice when compared with the young TBI group (p<0.05).
Figure 6. NADPH oxidase is increased in MG/MΦ in the aged TBI mice and is associated with reduced antioxidant enzyme expression.

Quantitative real-time PCR was used to assess the expression levels of SOD1 (A), GPX1 (B) p22phox (C) and gp91phox (D) in cortical tissue from young (3 month) and aged (24 month) mice at 24 h post-injury. SOD1 (A) expression levels were significantly reduced in aged sham mice when compared to young sham mice (p<0.01), and TBI in aged mice resulted in significantly reduced SOD1 levels when compared to young TBI mice (p<0.01). GPX1 (B) expression levels were significantly increased in the young TBI mice when compared to young sham mice (p<0.05), and TBI in aged mice resulted in significantly reduced GPX1 levels when compared to young TBI mice (p<0.05). p22phox (C) and gp91phox (D) expression levels were significantly increased in aged sham mice when compared to young sham mice (p<0.01), and TBI increased the expression of both subunits (p<0.001). The levels of p22phox and gp91phox were significantly higher in the aged TBI mice when compared to young TBI mice (p<0.001). Statistical analysis was by two-way ANOVA (injury x aging), followed by post-hoc adjustments using Student-Newman-Keuls Multiple Comparison test; +p<0.5, +++p<0.001, #p<0.05, ##p<0.01, ###p<0.001, ^^p<0.01, and ***p<0.001, where + = TBI 3 month vs. sham 3 month, * = TBI 24 month vs. sham 24 month, # = TBI 3 month vs. TBI 24 month, ^ = sham 3 month vs. sham 24 month. Bars represent mean ± s.e.m. (E) Triple immunofluorescence staining for gp91phox (red), Iba-1 (green) and ED1 (magenta) in the cortex of young and aged TBI mice at 7 d post injury. gp91phox expression was strongly up-regulated in ED1- and Iba-1-positive MG/MΦ that displayed a hypertrophic/bushy cellular morphology in the aged TBI mice when compared to young TBI mice. Bar = 200μm, inset bar = 25μm.
We recently demonstrated that NADPH oxidase was chronically expressed in reactive MG/MΦ up to 4 months after TBI (Byrnes et al., 2012), and therefore we sought to assess whether aging modulated NADPH oxidase expression following TBI. We measured the expression levels of p22phox and gp91phox, membrane bound subunits of the NADPH oxidase enzyme complex, in the injured cortex of young (3 month old) and aged (24 month old) TBI mice at 24 h post-injury. Aging alone resulted in a significant increase in p22phox and gp91phox mRNA (Figure 6C, D p<0.01), and there was a significant increase in p22phox and gp91phox levels in young and aged TBI mice when compared to age-matched sham-injured controls (p<0.001). Notably, the levels of p22phox and gp91phox were significantly higher in the aged TBI mice when compared to young TBI mice (p<0.001). We then performed triple immunofluorescence staining for reactive MG/MΦ (Iba-1/ED1-positive) that expressed NADPH oxidase (gp91phox-positive) in 7 d TBI tissue from young (3 month old) and aged (24 month old) TBI mice (Figure 6E). There was modest expression of gp91phox in young TBI mice that co-localized with Iba-1/ED1-positive MG/MΦ in the injured cortex. In contrast, gp91phox was highly expressed in the aged TBI mice and co-localized with Iba-1/ED1-positive MG/MΦ that displayed a reactive phenotype (bushy/amoeboid cell morphology; Figure 6E inset).
3.5 Aging results in increased lesion size and neurodegeneration in the hippocampus and thalamus after TBI
Finally, we used unbiased stereological techniques to quantify TBI-induced lesion size and neuronal loss in the hippocampus and thalamus of young (3 month old) and aged (24 month old) mice at 7 d post-injury. There was a significant increase in lesion volume in the aged TBI mice when compared to young TBI mice (Figure 7A; p<0.001). Furthermore, TBI resulted in significant neuronal loss in the CA1 (Figure 7B), CA3 (Figure 7C) and dentate gyrus (Figure 7D) subregions of the hippocampus in both young and aged mice (for each subregion: p<0.05 young TBI, p<0.01 aged TBI compared to contralateral cell counts). Notably, for each hippocampal subregion there was significantly increased neuronal loss in the aged TBI mice when compared to the young TBI mice (Figure 7B-D; p<0.05). Similarly, there was increased neuronal loss in the thalamus in the aged TBI mice when compared to the young TBI mice (Figure 7E; p<0.05). These data demonstrate that there was increased neurodegeneration in the hippocampus and thalamus of aged TBI mice when compared to young TBI mice and this was associated with increased tissue loss.
Figure 7. Aging results in larger lesion size and increased neurodegeneration in the hippocampus and thalamus after TBI.

(A) Stereological assessment of TBI-induced lesion size at 7 d post-injury in young and aged TBI mice. TBI resulted in a significantly larger lesion volume in aged TBI mice when compared with young TBI mice (p<0.001). Student’s t-test. (B, C and D) Unbiased stereological assessment of surviving neurons in the hippocampus at 7 d post-injury in young and aged TBI mice. TBI resulted in significant neuronal loss in the CA1 (B), CA3 (C) and dentate gyrus (D) sub-regions of the hippocampus in both young and aged mice (for each sub-region: p<0.05 young TBI, p<0.01 aged TBI compared to contralateral cell counts). In each hippocampal sub-region there was significantly increased neuronal loss in the aged TBI mice when compared to the young TBI mice (B–D; p<0.05). Statistical analysis was by one-way ANOVA followed by Student-Newman-Keuls Multiple Comparison Test, *p<0.05, ++p<0.01 and #p<0.05, where * = young ipsilateral vs. young contralateral; + = aged ipsilateral vs. aged contralateral and # = young ipsilateral vs. aged ipsilateral. (E) There was increased neuronal loss in the thalamus in the aged TBI mice when compared to the young TBI mice (p<0.05). Student’s t-test.
4. Discussion
Neuroinflammation, oxidative stress, and MG/MΦ activation are important secondary injury mechanisms that lead to progressive neuronal damage and tissue loss after TBI (Byrnes et al., 2012; Hall et al., 2010; Kumar and Loane, 2012). MG/MΦ in the aged brain are primed and have a ‘reactive’ phenotype (Miller and Streit, 2007), which is characterized by increased expression of M1 and pro-inflammatory markers such as ED1 (Wong et al., 2005), CD86 (Downer et al., 2010), and MHC II (Godbout et al., 2005; Loane et al., 2009a). In this study our stereological analysis demonstrated that the aged brain had an exaggerated MG/MΦ activation response to TBI, and that there were increased numbers of highly activated MG/MΦ displaying a bushy and hypertrophic cellular morphology and reduced numbers of resting MG/MΦ displaying the ramified cellular morphology in the cortex, hippocampus and thalamus of aged TBI mice when compared to young TBI mice. In addition, the expression levels of M1 (classical) MG/MΦ activation genes (IL-1β, TNFα, CD86, iNOS, CCL2 and CCL3) were significantly higher in aged TBI mice when compared to young TBI mice. These findings are consistent with previous reports of exaggerated MG/MΦ activation responses in the aged injured brain in experimental models of acute brain and peripheral nerve injury (Badan et al., 2003; Conde and Streit, 2006; Kyrkanides et al., 2001; Popa-Wagner et al., 2007; Sandhir et al., 2008), and suggest that MG/MΦ in the aged brain are primed to produce a more robust pro-inflammatory response after TBI. Pro-inflammatory mediators such as IL-1β, TNFα, and iNOS produced by M1-polarized MG/MΦ can react with superoxide free radicals produced within the highly oxidized milieu of the lesion microenvironment to generate ROS and other neurotoxic substances that may contribute to neuronal dysfunction and cell death (Block and Hong, 2005; Kumar and Loane, 2012). Notably, highly activated bushy and hypertrophic MG/MΦ were localized at sites of increased neurodegeneration in the hippocampus and thalamus of aged TBI mice and this was associated with significantly increased lesion sizes in these animals. These results support the hypothesis that the aged brain is more vulnerable to the damaging effects of TBI and that highly activated MG/MΦ that have an M1 pro-inflammatory phenotype contribute to exacerbated neurodegeneration in the aged TBI brain.
Recently we have demonstrated that NADPH oxidase plays a key role in MG/MΦ activation after TBI (Byrnes et al., 2012), and here we show that it is robustly expressed in highly activated MG/MΦ in the aged TBI brain at sites of increased oxidative damage and tissue loss. TBI resulted in exaggerated up-regulation of the NADPH oxidase subunits, p22phox and gp91phox, in the injured cortex of aged mice, and gp91phox was highly expressed in ED1-positive MG/MΦ that had hypertrophic and bushy cellular morphologies. There was some modest gp91phox+/Iba1-/ED1-cell staining observed in the injured cortex of aged TBI mice, and gp91phox has also been shown to be expressed at lower levels in neurons and astrocytes within 48 h of TBI (Dohi et al., 2010), which might explain low level non-MG/MΦ cell staining. However, the majority of gp91phox immunostaining co-localized with highly activated MG/MΦ at 7 d post-injury. The expression of antioxidant enzymes SOD1 and GPX1 were down-regulated with age, and in response to TBI aged animals were unable to up-regulate the expression of both antioxidant enzymes, thereby confirming impaired antioxidant capacity in aged TBI animals which was demonstrated in prior experimental studies (Ansari et al., 2008a; Ansari et al., 2008b). These data indicate an inverse correlation between regional O2− generation (by NADPH oxidase) and its scavenging within injured cortex of aged TBI animals. This suggests that increasing age disrupts the delicate balance between ROS generation and antioxidant buffering in the brain, thereby leading to a shift in the microenvironment to a more oxidative cellular milieu that may promote and enhance the damaging effects of M1 MG/MΦ after TBI. In support of this concept, in vitro studies have shown that redox state (i.e. ROS generated by NADPH oxidase) modulates MG/MΦ activation and function (Mander et al., 2006; Pawate et al., 2004). MG/MΦ are a chronic driver of neuroinflammation and ROS production culpable of progressive neuronal damage (Block et al., 2007), and NADPH oxidase in MG/MΦ is essential for initiating self-propogating cycles of MG/MΦ-mediated neurotoxicity in experimental models of neurodegeneration (Gao et al., 2003; Lull and Block, 2010; Qin et al., 2002; Wu et al., 2003). Moreover, a recent in vivo study demonstrated that NADPH oxidase plays a critical role in promoting and maintaining the M1 MG/MΦ activation phenotype in response to inflammatory challenges such as LPS or amyloid-β1–42 (Aβ1–42) (Choi et al., 2012). In these studies Choi and colleagues demonstrated that in response to inflammatory challenges pharmacological inhibition of NADPH oxidase or genetic ablation of its functional p47phox subunit switched MG/MΦ from an M1-polarized state (reduced M1 markers: TNFα and CCL2) to an M2-polarized state (increased M2 markers: Ym1 and IL-4Rα). Additionally, inhibition of NADPH oxidase significantly protects younger animals after ischemic brain injury (Chen et al., 2011; Chen et al., 2009) or TBI (Dohi et al., 2010; Zhang et al., 2012), and genetic ablation (gp91phox−/−) or pharmacological inhibition (apocynin) of NADPH oxidase results in reduced oxidative stress and MG/MΦ activation after injury. However, given our findings, inhibition of NADPH oxidase may represent an even more effective therapeutic approach to reduce oxidative stress and the M1 MG/MΦ activation state in the aged TBI brain.
In addition to the well described detrimental and neurotoxic effects of MG/MΦ activation, MG/MΦ also secrete anti-inflammatory cytokines and neurotrophic factors such as, IL-4, IL-10, and TGFβ, which play a key role in responding to injury and infection and maintaining homeostasis in the CNS (Loane and Byrnes, 2010; Lynch, 2009). Functional heterogeneity of MG/MΦ enables these cells to participate in repair and resolution processes after infection or injury to the brain (Colton, 2009). IL-4 promotes an M2a (alternative activation) MG/MΦ phenotype that is involved in tissue repair and ECM reconstruction, whereas IL-10 promotes an M2c (acquired deactivation) MG/MΦ phenotype that is immunosuppressive and is associated with uptake of apoptotic cells (Colton, 2009; Gordon, 2003). Both M2 MG/MΦ phenotypes reduce M1 pro-inflammatory cytokines leading to resolution of inflammation after injury or infection (Gordon, 2003; Mantovani et al., 2004). However, the M1/M2 phenotypic classification likely oversimplifies the complexity and functional consequences of MG/MΦ activation in vivo, which may be better represented as a continuum of activation between two extremes with the pro-inflammatory M1 state at one end and the anti-inflammatory M2 state at the other. Arg-1 and Mrc are characteristic antigens of M2a MG/MΦ. Arg-1 competes with iNOS for arginine to produce L-ornithine and urea, rather than NO (Munder et al., 1999), and activation of the Mrc promotes anti-inflammatory signaling that results in decreased IL-12 and TNFα, and increased IL-10 and IL-1ra expression (Chieppa et al., 2003; Taylor et al., 2005). TBI induced the expression of both M2a MG/MΦ genes, and Arg-1 and Mrc expression was further enhanced in the aged TBI brain. Similarly, both genes are transiently up-regulated during the acute phase MΦ response to moderate level spinal cord injury (Kigerl et al., 2009). It is possible that the increased expression profile of some M2a genes (Arg-1, Ym1, Mrc) in the aged TBI brain may reflect a failed effort to initiate an M2 repair response, as has been demonstrated in aged animals following peripheral LPS injection in a model of sickness behavior (Fenn et al., 2012). We also evaluated Fizz1 and Ym1 expression, both of which are associated with ECM reconstruction (Raes et al., 2002). Fizz1 increases collagen expression and myofibroblast differentiation (Liu et al., 2004), and it was significantly reduced in aged compared to young mice; in contrast to other M2a genes, TBI resulted in a significant reduction of Fizz1 expression in young animals. Fizz1 expression is dependent on IL-4 and IL-13 stimulation and is inhibited by IFNγ (Pesce et al., 2009). It has be found to play an important role in limiting the pathogenesis of Th2 cytokine mediated pulmonary inflammation, in part through the regulation of of CD4(+) T cell responses (Nair et al., 2009; Pesce et al., 2009). However, the functional role of Fizz1 in CNS injury is unknown.
Ym1 expression was increased by TBI and further increased in the aged TBI brain at 24 h post-injury. Despite being expressed in the brain there is limited information on its expression and function under physiological and pathophysiologcal conditions (Colton, 2009). Our immunohistochemical analysis in 7 d TBI tissue revealed that Ym1 was expressed in heterogeneous MG/MΦ populations displaying distinct cellular morphologies that were spatially separated in the injured cortex and hippocampus. Ym1 was highly expressed in perilesional regions of the injured cortex and these Ym1-positive cells assumed the typical morphology of highly activated and amoeboid MG/MΦ cells. In contrast, in subcortical regions distant from the lesion core Ym1 was expressed in cells displaying a ramified morphology with numerous highly branched processes. The heterogenous expression of Ym1 after TBI is consistent with prior observations from a stab wound brain injury model in which Ym1-positive cells assumed an amoeboid cellular morphology adjacent to the wound and whereas others had a ramified cellular morphology (Hung et al., 2002). In the current study, we did not discriminate whether Ym1 was expressed on resident MG or infiltrating MΦ. It is thought that amoeboid cells may represent the recruitment of MΦ derived from circulating monocytes, whereas solitary Ym1-positive cells with ramified morphologies may be MG derived directly from the resident population (Hung et al., 2002). However, further studies using EGFP-positive monocytes in irradiated bone marrow chimeric mice will be required to discriminate between the expression and function of Ym1 in resident MG or infiltrating MΦ after TBI. In our study, Ym1-positive amoeboid MG/MΦ were negative for ED1 but were localized adjacent to ED1-positive cells in areas surrounding the lesion core, suggesting that Ym1-positive MG/MΦ may have altered functional capacity that is not involved in phagocytosis. Notably, highly branched ramified Ym1-positive MG/MΦ were dispersed throughout the hippocampus and white matter of young TBI mice, whereas fewer of these cells were detected in aged TBI mice. These results indicate that Ym1 is highly expressed in ramified cells (i.e. resting MG/MΦ) in the young TBI brain, but is expressed in both the highly activated bushy/amoeboid and to a lesser extent in ramified MG/MΦ in the aged TBI brain.
M2c MG/MΦ activation promotes immunosuppression and is associated with uptake of apoptotic cells (Gordon, 2003; Mantovani et al., 2004). We found that the M2c MG/MΦ activation response to TBI was diminished in aged animals when compared to young. Although increased compared to aged matched sham controls, the M2c genes IL-4Rα, SOCS3, TGFβ, as well as STAT6 which is an important player in the IL-4 signal transduction pathway, were significantly lower in aged TBI mice when compared to young TBI mice. These data suggest that the aged brain has reduced capacity to promote an anti-inflammatory M2c MG/MΦ response after TBI, leading to a failure to antagonize the pro-inflammatory M1 MG/MΦ activation state and also diminishing the M2a-mediated repair potential of these cells. Similarly, in another study an inflammatory stimulus (peripheral LPS injection) did not increase IL-4Rα protein expression on the surface of MG/MΦ in aged mice (Fenn et al., 2012), and this was associated with a reduced sensitivity for M2 promoting effects of IL-4, thereby indicating that IL-4 regulation of MG/MΦ may be impaired in aged brain. Our M2c MG/MΦ activation data in the aged TBI brain is consistent with this hypothesis. Taken together, the M2 MG/MΦ marker data indicate that MG/MΦ undergo an age-dependent activation phenotype balance switch from favoring an anti-inflammatory M2 phenotype with repair capabilities in young TBI mice to a pro-inflammatory M1 phenotype that may contribute to increased post-traumatic neurodegeneration in aged TBI mice. Interestingly, a recent study demonstrated an age-dependent phenotypic change of MG/MΦ activation in the hippocampus of AD transgenic (PS1M146L/APP751SL) mice from an M2 activation state with Aβ phagocytic capabilities to a M1 and neurotoxic phenotype, which promoted high levels of soluble Aβ oligomers and significant pyramidal neuron degeneration (Jimenez et al., 2008).
In conclusion, the present studies demonstrate that MG/MΦ undergo an age-dependent phenotypic switch in activation status in response to TBI, which may contribute in part to the increased oxidative stress and neuronal loss that is associated with neurological impairments and poorer functional outcomes of aged TBI animals. Increased NADPH oxidase activity and a decline in antioxidant capacity in the aged brain results in a shift in redox state towards an oxidizing microenvironment, and this may promote and maintain MG/MΦ in a pro-inflammatory M1 activation state after TBI. Inhibition of NADPH oxidase-mediated redox signaling has been shown to be an important molecular switch for MG/MΦ polarization and shifts cells from an M1 to M2 phenotype in response to inflammatory challenges (Choi et al., 2012). Similarly, polarizing MG/MΦ towards an M2 phenotype after TBI, thereby promoting repair processes while limiting inflammatory-mediated secondary injury cascades, may be beneficial for the future treatment of TBI in the elderly.
Acknowledgments
We thank Dr. Shruti Kabadi for helpful discussion. This work is supported by a pilot award from the National Capital Area Rehabilitation Research Network (R24HD050845) (D.J.L.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson J, Sandhir R, Hamilton ES, Berman NE. Impaired expression of neuroprotective molecules in the HIF-1alpha pathway following traumatic brain injury in aged mice. J Neurotrauma. 2009;26:1557–66. doi: 10.1089/neu.2008.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008a;45:443–52. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. Journal of neurotrauma. 2008b;25:513–26. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- Badan I, Buchhold B, Hamm A, Gratz M, Walker LC, Platt D, Kessler C, Popa-Wagner A. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional recovery. J Cereb Blood Flow Metab. 2003;23:845–54. doi: 10.1097/01.WCB.0000071883.63724.A7. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, LeVine H, 3rd, Keller JN, Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal. 2010;12:1371–82. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42:341–8. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–72. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieppa M, Bianchi G, Doni A, Del Prete A, Sironi M, Laskarin G, Monti P, Piemonti L, Biondi A, Mantovani A, Introna M, Allavena P. Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J Immunol. 2003;171:4552–60. doi: 10.4049/jimmunol.171.9.4552. [DOI] [PubMed] [Google Scholar]
- Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. Journal of neurochemistry. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde JR, Streit WJ. Effect of aging on the microglial response to peripheral nerve injury. Neurobiology of aging. 2006;27:1451–61. doi: 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Coronado VG, Thomas KE, Sattin RW, Johnson RL. The CDC traumatic brain injury surveillance system: characteristics of persons aged 65 years and older hospitalized with a TBI. J Head Trauma Rehabil. 2005;20:215–28. doi: 10.1097/00001199-200505000-00005. [DOI] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. Journal of neuroinflammation. 2010;7:41. doi: 10.1186/1742-2094-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downer EJ, Cowley TR, Lyons A, Mills KH, Berezin V, Bock E, Lynch MA. A novel anti-inflammatory role of NCAM-derived mimetic peptide, FGL. Neurobiology of aging. 2010;31:118–28. doi: 10.1016/j.neurobiolaging.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta (GA): 2010. [Google Scholar]
- Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP. Lipopolysaccharide-induced interleukin (IL)-4 receptor-alpha expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav Immun. 2012;26:766–77. doi: 10.1016/j.bbi.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- Galbraith S. Head injuries in the elderly. Br Med J (Clin Res Ed) 1987;294:325. doi: 10.1136/bmj.294.6568.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003;23:6181–7. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Age-related mitochondrial changes after traumatic brain injury. J Neurotrauma. 2010;27:939–50. doi: 10.1089/neu.2009.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. 2005;19:1329–31. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, Jenkins LW, Lyeth BG, White-Gbadebo DM, Hayes RL. The effect of age on outcome following traumatic brain injury in rats. J Neurosurg. 1991;75:916–21. doi: 10.3171/jns.1991.75.6.0916. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, White-Gbadebo DM, Lyeth BG, Jenkins LW, Hayes RL. The effect of age on motor and cognitive deficits after traumatic brain injury in rats. Neurosurgery. 1992;31:1072–7. doi: 10.1227/00006123-199212000-00013. discussion 8. [DOI] [PubMed] [Google Scholar]
- Hung SI, Chang AC, Kato I, Chang NC. Transient expression of Ym1, a heparin-binding lectin, during developmental hematopoiesis and inflammation. J Leukoc Biol. 2002;72:72–82. [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–61. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Hanscom M, Loane DJ, Kharebava G, Murray MG, II, Cabatbat RM, Faden AI. CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics. 2012;9:405–21. doi: 10.1007/s13311-011-0095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, O’Banion MK, Whiteley PE, Daeschner JC, Olschowka JA. Enhanced glial activation and expression of specific CNS inflammation-related molecules in aged versus young rats following cortical stab injury. Journal of neuroimmunology. 2001;119:269–77. doi: 10.1016/s0165-5728(01)00404-0. [DOI] [PubMed] [Google Scholar]
- Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164:1315–26. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston DH, Lavery RF, Mosenthal AC, Knudson MM, Lee S, Morabito D, Manley GT, Nathens A, Jurkovich G, Hoyt DB, Coimbra R. Recovery at one year following isolated traumatic brain injury: a Western Trauma Association prospective multicenter trial. J Trauma. 2005;59:1298–304. doi: 10.1097/01.ta.0000196002.03681.18. discussion 304. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Deighan BF, Clarke RM, Griffin RJ, Lynch AM, Lynch MA. Interleukin-4 mediates the neuroprotective effects of rosiglitazone in the aged brain. Neurobiology of aging. 2009a;30:920–31. doi: 10.1016/j.neurobiolaging.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med. 2009b;15:377–9. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–65. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–56. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol. 2006;176:1046–52. doi: 10.4049/jimmunol.176.2.1046. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 2007;3:245–53. doi: 10.1017/S1740925X08000136. [DOI] [PubMed] [Google Scholar]
- Moor E, Shohami E, Kanevsky E, Grigoriadis N, Symeonidou C, Kohen R. Impairment of the ability of the injured aged brain in elevating urate and ascorbate. Exp Gerontol. 2006;41:303–11. doi: 10.1016/j.exger.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–7. [PubMed] [Google Scholar]
- Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, Swain GP, Yancopoulos GD, Valenzuela DM, Murphy A, Karow M, Stevens S, Pearce EJ, Artis D. Alternatively activated macrophage-derived RELM-{alpha} is a negative regulator of type 2 inflammation in the lung. J Exp Med. 2009;206:937–52. doi: 10.1084/jem.20082048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyszchuk G, He YY, Berman NE, Brooks WM. Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. Journal of neurotrauma. 2008;25:153–71. doi: 10.1089/neu.2007.0430. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. Journal of neuroscience research. 2004;77:540–51. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Pennings JL, Bachulis BL, Simons CT, Slazinski T. Survival after severe brain injury in the aged. Arch Surg. 1993;128:787–93. doi: 10.1001/archsurg.1993.01420190083011. discussion 93–4. [DOI] [PubMed] [Google Scholar]
- Pesce JT, Ramalingam TR, Wilson MS, Mentink-Kane MM, Thompson RW, Cheever AW, Urban JF, Jr, Wynn TA. Retnla (relmalpha/fizz1) suppresses helminth-induced Th2-type immunity. PLoS Pathog. 2009;5:e1000393. doi: 10.1371/journal.ppat.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner A, Carmichael ST, Kokaia Z, Kessler C, Walker LC. The response of the aged brain to stroke: too much, too soon? Curr Neurovasc Res. 2007;4:216–27. doi: 10.2174/156720207781387213. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. Journal of neurochemistry. 2002;83:973–83. doi: 10.1046/j.1471-4159.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh GhG. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. Journal of leukocyte biology. 2002;71:597–602. [PubMed] [Google Scholar]
- Rapoport MJ, Herrmann N, Shammi P, Kiss A, Phillips A, Feinstein A. Outcome after traumatic brain injury sustained in older adulthood: a one-year longitudinal study. Am J Geriatr Psychiatry. 2006;14:456–65. doi: 10.1097/01.JGP.0000199339.79689.8a. [DOI] [PubMed] [Google Scholar]
- Sandhir R, Berman NE. Age-dependent response of CCAAT/enhancer binding proteins following traumatic brain injury in mice. Neurochem Int. 2010;56:188–93. doi: 10.1016/j.neuint.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Experimental neurology. 2008;213:372–80. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Ford AL, Foulcher E, Airriess R. Central nervous system microglial cell activation and proliferation follows direct interaction with tissue-infiltrating T cell blasts. J Immunol. 1998;160:5320–30. [PubMed] [Google Scholar]
- Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- Soltys Z, Ziaja M, Pawlinski R, Setkowicz Z, Janeczko K. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–7. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Stocchetti N, Paterno R, Citerio G, Beretta L, Colombo A. Traumatic brain injury in an aging population. Journal of neurotrauma. 2012;29:1119–25. doi: 10.1089/neu.2011.1995. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends in immunology. 2005;26:104–10. doi: 10.1016/j.it.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Testa JA, Malec JF, Moessner AM, Brown AW. Outcome after traumatic brain injury: effects of aging on recovery. Arch Phys Med Rehabil. 2005;86:1815–23. doi: 10.1016/j.apmr.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Wong AM, Patel NV, Patel NK, Wei M, Morgan TE, de Beer MC, de Villiers WJ, Finch CE. Macrosialin increases during normal brain aging are attenuated by caloric restriction. Neuroscience letters. 2005;390:76–80. doi: 10.1016/j.neulet.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:6145–50. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]