

Abstract
Biophysical and structural characterization of G protein-coupled receptors (GPCRs) has been limited due to difficulties in expression, purification, and vitro stability of the full-length receptors. “Divide and conquer” approaches aimed at the NMR characterization of peptides corresponding to specific regions of the receptor have yielded insights into the structure and dynamics of GPCR activation and signaling. Though significant progress has been made in the generation of peptides that are composed of GPCR transmembrane domains, current methods utilize fusion protein strategies that require chemical cleavage and peptide separation via chromatographic means. We have developed an expression and purification system based on fusion to ketosteroid isomerase, thrombin cleavage, and tandem affinity chromatography that enables the solubilization, cleavage, and characterization in a single detergent system relevant for biophysical and structural characterization. We have applied this expression and purification system to the production and characterization of peptides of the adenosine receptor family of GPCRs in Escherichia coli. Herein, we demonstrate using a model peptide that includes extracellular loop 3, transmembrane domain 7, and a portion of the carboxy-terminus of the adenosine A2a receptor that the peptide is sufficiently pure for biophysical characterization, where it adopts α-helical structure. Furthermore, we demonstrate the utility of this system by optimizing the construct for thrombin processing and apply the system to peptides with more complex structures.
Keywords: Ketosteroid isomerase, Peptide, G-protein coupled receptor, Human A2a receptor, Human A2b receptor, Escherichia coli
Introduction
G protein-coupled receptors (GPCRs)1 are α-helical seven transmembrane domain proteins that play critical roles in cellular signaling in response to diverse extracellular ligands. Approximately 30% of current small molecule and biological drugs target GPCRs [1], where the effectiveness and specificity of these pharmaceutical therapies could be vastly improved by structure-based drug design. To date, medium to high resolution crystal structures of eight GPCRs have been determined, including rhodopsin [2], the β1 and β2 adrenergic receptors [3–7], the adenosine A2a receptor [8–10], the CXCR4 chemokine receptor [11], the dopamine D3 receptor [12], the histamine H1 receptor [13], and the M2 muscarinic receptor [14]. Opportunities for further development of pharmaceuticals that target GPCRs are considerable, as the sequencing of the human genome has identified upwards of 1000 GPCRs [15]; however, the expression, purification, and in vitro stability of full-length GPCRs have impeded the biophysical and structural characterization of this class of membrane proteins.
Recently, “divide and conquer” approaches, which involve the generation and analysis of peptides corresponding to GPCR transmembrane (TM) domains, have been utilized to overcome obstacles that prohibit characterization of full-length GPCRs. For example, structural characterization by NMR is more easily achieved and less constrained in the membrane mimetic environment than by crystallography. Furthermore, peptides derived from rhodopsin and characterized by NMR were shown to adopt secondary structures similar to those observed by X-ray crystallography [16–19]. In addition, studies of GPCR-derived peptides offer a unique opportunity: specific regions of the receptor, i.e. regions responsible for ligand binding, receptor activation or cellular signaling, can be characterized independently. Since these regions are often quite mobile and poorly resolved, this approach complements high-resolution crystallography. In fact, this piecemeal approach to GPCR characterization has led to insights into the structure of full-length GPCRs, including rhodopsin [16–19], Ste2p, the α-factor receptor of Saccharomyces cerevisiae [20–31], the neurokinin 1 (NK1) receptor [32], the adenosine A2a receptor [33–37], the μ-opioid receptor [38,39], and the cannabinoid CB2 receptor [40].
Only recently have GPCR peptides that are composed of more than a single TM been characterized by this approach, where the cost of chemical synthesis of isotopically labeled peptides (2H, 13C, 15N) and the inherent hydrophobic character of TM peptides have been the major obstacles. However, characterization of multiple transmembrane domains of the μ-opioid receptor, the cannabinoid CB2 receptor, and yeast Ste2p have been achieved through expression in Escherichia coli of fusion proteins containing the desired peptide [30,38–40]. In fact, large quantities of the isotopically labeled fusion protein/peptide can be generated relatively inexpensively by expression in media containing 2H, 13C, and 15N as sole sources for hydrogen, carbon, and nitrogen. After purification and liberation of the GPCR peptide from the fusion protein, these multi-TM GPCR peptides have shown highly α-helical secondary structure in trifluoro-ethanol/water and a variety of detergents [30,38–40] and provide motivation for further investigations into the “divide and conquer” approach.
In general, expression of GPCR peptides in E. coli is achieved by constructs with multiple peptides in tandem or by fusion to carrier proteins that protect the peptide from intracellular degradation [41]. A number of fusion protein expression systems are commercially available, including those with maltose binding protein, glutathione S-transferase, small ubiquitin-like modifier, and thioredoxin. These carrier proteins not only promote solubility and high expression levels but in some cases also provide convenient means for purification by affinity chromatography. Other carrier proteins, including ketosteroid isomerase [42], the N-terminal domain of S. nuclease (SFC120) [43], fusion to E. coli TrpE [44], and the anti-apoptotic Bcl-2 family protein, Bcl-XL [45], promote insoluble expression and readily form inclusion bodies. Inclusion body expression often results in the highest expression yields [46,47], provides a simple procedure for recovery through lysis and centrifugation, and even permits the production of toxic peptides [48,49]. Due to their inherent hydrophobic character, GPCR TM peptides are almost exclusively expressed as fusion proteins with carriers that drive expression to inclusion bodies.
Though required for the generation of peptides in E. coli, removing the carrier protein is not always a trivial process. The most common methods for liberating peptides from fusion proteins involve chemical cleavage through CNBr [42] or acid hydrolysis [50] and are limited to peptides that do not contain methionine or the acid labile Asp-Pro sequence, respectively, or require their modification by site-directed mutagenesis. Additionally, the harsh conditions under which chemical cleavage is achieved requires purification of the cleavage products by HPLC, where hydrophobic peptides are known to result in low yield [44,51], poor resolution, and low purity [51]. Alternatively, the peptide may be liberated from the carrier protein through incorporation of a protease-specific cleavage site, including those for enterokinase [52], factor Xa 11, tobacco etch virus (TEV) protease [53], and thrombin [54,55], and combined with purification strategies that utilize detergents or chaotropes.
We have developed a robust system for the high-level expression and purification of GPCR peptide fragments in E. coli that exploits methods previously developed for solubilizing and recovering inclusion bodies with the zwitterionic detergent fos-choline 16 [56]. The engineered expression system utilizes an N-terminal ketosteroid isomerase (KSI) domain, redundant Strep-Tactin affinity tags, a thrombin cleavage site, and a nickel affinity tag to enable the expression and rapid purification of receptor peptides for biophysical and structural characterization.
Results
A peptide representing extracellular loop 3, transmembrane domain 7, and a portion of the C-terminus of hA2aR was used as a model peptide for developing peptide expression and purification protocols (Fig. 1). Unlike many other G protein-coupled receptors (GPCRs), extensive biophysical and structural characterization of the human adenosine A2a receptor have been accomplished through biophysical characterization of hA2aR TM peptides [33,35–37] and the full-length receptor [57,58] as well as through X-ray crystallography [9,10,59]. Completing the biophysical characterization of the hA2aR TM 7 peptide and comparing the results with the literature would validate the peptide expression and purification system and motivate its application to other GPCR-derived peptides.
Fig. 1.

(A) Topology diagram of hA2aR denoting the model peptide hA2aR TM 7. The hA2aR TM 7 peptide is highlighted in gold, and includes residues 259–316. (B) Schematic of the expression construct highlighting design elements, including redundant Strep-tag II affinity tags, the ketosteroid isomerase carrier protein, the thrombin cleavage site, the GPCR peptide, and a His6 affinity tag. The glycine-rich linker downstream of the thrombin cleavage site has been varied by the number of GSG repeats, where n is 0, 1, 2, 4, or 8.
Designing the expression construct
There have been a number of fusion proteins used for the expression and purification of GPCR-derived peptides, including those that promote solubility or insolubility of the fusion protein [22–31,38–40]. Due to the high hydrophobic character of GPCR TM peptides, carrier proteins designed to promote soluble expression of the fusion protein oftentimes fail in this regard and instead readily form inclusion bodies. For example, initial attempts in our lab at expressing GPCR TM peptides that utilized thioredoxin, a carrier protein that promotes soluble expression, showed that the fusion protein readily formed inclusion bodies in E. coli (data not shown). Therefore, in lieu of choosing a carrier protein that promotes soluble expression, we pursued fusion proteins that utilize ketosteroid isomerase (KSI), which promotes insoluble expression in inclusion bodies in E. coli.
The commercially available vector, pET-31b (Merck KGaA, Germany), was used as a starting point for developing the expression system; this construct encodes for an N-terminal KSI leader and contains an in-frame His6 tag for facile immobilized metal affinity chromatography (IMAC) purification. Directing the fusion protein to inclusion bodies exploits our previous success with the recovery of the human neurokinin NK1 receptor, a GPCR, from inclusion bodies in E. coli [56], where protocols for expression, detergent solubilization, and IMAC purification have already been optimized.
Unlike our previous work, however, fusion proteins carrying GPCR-derived peptides require additional preparation and purification steps: the GPCR peptide must be liberated from the fusion protein and then isolated. As previously described, common methods for liberating the peptide include chemical cleavage through CNBr or acid hydrolysis at susceptible sites. This approach, however, does not utilize the detergent-solubilized state of the fusion protein and requires purification by HPLC, which has confounded the recovery of highly hydrophobic peptides [44,51]. Thus, we sought alternatives to chemical cleavage that would permit the cleavage and recovery of fusion proteins in a single detergent system. Our previous work with solubilizing the NK1 receptor [56] and recent evidence from a systematic screen of 96 detergents identified that the fos-choline series of detergents (FC9–16) was the most effective at solubilizing human chemokine receptors from E. coli [60]. Therefore, we used fos-choline 16 as the detergent of choice and the previously developed expression and purification protocols [56] as the basis for developing peptide expression and purification methods.
Site-specific proteases, which cleave at specific sequences that are designed into the expression construct through recombinant DNA methods, are an alternative to chemical cleavage. And, importantly, thrombin was determined to be insensitive to a panel of 94 detergents, including the fos-choline series (FC10–16) [61]. Thus, a thrombin cleavage site (LVPRGS) was incorporated between the KSI carrier protein and the peptide of interest in the expression construct. In order to clarify the peptide of interest from undigested fusion protein, we have incorporated two Strep-tag II sequences into the construct design: one on the N-terminus of KSI and one immediately upstream of the thrombin cleavage site. Any undigested fusion protein carrying the Strep-tag II is bound upon exposure to Strep-Tactin resin, whereas the GPCR peptide passes unimpeded through the column and is collected in the flow-through fraction. This clarification step ensures the purity of the GPCR peptide for biophysical and structural characterization.
Initial expression studies using this construct resulted in IMAC purification yields of ~15–20 mg/L fusion protein per liter of culture, which is common for KSI-driven fusion protein expression [42]. However, liberation of the hA2aR TM 7 peptide from the fusion protein limited overall peptide yields and further optimization of this processing step was investigated to improve yields.
Thrombin processing efficiency
Even in cases where the detergent does not influence protease activity, ineffective processing of fusion proteins by the protease can limit high peptide yields. Protease processing yields are commonly in the range of 60–70% [62] but may be improved by incorporation of glycine rich linkers flanking the cleavage site [63]. A fusion protein of hA2aR TM 7 that lacked a glycine-rich linker was produced and purified by IMAC; this peptide was tested for thrombin processing and found to be 65% cleaved after 24 h. at 4 °C (Fig. 2). In order to improve the cleavage yields, we incorporated glycine rich linkers G(GSG)8G upstream and downstream of the thrombin cleavage site. We then determined the effect of different linker lengths on the thrombin processing efficiency of purified fusion proteins that differed only in the downstream glycine rich linker, including G(GSG)nG linkers of n = 8,4,2,1,0 (Fig. 2), as thrombin processing would remove the upstream linker.
Fig. 2.
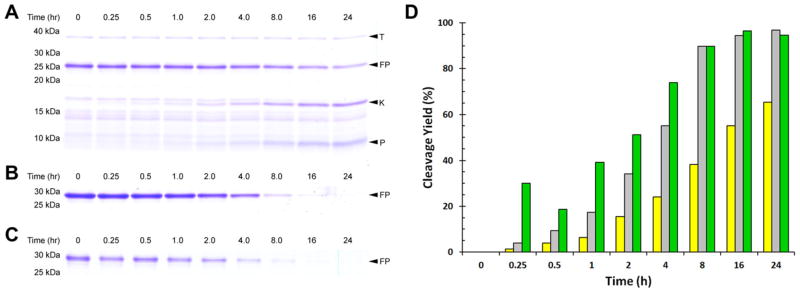
Thrombin processing efficiency differs significantly for fusion proteins with different linkers. SDS–PAGE (16% acrylamide) shows thrombin processing of Strep-KSI-Strep- Thrombin-hA2aR TM 7 -His6 (A), Strep-KSI-G(GSG)8G-Strep-Thrombin-GG-hA2aR TM 7 -His6 (B), and Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 -His6 (C). Quantification of fusion protein cleavage for Strep-KSI-Strep-Thrombin-hA2aR TM 7 -His6 (yellow), Strep-KSI-G(GSG)8G-Strep-Thrombin-GG-hA2aR TM 7 -His6 (grey), and Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 -His6 (green) (D) shows that thrombin processing efficiency is improved with a glycine-rich linker flanking the cleavage site. Thrombin (T); fusion protein (FP); KSI-containing cleavage product (K); and hA2aR TM 7 peptide (P) are indicated with arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Achieving high thrombin processing yields with only very short glycine rich linkers ensured that contributions of the artificial linker to biophysical and structural characterization are minimized. We, therefore, pursued expression and purification of Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 that when released from the carrier protein would result in a peptide corresponding to GSLKGGSGGVD-hA2aR259-316-LEHHHHHH.
Purification of hA2aR TM 7
We purified hA2aR TM 7 for biophysical characterization and validated protocols for expressing and purifying GPCR peptides in E. coli. One liter of E. coli Rosetta(DE3)/pLysS culture expressing Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 was grown and harvested. Inclusion bodies were extracted, solubilized with fos-choline 16, and purified by immobilized metal affinity chromatography (IMAC) through selective affinity of the His6-tag for Ni–NTA resin. Samples were taken after each step of the IMAC purification and analyzed by SDS–PAGE and Coomassie staining (Fig. 3). Following elution, Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 appeared as a single band on Coomassie-stained SDS–PAGE. Western blot analysis using an anti-His6 antibody confirmed the identification of the ~29 kDa band.
Fig. 3.

Analysis of Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7-His6 purification by IMAC. SDS–PAGE (12% acrylamide) shows samples from each step of the purification corresponding to 1 OD cells. Solubilized protein following extraction of insoluble fraction and inclusion bodies with Fos-choline 16 (IB); flow through (FT), 20 mM imidazole wash (W1); 30 mM imidazole wash (W2); 50 mM imidazole wash (W3); 500 mM imidazole elutions (E1, E2, E3). Full-length fusion protein (FP) is indicated with an arrow.
4 mg Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 was digested in 10 mL thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline 16 at 4 °C with 25 U/mL thrombin. Thrombin digestion resulted in 43% cleavage of the fusion protein in this case. Other 1L scale studies showed that fresh thrombin and detergent concentration were critical for best yields (data not shown). Thus, we believe the uncommonly low cleavage yield compared to our earlier success was the result of the thrombin preparation or ineffective reduction of the detergent concentration for this particular preparation. Despite the low cleavage yield, the uncut fusion protein and hA2aR TM 7 -His6 peptide that contained the His6 tag were readily separated from thrombin and the KSI-containing digestion product by IMAC. His-tagged proteins were eluted in thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline 16 and 500 mM imidazole (Fig. 4A). Fractions enriched in uncut Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 -His6 and hA2aR TM 7 -His6 peptide were combined and separated by differential affinity for Strep-Tactin resin (Fig. 4B). Flow-through fractions were collected and analyzed by SDS–PAGE and Coomassie staining. Fractions enriched in pure peptide were combined and concentrated using a centrifugal concentrator. The hA2aR TM 7 peptide was >95% pure as judged by SDS–PAGE and permitted biophysical characterization by circular dichroism (CD) spectroscopy. A summary of the processing and purification steps leading to the final peptide yield appears in Table 3.
Fig. 4.
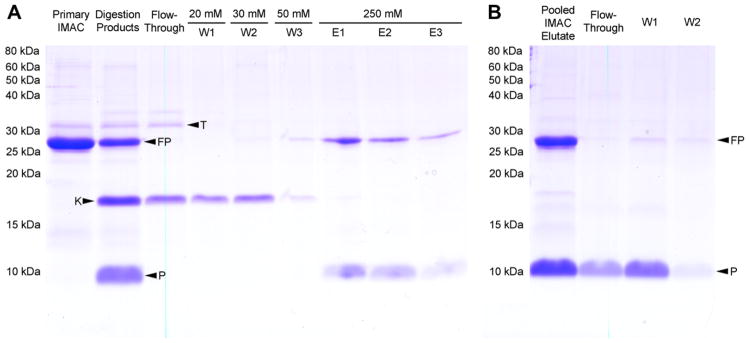
SDS–PAGE (16% acrylamide) shows the purification of GGSGG-hA2aR TM 7 after thrombin cleavage by a second IMAC step (A) and Strep-Tactin affinity (B). Dialyzed fusion protein was digested with thrombin and the cleavage products were separated by IMAC. Samples of each purification step were analyzed including undigested fusion protein (primary IMAC), digestion products, flow through, 20 mM imidazole wash (W1); 30 mM imidazole wash (W2); 50 mM imidazole wash (W3); 250 mM imidazole elutions (E1, E2, E3). IMAC elutions consisting of uncleaved fusion protein and hA2aR TM 7 peptide from the pooled eluate (E1–E3) were separated by affinity to high-capacity Strep-Tactin resin. Fractions collected from the flow-through and column washes W1 and W2 representing 3 mL thrombin cleavage buffer supplemented with FC16 are enriched in peptide and not uncleaved fusion protein. Thrombin (T); fusion protein (FP); KSI-containing cleavage product (K); and hA2aR TM 7 peptide (P) are indicated with arrows.
Table 3.
Summary of thrombin cleavage and overall GGSGG-hA2aR TM 7-His6 purification yields for large-scale preparation from Strep-KSI-G(GSG)8-Strep-Thrombin-GGSGG-hA2aR TM 7-His6. Note that only a portion of the total fusion protein was treated during thrombin step (equivalent to 8 mg inclusion body extract).
| Total protein (mg) | Fusion protein (mg) | Peptide (mg) | Step yield of peptide (%) | Overall peptide yield (%) | Purity | Relative purification | |
|---|---|---|---|---|---|---|---|
| Inclusion body extract | 39 | 19 | (6.3)a | N/A | 16 | ||
| Nickel purification | 17 | 17 | (5.7) | 90 | 90 | 33.5 | 2.1 |
| Thrombin cleavage | 4 | 4 | (1.3) | 43 | 39 | 32.5 | 2.0 |
| Nickel purification | 2.6b | 0.52 | 90 | 35 | 20 | 1.2 | |
| Streptactin purification | 0.33 | 0.33 | 65 | 23 | >99 | 6.2 |
Parentheses indicate that peptide mass was a calculated value based on mass fraction of peptide in the fusion protein.
Thrombin cleavage yield based on quantification of SDS–Page and Coomassie staining of fusion protein before and after thrombin digestion.
β-mercaptoethanol (β-ME) was included throughout the purification of hA2aR TM 7 to reduce disulfide bonds and promote fusion protein and peptide monomer formation, since the hA2aR TM 7 peptide contains two cysteines known to form a disulfide bond. However, β-ME has been known to confound secondary structure characterization by CD spectroscopy due to inherent optical properties, and was therefore removed prior to CD characterization of the peptide. Eliminating the reducing agent resulted in the formation of reversible peptide dimers (Fig. 5A). Peptides dimers were reduced by treatment with DTT indicating that peptide dimerization is the result of intermolecular disulfide bonds.
Fig. 5.
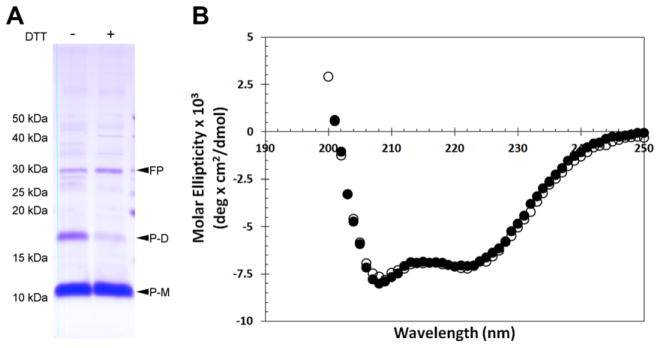
Circular dichroism spectroscopy of A2aR TM 7 indicates α-helical secondary structure. SDS–PAGE (16% acrylamide) shows that purified and dialyzed hA2aR TM 7 peptide forms dimers through a disulfide bond, where addition of DTT reduces these intermolecular bonds (A). Purified hA2aR TM 7 was diluted to 41.4 μM in 20mM phosphate buffer, pH 7.4, 0.1% Fos-choline 16 with (closed circles) and without (open circles) 5 mM TCEP and characterized by CD (B). Fusion protein (FP); monomeric hA2aR TM 7 peptide (P-M); putative dimeric hA2aR TM 7 peptide (P-D) are indicated with arrows.
Secondary structure
The secondary structure of hA2aR TM 7 was investigated using circular dichroism (CD) spectroscopy. In order to determine the secondary structure of hA2aR TM 7 of both the monomeric, reduced and dimeric peptide by CD spectroscopy, samples were obtained in phosphate buffer and fos-choline 16 with or without the reducing agent TCEP (Fig. 5B), which previously has been shown to fully reduce the disulfide bonds in the full-length hA2aR at the concentration tested [64]. CD spectra show that hA2aR TM 7 exhibits α-helical structure that is signified by two distinct minima at 208 and 222 nm, in either the presence or absence of TCEP (Fig. 5B). Addition of TCEP did not result in a significant change to spectra, indicating that the secondary structure of intermolecular disulfide-bonded hA2aR TM 7 dimers occupy similar secondary structure to their monomeric counterparts. Using this spectrum, the α-helical content of the hA2aR TM 7 peptide was estimated to be ~17% (Eq. (2), Materials and Methods) for both buffer conditions.
Analysis of expression for TM peptides derived from the adenosine family of GPCRs
In order to determine whether the expression system could be easily applied to more complex GPCR peptides, GPCR peptides corresponding to hA2aR TM 7, hA2aR TM 6–7, hA2aR TM 5–7, and hA2aR TM 6 were subcloned into the pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G vector. Each was transformed into the Rosetta(DE3)/pLysS cell line and grown in 250 mL cultures. For each culture, samples of cellular lysate, including soluble and insoluble proteins, were analyzed by SDS–PAGE (Fig. 6A). Of the five fusion proteins containing hA2aR transmembrane peptides, only fusion proteins containing peptides of hA2aR TM 7 and hA2aR TM 6 were expressed in Rosetta(DE3)/pLysS at levels sufficient for detection by SDS–PAGE and Coomassie staining (Fig. 6A). Western blot analysis using an anti-His6 antibody confirmed that bands identified by Coomassie staining are indeed the fusion proteins, and also shows that fusion proteins corresponding to hA2aR TM 6–7 and hA2aR TM 5–7 peptides are expressed, albeit at much lower levels (Fig. 6B). In addition to lower per cell expression levels, expression of fusion proteins corresponding to hA2aR TM 6–7, hA2aR TM 5–7 and to a moderate extent hA2aR TM 6 peptides exhibited severe growth inhibition and in some cases modest cell death, as determined by evaluating doubling times based on changes to optical density post-induction.
Fig. 6.
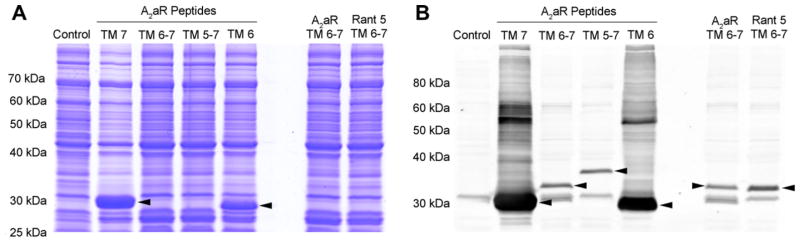
Expression of hA2aR TM peptides in E. coli is TM-specific. SDS–PAGE (12% acrylamide) of cellular proteins corresponding to 1 OD cells analyzed by Coomassie staining (A) and anti-His6 Western blot (B) shows samples obtained from strains expressing Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G fusion protein with peptides corresponding to hA2aR TM 7, TM 6–7, TM 5–7 and TM 6. A previously identified mutation in hA2aR TM 6 that stabilizes the antagonist-bound form of hA2aR (Rant 5) [65] had little effect on overall expression of the hA2aR TM 6–7 fusion protein. Arrows indicate the fusion protein.
Expression of particularly flexible or disordered peptides in E. coli may have ill effects despite KSI-driven expression to inclusion bodies. For example, structural flexibility of the peptide may promote soluble expression and inhibit KSI-driven inclusion body formation or alternatively, elicit cellular responses that ultimately lead to degradation of the fusion protein. Mutations to hA2aR created by alanine-scanning mutagenesis and selected for improvements in thermal stability and either agonist or antagonist conformational homogeneity have previously been identified [65]. Rant 5 (an antagonist-stabilized mutant of hA2aR) that contains a single mutation within TM 6 (V239A) was used to generate a TM 6–7 peptide, with the hypothesis that we might observe improved fusion protein expression yields by reducing TM 6 structural flexibility. However, the fusion protein for Rant 5 TM 6–7 was not observed by SDS–PAGE and only modestly improved expression yields over wild-type hA2aR TM 6–7 as determined via Western blot analysis (Fig. 6). Therefore, fusion protein yields of hA2aR TM 6–7 and TM 5–7 may not lead to the yields required for structural characterization.
The limited expression yields observed here for fusion proteins of hA2aR TM 6–7 may be GPCR-specific and not a limitation of the expression system as multi-TM [30,38–40,66] and full-length GPCRs [56,62,66] have been expressed in E. coli with high yields. To determine whether limitations to expression yields of fusion proteins of hA2aR TM 6–7 were specific to hA2aR, fusion protein constructs were generated for TM 7 and TM 6–7 peptides of hA1R and hA2bR. Analysis of expression via SDS–PAGE and Coomassie staining (Fig. 7A) as well as Western blot assay (Fig. 7B) showed that all fusion proteins for TM 7 peptides tested exhibited high expression yields; however, only the fusion protein corresponding to hA2bR TM 6–7 maintained high expression yields. The fusion protein corresponding to hA1R TM 6–7 exhibited severely diminished expression yields and cells expressing this fusion protein grew poorly post-induction, where the OD600 at harvest was ~0.5. Small-scale studies suggested thrombin cleavage of these peptides was efficient in the designed module, so long as expression was sufficient, indicating expression was the limiting step in the final peptide yields.
Fig. 7.
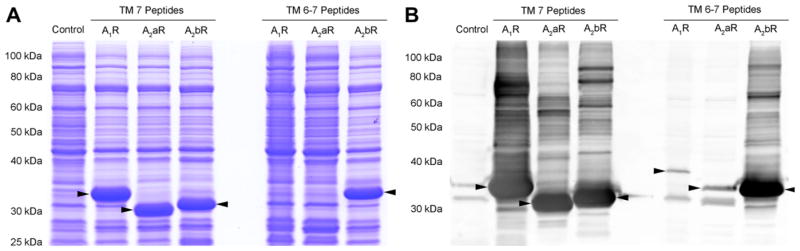
Reduced expression yields of hA2aR TM 6–7 are also observed in A1R TM 6–7 but not for A2bR TM 6–7. SDS–PAGE (12% acrylamide) of cellular proteins corresponding to 1 OD cells analyzed by Coomassie staining (A) and anti-His6 Western blot (B) shows samples obtained from strains expressing Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G fusion protein with peptides corresponding to hA1R, hA2aR and hA2bR TM 7 as well as hA1R, hA2aR and hA2bR TM 6–7 as indicated. All TM 7 peptides analyzed express at a high level, whereas only hA2bR TM 6–7 express at a high level. Arrows indicate the fusion protein.
Discussion
We have developed an expression and purification system for the generation of GPCR-derived peptides of the adenosine family of receptors based on fusion to ketosteroid isomerase. The developed approach utilizes the solubilization, thrombin cleavage, and tandem affinity purification of GPCR peptides in a single detergent system and offers an alternative to current methods for purifying GPCR-derived peptides that are based on chemical cleavage and HPLC.
In all cases, thrombin processing efficiency of fusion proteins with glycine rich linkers was improved over fusion proteins without the linker and resulted in efficiencies up to 95% in 24 h. or less on small-scale preparations (Fig. 2). Furthermore, this thrombin-based strategy for liberating peptides from the KSI carrier appeared to be general, as all adenosine TM peptides tested were efficiently cleaved by this method (data not shown). Because the purification strategy avoids the dependence on chemical cleavage for liberating the GPCR peptide from the fusion protein, GPCR peptides in this system are ensured to represent domains within the wild-type receptor without the need for modifying the primary sequence, which adds an additional advantage over currently used peptide production systems.
We have expressed and purified a model peptide of the human adenosine A2a receptor comprised of extracellular loop 3, transmembrane domain 7, and a portion of the C-terminus that contains helix 8 at larger scale to validate the expression and purification system. IMAC purification resulted in a yield of ~17 mg pure fusion protein per liter of culture and enabled further processing and purification steps prior to characterization of the hA2aR TM 7 peptide. In this case, yields were sufficient for further processing, but may be improved by sonication of recovered inclusion bodies in the presence of detergent, as batch-to-batch purification yields varied significantly, from 10 to 50 mg per liter of culture.
Characterization of the model peptide by CD spectroscopy showed that the peptide adopted α-helical structure in fos-choline 16 micelles consistent with the full-length receptor. Based on the spectra obtained, the α-helical content of the hA2aR TM 7 peptide was estimated to be ~17% (Eq. (2), Materials and Methods) for both buffer conditions investigated. This differs from the expected content of the hA2aR TM 7 peptide based on the hA2aR crystal structure (49%) [9] and studies of hA2aR TM 7 peptides in SDS micelles (>70%) and TFE (~70%) [37]. However, SDS and TFE both promote helix formation and TM 7 in the context of other helices may retain more helical content. Alternatively, these differences may result from an inability of the hydrophobic potential of the fos-choline detergent alone to stabilize helical structure. This observation motivates production and biophysical characterization of peptides with greater complexity, i.e. peptides that contain multiple α-helical TM domains, where helix-helix interactions have been shown to promote helix stability [67].
The expression system was extended to peptides of A2aR containing multiple transmembrane domains. Of the five fusion proteins containing hA2aR transmembrane peptides, expression of multidomain peptides containing either helix 5 or 6 resulted in much lower expression yield and growth inhibition and sometimes cell death.
It remains unclear as to why the addition of hA2aR TM 6 to the well-expressing peptide hA2aR TM 7 caused severely decreased expression levels and cell viability, where the final OD600 was ~0.5; however, the recent crystal structures of hA2aR [9,10,59] and biophysical characterization of synthesized peptides corresponding to TM peptides of hA2aR [33,35–37] yield some clues. These studies suggest that hA2aR TM 6, especially the intracellular portion of this TM, is inherently flexible. This structural flexibility contributes to GPCR activation by accommodating structural rearrangements within the GPCR helical core, but may contribute to instability or affect folding into inclusion bodies in vivo.
In contrast to the observations for A2aR, the fusion protein corresponding to hA2bR TM 6–7 maintained high expression yields. However, hA1R TM 6–7 also exhibited severely diminished expression yields and cells expressing this fusion protein grew poorly post-induction. Peptides corresponding to hA1R TM 6–7 and hA2aR TM 6–7 share the conserved CPXC motif [68] shown to form a disulfide bond in hA2aR [9,10,59], whereas hA2bR TM 6–7 does not. Mutations that eliminate this conserved motif (APXA) did not improve fusion protein yields for the pET-31b construct of these peptides (data not shown). It remains unclear why expression yields of KSI fusion proteins corresponding to hA1R and hA2aR TM 6–7 are severely limited whereas other peptides and full-length GPCRs expressed in this system are unaffected (data not shown).
These results highlight the importance of exploring multiple fusion protein constructs for optimal peptide expression yields; yet, the general approach developed here for liberating and purifying the peptide of interest are applicable in any system.
Materials and methods
Generating the expression system
DNA encoding a Strep-tag II affinity tag (WSHPQFEK), which enables purification by affinity for Strep-Tactin affinity resin, was introduced on the N-terminus of ketosteroid isomerase (KSI) by polymerase chain reaction (PCR) of KSI from pET-31b and cloned into pET-23b after NdeI and BamHI restriction digest to form pET-23b-Strep-KSI-His6 (primers listed in Table 1). A second Strep-tag II affinity tag and a thrombin cleavage site (LVPRGS) were incorporated on the C-terminus of Strep-KSI by ligating annealed oligos into KpnI and AflII digested pET-23b-Strep-KSI-His6 to form pET-23b-Strep-KSI-Strep-Thrombin-His6. DNA encoding the glycine-rich linker G(GSG)8G was then incorporated between the Strep-KSI and Strep-Thrombin cleavage site by cloning annealed oligos into BamHI and KpnI digested pET-23b-Strep-KSI-Strep-Thrombin-His6 to form pET-23b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6. DNA encoding Strep-KSI-G(GSG)8G-Strep-Thrombin was shuttled into pET-31b by NdeI and XhoI digest. A second G(GSG)8G protein linker was incorporated after the thrombin cleavage site in pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 by ligating annealed oligos into AflII and SalI restriction sites to form pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6. A summary of the cloning strategy appears in Table 2. DNA encoding for peptides of the adenosine A2a receptor, adenosine A1 receptor, and adenosine A2b receptor were then introduced into pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6by SalI and XhoI restriction digest. Tables 4–6 contain details of the cloning strategy and the peptides developed.
Table 1.
Primer sequences used in developing the expression constructs.
| Forward primers | |
| FPa Strep-KSI | GATATACATATGTGGAGCCACCCGCAGTTCGAAAAGATGCATACCCCAGAACACATCAC |
| FP A2aR 259 | GCAGCGTCGACTGCCCCGACTGCAGC |
| Reverse primers | |
| RP KSI | GAATTCGGATCCGCATGCGTGAATATTCTTCTCGC |
| RP A2aR 316 | GTGTACTCGAGTGCCTTGAAAGGTTCTTGCTGCCTC |
| Forward oligonucleotide | |
| FO Strep-Thrombin | GATCCATAGCAGGTACCTGGAGCCACCCGCAGTTCGAAAAGCTGGTGCCGCGCGGCAGCCTTAAGGCAG |
| FO B G(GSG)8G | GATCCGGTGGGTCTGGCGGATCTGGTGGGAGTGGCGGATCAGGTGGGAGCGGCGGATCCGGTGGGTCCGGTGGCTCGGGTGGGGGTAC |
| FO A G(GSG)8G | TTAAGGGTGGGTCTGGCGGATCTGGTGGGAGTGGCGGATCAGGTGGGAGCGGCGGATCCGGTGGGTCCGGTGGCTCGGGTGGGG |
| FO A G(GSG)4G | TTAAGGGTGGGTCTGGCGGATCTGGTGGGAGTGGCGGATCAGGTGGAG |
| FO A G(GSG)2G | TTAAGGGTGGGTCTGGCGGATCTGGTGGAG |
| FO A G(GSG)1G | TTAAGGGTGGGTCTGGCGGAGTCGACAAGCTTGCGGCCGCAC |
| FO A G(GSG)0G | TTAAGGGTGGAGTCGACAAGCTTGCGGCCGCAC |
| Reverse oligonucleotide | |
| RO Strep-Thrombin | TCGACTGCCTTAAGGCTGCCGCGCGGCACCAGCTTTTCGAACTGCGGGTGGCTCCAGGTACCTGCTATG |
| RO K G(GSG)8G | CCCCACCCGAGCCACCGGACCCACCGGATCCGCCGCTCCCACCTGATCCGCCACTCCCACCAGATCCGCCAGACCCACCG |
| RO S G(GSG)8G | TCGACCCCACCCGAGCCACCGGACCCACCGGATCCGCCGCTCCCACCTGATCCGCCACTCCCACCAGATCCGCCAGACCCACCC |
| RO S G(GSG)4G | TCGACTCCACCTGATCCGCCACTCCCACCAGATCCGCCAGACCCACCC |
| RO S G(GSG)2G | TCGACTCCACCAGATCCGCCAGACCCACCC |
| RO X G(GSG)1G | TCGAGTGCGGCCGCAAGCTTGTCGACTCCGCCAGACCCACCC |
| RO X G(GSG)0G | TCGAGTGCGGCCGCAAGCTTGTCGACTCCACCC |
Forward primer (FP) reverse primer (RP), forward oligonucleotide (FO) and reverse oligonucleotide (RO). NdeI (CATATG), BamHI (GTCGAC), KpnI (GGTACC), AflII (CTTAAG), SalI (GTCGAC), and XhoI (CTCGAG) restriction sites are underlined.
Table 2.
Cloning strategy for developing the expression constructs. Plasmids purchased from UMR cDNA resource center. Forward primer (FP) reverse primer (RP), forward oligonucleotide (FO) and reverse oligonucleotide (RO) sequences are listed in Table 1.
| Vector | Primers/oligonucleotides | Template/parental vector | Destination vector | Restriction sites |
|---|---|---|---|---|
| pET23b-Strep-KSI-His6 | FP Strep-KSI, RP KSI | pET31b | pET-23b | NdeI, BamHI |
| pET23b-Strep-KSI-Strep-Thrombin-His6 | FO Strep-Thrombin, RO Strep-Thrombin | – | pET-23b-Strep-KSI-His6 | BamHI, SalI |
| pET23b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | FO B G(GSG)8G, RO K G(GSG)8G | – | pET23b-Strep-KSI-Strep-Thrombin-His6 | BamHI, KpnI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | – | pET23b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | pET31b | NdeI, SalI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | FO A G(GSG)8G, RO S G(GSG)8G | – | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | AflII, SalI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 7-His6 | FP A2aR 259, RP A2aR 316 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)4G-hA2aR TM 7-His6 | FO A G(GSG)4G, RO S G(GSG)4G | – | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 7-His6 | AflII, SalI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)2G-hA2aR TM 7-His6 | FO A G(GSG)2G, RO S G(GSG)2G | – | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 7-His6 | AflII, SalI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)1G-His6 | FO A G(GSG)1G, RO X G(GSG)1G | – | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | AflII, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)1G-hA2aR TM 7-His6 | FP A2aR 259, RP A2aR 316 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)1G-His6 | SalI, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)0G-His6 | FO A G(GSG)0G, RO X G(GSG)0G | – | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 | AflII, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)0G-hA2aR TM 7-His6 | FP A2aR 259, RP A2aR 316 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)0G-His6 | SalI, XhoI |
Table 4.
Cloning strategy for peptide expression constructs of the adenosine family of receptors. Plasmids purchased from UMR cDNA resource center. Forward primer (FP) and reverse primers (RP) listed in Table 5.
| Vector | Primers/oligonucleotides | Template vector | Destination vector | Restriction sites |
|---|---|---|---|---|
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 6–7-His6 | FP A2aR 205, RP A2aR 316 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 5–7-His6 | FP A2aR 170, RP A2aR 316 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2aR TM 6-His6 | FP A2aR 205, RP A2aR 265 | pCDNA 3.1 hA2aR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-Rant 5 TM 6–7-His6 | FP A2aR 205, RP A2aR 316 | pRG/III-hs-MBP-Rant 5** | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA1R TM 7-His6 | FP A1R 260, RP A1R 326 | pCDNA3.1-hA1R* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA1R TM 6–7-His6 | FP A1R 208, RP A1R 326 | pCDNA3.1-hA1R* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2bR TM 7-His6 | FP A2bR 260, RP A2bR 332 | pCDNA3.1-hA2bR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
| pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-hA2bR TM 6–7-His6 | FP A2bR 211, RP A2bR 332 | pCDNA3.1-hA2bR* | pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-His6 | SalI, XhoI |
Table 6.
Primers used for generating peptides of the adenosine family of GPCRs. SalI to (GTCGAC) and XhoI (GTCGAC) restriction sites used for sub-cloning are underlined. A silent mutation was created in A2bR TM 6–7 to remove an XhoI restriction site, which is marked in bold.
| Forward primers | |
| FP A2aR 259 | GCAGCGTCGACTGCCCCGACTGCAGC |
| FP A2aR 205 | CAATAAGTCGACCGACGTCAGCTGAAGCAGATG |
| FP A2aR 170 | GTACGGTCGACGAGGATGTGGTCCCCATGAACTAC |
| FP A1R 260 | CAATAAGTCGACTGCCCGTCCTGCCAC |
| FP A1R 208 | CAAGCAGTCGACCGCAAGCAGCTCAACAAGAAG |
| FP A2bR 260 | GTACGGTCGACCAGCCAGCTCAGGGTAAAAATAAG |
| FP A2bR 211 | GTGAGGTCGACAGGCAGCTTCAGCGCACTGAGCTGATGGACCACTCAAGGACCACCCTCCAGCG |
| Reverse primers | |
| RP A2aR 316 | GTGTACTCGAGTGCCTTGAAAGGTTCTTGCTGCCTC |
| RP A2aR 265 | GTGTGCTCGAGGGCGTGGCTGCAGTCG |
| RP A1R 326 | TGATACTCGAGGTCATCAGGGCGCTCTTCTGG |
| RP A2bR 332 | GTGTGCTCGAGTAGTCCGACACCGAGAGCAG |
A model peptide of A2aR (AA 259–316) composed of extracellular loop 3, transmembrane domain 7, and a portion of the C-terminus that includes the membrane-associated helix 8 (Fig. 1), was devised based on the design of a 73-residue peptide of the S. cerevisiae alpha-factor pheromone receptor (Ste2p) that previously has been characterized by NMR [22]. In previous work on synthetic peptides of hA2aR, TM 7 was shown to have high α-helical content and biophysical characterization of this peptide could serve to validate the expression and purification protocols developed.
Optimizing the protein linker for A2aR TM 7
In order to optimize thrombin digestion, constructs of hA2aR TM 7 that differ in protein linker length were generated by digesting pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G-A2aR TM 7 with AflII and SalI and ligating annealed oligos corresponding to G(GSG)4G and G(GSG)2G linkers (Fig. 1B). For linkers of GGSGG and GG, annealed oligos of GGSGG and GG were ligated into pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-His6 previously digested with AflII and XhoI restriction sites. DNA encoding for A2aR TM 7 was then introduced into pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-His6 and pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-GG-His6at SalI and XhoI restriction sites.
Expression and immobilized metal affinity chromatography (IMAC) purification
Chemically competent Rosetta(DE3)/pLysS (Novagen) were transformed with the pET-31b vector containing the GPCR transmembrane peptide of interest. Transformants were selected by growth on Luria–Bertani (LB) plates supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Cells were grown overnight at 37 °C in 50 mL LB liquid media supplemented with 50 μg/mL ampicillin and 34 μg/mL chloramphenicol. The overnight culture was used to inoculate 1 L fresh media at an OD600 of ~0.1 and grown at 37 °C. When the optical density reached ~0.6, isopropyl-β-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce expression. Cells were grown for four hours post-induction and were then harvested by centrifugation for 5 min at 10,000g. The cell pellet was resuspended in 20 mL lysis buffer (50 mM sodium phosphate, pH 8.0; 10 mM EDTA, 0.1% Triton X-100; 100 μg/mL lysozyme; complete EDTA-free protease inhibitors (Roche)). The suspension was treated with three freeze/thaw cycles (−80/37 °C). After the third thaw, the suspension was supplemented with MgCl2 and DNAse (Deoxyribonuclease I, Sigma, St. Louis MO) to final concentrations of 20 mM and 20 μg/mL, respectively. Samples were incubated at 25 °C for 20 min and then were centrifuged at 16,000g for 10 min to separate inclusion bodies from soluble proteins. The pellet was treated with 20 mL binding buffer (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10% glycerol; 5 mM β-mercaptoethanol (β-ME); 20 mM imidazole) supplemented with 1% (w/v) fos-choline 16 for 1 h. at 25 °C to solubilize inclusion bodies. After centrifuging for 10 min at 16,000g, the supernatant containing solubilized inclusion bodies was collected and the pellet containing insoluble material was discarded.
Prior to purification, 5 mL of packed Ni–NTA resin (Qiagen) was pre-equilibrated for 1 h. at 25 °C with 20 mL binding buffer containing 0.1% fos-choline 16 and 20 mM imidazole. The resin was centrifuged 3,220g for 3 min and the equilibration buffer was removed. The resin was incubated in batch mode with solubilized inclusion bodies at 4 °C and tumbled gently on an end-over-end mixer overnight. The resin was centrifuged at 3,220g for 3 min and the supernatant containing unbound protein was removed. The resin was washed with 20 mL binding buffer containing 0.1% fos-choline 16 and 20 mM imidazole, then with 20 mL binding buffer containing 0.1% fos-choline 16 and 30 mM imidazole. The resin was then washed with 10 mL binding buffer containing 0.1% fos-choline 16 and 50 mM imidazole. Purified fusion proteins were eluted with 5 mL binding buffer containing 0.1% fos-choline 16 and 500 mM imidazole. Samples taken throughout the purification were analyzed by SDS–PAGE.
Thrombin cleavage
As determined by SDS–PAGE analysis and Coomassie staining, elutions enriched in purified fusion protein were pooled and dialyzed against thrombin cleavage buffer (20 mM Tris–HCl, pH 8.0; 150 mM NaCl; 2.5 mM CaCl2) supplemented with 5 mM β-ME and 0.0106% fos-choline 16. The concentration of dialyzed fusion proteins were determined by UV spectroscopy, where the extinction coefficient for the fusion protein at 280 nm (ε280) was calculated based on the Gill and von Hippel method [69]. Fusion proteins were diluted to 0.4 mg/mL in thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline and digested at 4 °C with 25 U/mL thrombin (Merck KGaA, Germany) on an end-over-end mixer. Samples corresponding to time = 0, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, 16 and 24 h after addition of thrombin were removed, quenched with 5x non-reducing sample buffer (163 mM Tris–HCl, pH 6.8, 0.25 mg/ml Bromophenol blue, 0.5% SDS, 50% glycerol), and stored at 4 °C until analysis by SDS–PAGE.
Twenty microliters of each sample were loaded on 16% poly-acrylamide gels and electrophoresed at 25 °C at 150 V for 1.5 h in SDS running buffer (25 mM Tris–HCl, 192 mM Glycine, 1% SDS, pH 8.3). Following electrophoresis, gels were subjected to Coomassie staining (10% acetic acid, 10% 2-propanol, 0.25 mg/mL Coomassie blue for 4 h followed by 10% acetic acid, 0.25 mg/mL Coomassie blue for 4 h). Gels were destained with 10% acetic acid. Gel images were recorded at a resolution of 650 dpi using an Epson flatbed scanner. In order to determine thrombin processing efficiency, images were analyzed using ImageJ software, where the percentage of fusion protein remaining at a given time was determined by the ratio of optical densities of the fusion protein at t = x, where x = 0, 0.25, 0.5, 1, 2, 4, 8, 16, 24 h and t = 0 h.
Purification of hA2aR TM 7
Expression of pET31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 -His6 in Rosetta(DE3)/pLysS and IMAC purification of Strep-KSI-G(GSG)8G-Strep-Thrombin-GGSGG-hA2aR TM 7 -His6 was performed as described previously. IMAC-purified fusion protein was extensively dialyzed against thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline 16 and the concentration of the dialyzed fusion protein was determined by UV spectroscopy. The fusion protein was then diluted to a concentration of 0.4 mg/mL in 10 mLs of thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline 16 and digested at 4 °C for 24 h with 25 U/mL thrombin on an end-over-end mixer. Thrombin cleavage products that contained the His6-tag (uncleaved fusion protein and hA2aR TM 7) were purified by IMAC. The thrombin cleavage products were permitted to bind 1 mL Ni–NTA that had previously been equilibrated with thrombin cleavage buffer supplemented with 5 mM β-ME and 0.0106% fos-choline 16 for 2 h. The resin was centrifuged 3,220g for 3 min and the supernatant containing unbound protein was removed. The resin was washed with 10 mL thrombin cleavage buffer containing 5 mM β-ME, 0.0106% fos-choline 16, and 20 mM imidazole, then with 10 mL thrombin cleavage buffer containing 5 mM β-ME, 0.0106% fos-choline 16, and 30 mM imidazole. The resin was then washed with 10 mL thrombin cleavage buffer containing 5 mM β-ME, 0.0106% fos-choline 16, and 50 mM imidazole. IMAC-purified cleavage products were eluted with 1 mL thrombin cleavage buffer containing 5 mM β-ME, 0.0106% fos-choline 16, and 250 mM imidazole. Samples taken through the purification were analyzed by SDS–PAGE and proteins were visualized with Coomassie staining.
Elutions enriched in hA2aR TM 7 peptide were combined and further purified from uncleaved fusion protein by differential affinity for Strep-Tactin resin. A 6 mL Kimble Chase Kontes FlexColumn was packed with 3 mL high-capacity Strep-Tactin resin (IBA-GmBH, Germany), washed extensively with water, and equilibrated by gravity flow with 40 mL thrombin cleavage buffer containing 5 mM β-ME and 0.0106% fos-choline 16. Three column volumes from the second IMAC purification that were enriched in hA2aR TM 7 peptide were applied directly to the Strep-Tactin column. Fractions representing the column flow-through and additional washes were collected in 3 mL fractions. Samples taken throughout the purification were analyzed by SDS–PAGE and Coomassie staining.
Those fractions enriched in purified hA2aR TM 7 peptide were pooled and concentrated using an Amicon centrifugal concentrator with a molecular weight cutoff of 3.5 kDa resulting in a final yield of ~ 0.55 mg/mL in 600 μL.
Circular dichroism
The secondary structure of GGSGG-hA2aR TM 7 -His6 in fos-choline 16 was characterized by circular dichroism spectroscopy. The purified peptide was dialyzed against 20 mM phosphate buffer, pH 7.4 containing 0.1% fos-choline 16 at 4 °C. This sample was then incubated with either buffer alone or buffer containing 5 mM Tris[2-carboxyethyl] phosphine (TCEP) for 1 h. at room temperature. The concentration of hA2aR TM 7 analyzed by circular dichroism was 41.4 μM.
Far-UV CD spectra were obtained using a Jasco-J-810 spectropolarimeter with Peltier thermal control. All measurements were taken at 25 °C using a 0.1 cm path-length quartz cuvette. Spectra were collected from 190 to 250 nm using one nm step resolution and four s integration time. The appropriate reference CD spectra have been subtracted from the reported CD spectra for GGSGG-hA2aR TM 7. CD spectra are expressed in mean molar ellipticities [Θ] (deg cm2dmol−1):
| (1) |
where Θabs is the observed ellipticity (mdeg), l is the optical path-length (cm), c is the protein concentration (M) and n is the number of residues in the protein. The percentage of α-helical content was estimated from the observed molar ellipticity at 222 nm ([Θ]222):
| (2) |
Applying the expression and purification system to other GPCR peptides
In order to confirm that the expression system is a general method for production of GPCR transmembrane peptides, the fusion protein system was extended to peptides consisting of single and multiple transmembrane domains of hA2aR and further validated on peptides of hA1R and A2bR.
Constructs that differed in the GPCR peptide were generated by digesting pET-31b-Strep-KSI-G(GSG)8G-Strep-Thrombin-G(GSG)8G with SalI and XhoI and ligating similarly digested PCR products representing transmembrane peptides of hA2aR, hA1R, and A2bR. Constructs confirmed by DNA sequencing were transformed into Rosetta(DE3)/pLysS and selected for growth on LB plates supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Control Rosetta(DE3)/pLysS and Rosetta(DE3)/pLysS containing a plasmid for GPCR peptide expression were grown overnight in 10 mL LB media containing either 34 μg/mL chloramphenicol or 50 μg/mL ampicillin and 34 μg/mL chloramphenicol, respectively, in a 37 °C water shaker. The overnight culture was used to inoculate 250 mL fresh LB media supplemented with the appropriate antibiotics at an OD600 of ~0.1 and grown at 37 °C. When the optical density reached 0.6, expression was induced by the addition of 1 mM IPTG and permitted to grow for 4 h post-induction. Aliquots corresponding to 100 mL of the culture were harvested by centrifugation for 5 min at 10,000g. Cell pellets were then resuspended in 1 mL solubilization buffer (20 mM Tris–HCl, pH 8.0 and 1% SDS), sonicated to homogeneity, and centrifuged at 16,000g for 10 min to remove any remaining insoluble material. Samples were analyzed by SDS–PAGE and subsequent Western blot analysis.
Western blot analysis
Following electrophoresis, proteins were transferred from the gel to a 0.2 μm nitrocellulose membrane at 50 V and 4 °C for 2 h in transfer buffer (25 mM Tris–HCl, 192 mM glycine, 20% methanol, pH 8.2). Proteins present on the membrane were detected using a primary antibody of anti-His6 IgG antibody (Covance, Princeton, NJ) at a dilution of 1:1000 in Tris-buffered saline (20 mM Tris–HCl, 150 mM NaCl, pH 7.6) containing 0.1% Tween 20 and 5% non-fat milk incubated overnight at 4 °C. The secondary antibody used for detection was Alexa Fluor® 633 goat anti-mouse IgG diluted to 1:5000. Resulting data were imaged with a Typhoon 9600™ variable mode imager (GE Bioscience, Piscataway, NJ).
Table 5.
Description of peptides explored in this work. Forward primer (FP) and reverse primers (RP) listed in Table 5.
| GPCR Peptide | AA | Molecular weighta | Oligonucleotides | PCR Template |
|---|---|---|---|---|
| hA2aR TM 7 | 259–316 | 6962.198 | FP A2aR 259, RP A2aR 316 | pCDNA 3.1 hA2aR |
| hA2aR TM 6–7 | 205–316 | 13152.621 | FP A2aR 205, RP A2aR 316 | pCDNA 3.1 hA2aR |
| hA2aR TM 5–7 | 170–316 | 17435.893 | FP A2aR 170, RP A2aR 316 | pCDNA 3.1 hA2aR |
| hA2aR TM 6 | 205–265 | 6922.217 | FP A2aR 205, RP A2aR 265 | pCDNA 3.1 hA2aR |
| Rant 5 TM 6–7 | 205–316 | 12968.379 | FP A2aR 205, RP A2aR 316 | pRG/III-hs-MBP-Rant 5 [65] |
| hA1R TM 7 | 260–326 | 7815.028 | FP A1R 260, RP A1R 326 | pCDNA 3.1 hA1R |
| hA1R TM 6–7 | 208–326 | 13717.173 | FP A1R 208, RP A1R 326 | pCDNA 3.1 hA1R |
| hA2bR TM 7 | 260–332 | 8087.404 | FP A2bR 260, RP A2bR 332 | pCDNA 3.1 hA2bR |
| hA2bR TM 6–7 | 211–332 | 13689.100 | FP A2bR 211, RP A2bR 332 | pCDNA 3.1 hA2bR |
Molecular weights are based on the theoretical molecular weights calculated from the peptide primary sequence [70] and correspond to the peptide alone without contributions from the expression cassette.
Acknowledgments
The authors would like to thank Dr. Asokhan Anbanandam, Professor Tatyana Polenova, and Dr. David Raden for useful discussions during the initial development of this project. The authors would also like to thank Drs. Chris Tate (University of Cambridge, UK) and Paula Booth (University of Bristol, UK) for the Rant 5 plasmid. These studies were supported by NIH P20 RR15588 (ASR) and NIH T32 GH08550 (ZTB).
Footnotes
Abbreviations used: GPCR, G protein-coupled receptor; TM, transmembrane; HPLC, high performance liquid chromatography; IMAC, immobilized metal affinity chromatography; A2aR, adenosine A2a receptor; NK1R, neurokinin 1 receptor; KSI, ketosteroid isomerase; Strep, Strep-tag II affinity tag; PCR, polymerase chain reaction; IPTG, isopropyl-β-thiogalactopyranoside; β-ME, β-mercaptoethanol; TCEP, Tris [2-carboxyethyl] phosphine; FC, fos-choline.
References
- 1.Hopkins AL, Groom CR. The druggable genome. Nature Reviews Drug Discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 2.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 3.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature. 2007;450:383–U384. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 4.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta(2)-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a beta(1)-adrenergic G-protein-coupled receptor. Nature. 2008;454:486-U482. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen SGF, DeVree BT, Zou YZ, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta(2) adrenergic receptor-Gs protein complex. Nature. 2011;477:549-U311. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu F, Wu HX, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A(2A) adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens RC, Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP. The 2. 6 Angstrom Crystal Structure of a Human A(2A) Adenosine Receptor Bound to an Antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tate CG, Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW. Agonist-bound adenosine A(2A) receptor structures reveal common features of GPCR activation. Nature. 2011;474:521-U154. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu BL, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien EYT, Liu W, Zhao QA, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H(1) receptor complex with doxepin. Nature. 2011;475:65–U82. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. 2012. advance online, publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XQH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang JH, Miklos GLG, Nelson C, Broder S, Clark AG, Nadeau C, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng ZM, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge WM, Gong FC, Gu ZP, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke ZX, Ketchum KA, Lai ZW, Lei YD, Li ZY, Li JY, Liang Y, Lin XY, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue BX, Sun JT, Wang ZY, Wang AH, Wang X, Wang J, Wei MH, Wides R, Xiao CL, Yan CH, Yao A, Ye J, Zhan M, Zhang WQ, Zhang HY, Zhao Q, Zheng LS, Zhong F, Zhong WY, Zhu SPC, Zhao SY, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An HJ, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi HY, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays AD, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu XJ, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen MY, Wu D, Wu M, Xia A, Zandieh A, Zhu XH. The sequence of the human genome. Science. 2001;291:1304. [Google Scholar]
- 16.Albert AD, Yeagle PL. Domain approach to three-dimensional structure of rhodopsin using high-resolution nuclear magnetic resonance. Vertebrate Phototransduction and the Visual Cycle, Part A. 2000;315:107. doi: 10.1016/s0076-6879(00)15838-0. [DOI] [PubMed] [Google Scholar]
- 17.Yeagle PL, Albert AD. Structure of the G-protein-coupled receptor, rhodopsin: a domain approach. Biochemical Society Transactions. 1998;26:520–531. doi: 10.1042/bst0260520. [DOI] [PubMed] [Google Scholar]
- 18.Yeagle PL, Choi G, Albert AD. Studies on the structure of the G-protein-coupled receptor rhodopsin including the putative G-protein binding site in unactivated and activated forms. Biochemistry. 2001;40:11932–11937. doi: 10.1021/bi015543f. [DOI] [PubMed] [Google Scholar]
- 19.Yeagle PL, Albert AD. Use of nuclear magnetic resonance to study the three-dimensional structure of rhodopsin, G Protein Pathways, Pt a. Receptors. 2002;343:223–231. doi: 10.1016/s0076-6879(02)43138-2. [DOI] [PubMed] [Google Scholar]
- 20.Naider F, Arshava B, Taran I, Xie HB, Becker JM. High resolution NMR analysis of the seven transmembrane domains of a heptahelical receptor in organic-aqueous medium. Biopolymers. 2002;64:161–176. doi: 10.1002/bip.10157. [DOI] [PubMed] [Google Scholar]
- 21.Naider F, Arshava B, Liu SF, Jiang HL, Breslav M, Becker JM. Structure of segments of a G protein-coupled receptor: CD and NMR analysis of the Saccharomyces cerevisiae tridecapeptide pheromone receptor. Biopolymers. 1998;46:343–357. doi: 10.1002/(SICI)1097-0282(199811)46:6<343::AID-BIP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 22.Naider F, Estephan R, Englander J, Arshava B, Samples KL, Becker JM. Biosynthesis and NMR analysis of a 73-residue domain of a Saccharomyces cerevisiae G protein-coupled receptor. Biochemistry. 2005;44:11795–11810. doi: 10.1021/bi0507231. [DOI] [PubMed] [Google Scholar]
- 23.Naider F, Arshava B, Ding FX, Arevalo E, Becker JM. Peptide fragments as models to study the structure of a G-protein coupled receptor: the alpha-factor receptor of Saccharomyces cerevisiae. Biopolymers. 2001;60:334–350. doi: 10.1002/1097-0282(2001)60:5<334::AID-BIP10175>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 24.Naider F, Khare S, Arshava B, Severino B, Russo J, Becker JM. Synthetic peptides as probes for conformational preferences of domains of membrane receptors. Biopolymers. 2005;80:199–213. doi: 10.1002/bip.20183. [DOI] [PubMed] [Google Scholar]
- 25.Naider F, Neumoin A, Arshava B, Becker J, Zerbe O. NMR studies in dodecylphosphocholine of a fragment containing the seventh transmembrane helix of a g-protein-coupled receptor from Saccharomyces cerevisiae. Biophysical Journal. 2007;93:467–482. doi: 10.1529/biophysj.106.103770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker JM, Umanah GKE, Huang LY, Maccarone JM, Naider F. Changes in conformation at the cytoplasmic ends of the fifth and sixth transmembrane helices of a yeast G protein-coupled receptor in response to ligand binding. Biochemistry. 2011;50:6841–6854. doi: 10.1021/bi200254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbe O, Neumoin A, Cohen LS, Arshava B, Tantry S, Becker JM, Naider F. Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophysical Journal. 2009;96:3187–3196. doi: 10.1016/j.bpj.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zerbe O, Zou C, Naider F. Biosynthesis and NMR-studies of a double transmembrane domain from the Y4 receptor, a human GPCR. Journal of Biomolecular NMR. 2008;42:257–269. doi: 10.1007/s10858-008-9281-z. [DOI] [PubMed] [Google Scholar]
- 29.Naider F, Arevalo E, Estephan R, Madeo J, Arshava B, Dumont M, Becker JM. Biosynthesis and biophysical analysis of domains of a yeast G protein-coupled receptor. Biopolymers. 2003;71:516–531. doi: 10.1002/bip.10491. [DOI] [PubMed] [Google Scholar]
- 30.Naider F, Estephan R, Englander J, Babu VVS, Arevalo E, Samples K, Becker JM. Sexual conjugation in yeast: a paradigm to study G-protein-coupled receptor domain structure. Biopolymers. 2004;76:119–128. doi: 10.1002/bip.10567. [DOI] [PubMed] [Google Scholar]
- 31.Reddy AP, Tallon MA, Becker JM, Naider F. Biophysical studies on fragments of the alpha-factor receptor protein. Biopolymers. 1994;34:679–689. doi: 10.1002/bip.360340510. [DOI] [PubMed] [Google Scholar]
- 32.Berlose JP, Convert O, Brunissen A, Chassaing G, Lavielle S. 3-dimensional structure of the highly conserved 7th transmembrane domain of G-protein-coupled receptors. European Journal of Biochemistry. 1994;225:827–843. doi: 10.1111/j.1432-1033.1994.0827b.x. [DOI] [PubMed] [Google Scholar]
- 33.Thevenin D, Roberts MF, Lazarova T, Robinson CR. Identifying interactions between transmembrane helices from the adenosine A(2A) receptor. Biochemistry. 2005;44:16239–16245. doi: 10.1021/bi051422u. [DOI] [PubMed] [Google Scholar]
- 34.Thevenin D, Lazarova T. Stable interactions between the transmembrane domains of the adenosine A(2A) receptor. Protein Science. 2008;17:1188–1199. doi: 10.1110/ps.034843.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson CR, Thevenin D, Lazarova T, Roberts MF. Oligomerization of the fifth transmembrane domain from the adenosine A(2A) receptor. Protein Science. 2005;14:2177–2186. doi: 10.1110/ps.051409205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazarova T, Roberts MF, Brewin KA, Robinson CR. Interactions between transmembrane helices in folding and assembly of adenosine A2a receptor. Biophysical Journal. 2004;86:253a–253a. [Google Scholar]
- 37.Robinson CR, Lazarova T, Brewin KA, Stoeber K. Characterization of peptides corresponding to the seven transmembrane domains of human adenosine A(2)a receptor. Biochemistry. 2004;43:12945–12954. doi: 10.1021/bi0492051. [DOI] [PubMed] [Google Scholar]
- 38.Kerman A, Ananthanarayanan VS. Expression and spectroscopic characterization of a large fragment of the mu-opioid receptor. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2005;1747:133–140. doi: 10.1016/j.bbapap.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Ananthanarayanan VS, Kerman A. Conformation of a double-membrane-spanning fragment of a G protein-coupled receptor: Effects of hydrophobic environment and pH (vol 1768, pg 1199. Biochimica Et Biophysica Acta-Biomembranes. 2007;1768:2342–2342. doi: 10.1016/j.bbamem.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Xie XQ, Zheng HA, Zhao J, Sheng WY. A transmembrane helix-bundle from G-protein coupled receptor CB2: biosynthesis, purification, and NMR characterization. Biopolymers. 2006;83:46–61. doi: 10.1002/bip.20526. [DOI] [PubMed] [Google Scholar]
- 41.Shen SH. Multiple joined genes prevent product degradation in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences. 1984;81:4627–4631. doi: 10.1073/pnas.81.15.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuliopulos A, Walsh CT. Production, purification, and cleavage of tandem repeats of recombinant peptides. Journal of the American Chemical Society. 1994;116:4599–4607. [Google Scholar]
- 43.Osborne MJ, Su ZD, Sridaran V, Ni F. Efficient expression of isotopically labeled peptides for high resolution NMR studies: application to the Cdc42/Rac binding domains of virulent kinases in Candida albicans. Journal of Biomolecular Nmr. 2003;26:317–326. doi: 10.1023/a:1024043620972. [DOI] [PubMed] [Google Scholar]
- 44.Jones DH, Ball EH, Sharpe S, Barber KR, Grant CWM. Expression and membrane assembly of a transmembrane region from Neu. Biochemistry. 2000;39:1870–1878. doi: 10.1021/bi992495e. [DOI] [PubMed] [Google Scholar]
- 45.Kelly JW, Yonemoto IT, Wood MR, Balch WE. A general strategy for the bacterial expression of amyloidogenic peptides using BCL-XL-1/2 fusions. Protein Science. 2009;18:1978–1986. doi: 10.1002/pro.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lilie H, Schwarz E, Rudolph R. Advances in refolding of proteins produced in E. coli. Current Opinion in Biotechnology. 1998;9:497–501. doi: 10.1016/s0958-1669(98)80035-9. [DOI] [PubMed] [Google Scholar]
- 47.Clark ED. Protein refolding for industrial processes. Current Opinion in Biotechnology. 2001;12:202–207. doi: 10.1016/s0958-1669(00)00200-7. [DOI] [PubMed] [Google Scholar]
- 48.Jerala R, Majerle A, Kidric J. Production of stable isotope enriched antimicrobial peptides in Escherichia coli: an application to the production of a N-15-enriched fragment of lactoferrin. Journal of Biomolecular NMR. 2000;18:145–151. doi: 10.1023/a:1008362401928. [DOI] [PubMed] [Google Scholar]
- 49.Cipakova I, Gasperik J, Hostinova E. Expression and purification of human antimicrobial peptide, dermcidin, in Escherichia coli. Protein Expression and Purification. 2006;45:269–274. doi: 10.1016/j.pep.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Jerala R, Zorko M, Japelj B, Hafner-Bratkovic I. Expression, purification and structural studies of a short antimicrobial peptide. Biochimica Et Biophysica Acta-Biomembranes. 2009;1788:314–323. doi: 10.1016/j.bbamem.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 51.Engelman DM, Fisher LE. High-yield synthesis and purification of an alpha-helical transmembrane domain. Analytical Biochemistry. 2001;293:102–108. doi: 10.1006/abio.2001.5122. [DOI] [PubMed] [Google Scholar]
- 52.Seong BL, Choi SI, Song HW, Moon JW. Recombinant enterokinase light chain with affinity tag: expression from Saccharomyces cerevisiae and its utilities in fusion protein technology. Biotechnology and Bioengineering. 2001;75:718–724. doi: 10.1002/bit.10082. [DOI] [PubMed] [Google Scholar]
- 53.Parks TD, Leuther KK, Howard ED, Johnston SA, Dougherty WG. Release of proteins and peptides from fusion proteins using a recombinant plant-virus proteinase. Analytical Biochemistry. 1994;216:413–417. doi: 10.1006/abio.1994.1060. [DOI] [PubMed] [Google Scholar]
- 54.Sticha KRK, Sieg CA, Bergstrom CP, Hanna PE, Wagner CR. Overexpression and large-scale purification of recombinant hamster polymorphic arylamine N-acetyltransferase as a dihydrofolate reductase fusion protein. Protein Expression and Purification. 1997;10:141–153. doi: 10.1006/prep.1997.0734. [DOI] [PubMed] [Google Scholar]
- 55.Vothknecht UC, Kannangara CG, von Wettstein D. Expression of catalytically active barley glutamyl tRNA(Glu) reductase in Escherichia coli as a fusion protein with glutathione S-transferase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9287–9291. doi: 10.1073/pnas.93.17.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson AS, Bane SE, Velasquez JE. Expression and purification of milligram levels of inactive G-protein coupled receptors in E. coli. Protein Expression and Purification. 2007;52:348–355. doi: 10.1016/j.pep.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson AS, O’Malley MA, Naranjo AN, Lazarova T. Analysis of adenosine A(2)a receptor stability: effects of ligands and disulfide bonds. Biochemistry. 2010;49:9181–9189. doi: 10.1021/bi101155r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson AS, O’Malley MA, Lazarova T, Britton ZT. High-level expression in Saccharomyces cerevisiae enables isolation and spectroscopic characterization of functional human adenosine A(2)a receptor. Journal of Structural Biology. 2007;159:166–178. doi: 10.1016/j.jsb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stevens RC, Xu FX, Wu HX, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V. Structure of an agonist-bound human A(2A) adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren H, Yu D, Ge B, Cook B, Xu Z, Zhang SG. High-level production, solubilization and purification of synthetic human GPCR chemokine receptors CCR5, CCR3, CXCR4 and CX3CR1. PLoS ONE. 2009;4(2):e4509. doi: 10.1371/journal.pone.0004509. http://dx.doi.org/10.1371/journal.phone.0004509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiener MC, Vergis JM. The variable detergent sensitivity of proteases that are utilized for recombinant protein affinity tag removal. Protein Expression and Purification. 2011;78:139–142. doi: 10.1016/j.pep.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baneres JL, Mesnier D, Martin A, Joubert L, Dumuis A, Bockaert J. Molecular characterization of a purified 5-HT4 receptor – a structural basis for drug efficacy. Journal of Biological Chemistry. 2005;280:20253–20260. doi: 10.1074/jbc.M412009200. [DOI] [PubMed] [Google Scholar]
- 63.Cross TA, Hu J, Qin H, Li C, Sharma M, Gao FP. Structural biology of transmembrane domains: Efficient production and characterization of transmembrane peptides by NMR. Protein Science. 2007;16:2153–2165. doi: 10.1110/ps.072996707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Malley MA, Naranjo AN, Lazarova T, Robinson AS. Analysis of adenosine A(2)a receptor stability: effects of ligands and disulfide bonds. Biochemistry. 2010;49:9181–9189. doi: 10.1021/bi101155r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tate CG, Magnani F, Shibata Y, Serrano-Vega MJ. Co-evolving stability and conformational homogeneity of the human adenosine A(2a) receptor. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10744–10749. doi: 10.1073/pnas.0804396105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naider F, Cohen LS, Arshava B, Estephan R, Englander J, Kim H, Hauser M, Zerbe O, Ceruso M, Becker JM. Expression and biophysical analysis of two double-transmembrane domain-containing fragments from a yeast G protein-coupled receptor. Biopolymers. 2008;90:117–130. doi: 10.1002/bip.20950. [DOI] [PubMed] [Google Scholar]
- 67.Neumoin A, Cohen LS, Arshava B, Tantry S, Becker JM, Zerbe O, Naider F. Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophysical Journal. 2009;96:3187–3196. doi: 10.1016/j.bpj.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park HS, Kim C, Kang YK. Preferred conformations of cyclic Ac-Cys-Pro-Xaa-Cys-NHMe peptides: a model for chain reversal and active site of disulfide oxidoreductase. Biophysical Chemistry. 2003;105:89–104. doi: 10.1016/s0301-4622(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 69.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 70.Stoscheck CM. Quantitation of Protein. Methods in Enzymology. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]