

Abstract
The striatum is critically involved in the selection of appropriate actions in a constantly changing environment. The spiking activity of striatal spiny projection neurons (SPNs), driven by extrinsic glutamatergic inputs, is shaped by local GABAergic and cholinergic networks. For example, it is well established that different types of GABAergic interneurons, activated by extrinsic glutamatergic and local cholinergic inputs, mediate powerful feedforward inhibition of SPN activity. In this study, using mouse striatal slices, we show that glutamatergic and cholinergic inputs exert direct inhibitory regulation of SPN activity via activation of metabotropic glutamate receptors (mGluRs) and muscarinic acetylcholine receptors. While pressure ejection of the group I mGluR (mGluR1/5) agonist DHPG [(S)-3,5-dihydroxyphenylglycine] equally engages both mGluR1 and mGluR5 subtypes, the mGluR-dependent component of IPSCs elicited by intrastriatal electrical stimulation is almost exclusively mediated by the mGluR1 subtype. Ca2+ release from intracellular stores specifically through inositol 1,4,5-triphospahte receptors (IP3Rs) and not ryanodine receptors (RyRs) mediates this form of inhibition by gating two types of Ca2+-activated K+ channels (i.e., small-conductance SK channels and large-conductance BK channels). Conversely, spike-evoked Ca2+ influx triggers Ca2+ release solely through RyRs to generate SK-dependent slow afterhyperpolarizations, demonstrating functional segregation of IP3Rs and RyRs. Finally, IP3-induced Ca2+ release is uniquely observed in SPNs and not in different types of interneurons in the striatum. These results demonstrate that IP3-mediated activation of SK and BK channels provides a robust mechanism for glutamatergic and cholinergic inputs to selectively suppress striatal output neuron activity.
Introduction
The output from the striatum to downstream basal ganglia structures plays a critical role in the selection and planning of actions (Graybiel et al., 1994). Medium-sized, GABAergic spiny projection neurons (SPNs), which constitute ∼95% of striatal neurons, provide the sole output of the striatum. In vivo intracellular recordings have revealed that membrane potential of SPNs shifts from a quiescent hyperpolarized DOWN state (approximately −70 to −90 mV) to a depolarized UP state (approximately −50 to −60 mV), during which action potential (AP) firing occurs (Wilson and Kawaguchi, 1996; Stern et al., 1998). Coordinated excitatory inputs mainly from the cerebral cortex drive the transition to the UP state, which lasts for hundreds of milliseconds to seconds before switching back to the DOWN state. During the UP state, the balance between depolarizing cortical and thalamic inputs and hyperpolarizing voltage-gated K+ conductances, together with inputs from local GABAergic and cholinergic networks, are thought to determine whether and when SPNs fire APs (Surmeier et al., 2007; Tepper et al., 2008). Single-unit recordings in behaving animals have shown that SPNs differentially exhibit increases and decreases, or pauses, in firing before and during performance of actions (Hikosaka et al., 1989; Jin and Costa, 2010; Krause et al., 2010). However, the underlying mechanisms of the pauses and whether they can occur in the UP state or represent transitions to the DOWN state remain unclear.
SPN firing is also shaped by two types of Ca2+-activated K+ channels, small-conductance SK channels and large-conductance BK channels (Pineda et al., 1992; Hopf et al., 2010). Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) triggered by each AP activates these channels, thereby generating afterhyperpolarizations (AHPs) that control AP duration and interspike intervals. AP-evoked Ca2+ transients can be further amplified by a process called Ca2+-induced Ca2+ release (CICR) via two types of Ca2+-sensitive receptors [i.e., inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs)] located on intracellular Ca2+ stores (Berridge, 1998). Furthermore, neurotransmitter inputs coupled to G-protein-dependent generation of second messengers elicit IP3R- and/or RyR-dependent Ca2+ release to activate SK and BK channels in a variety of neurons in the CNS (Fiorillo and Williams, 2000; Morikawa et al., 2003; Gulledge and Stuart, 2005; Canepari and Ogden, 2006; Hagenston et al., 2008; Power and Sah, 2008; El-Hassar et al., 2011). Although group I metabotropic glutamate receptors (mGluRs) (mGluR1/5) and the M1-subtype of muscarinic acetylcholine receptors (mAChRs), which are coupled to the phospholipase C (PLC)–IP3 cascade, are expressed in virtually all SPNs (Yan et al., 2001; Gubellini et al., 2004), the role of Ca2+ store-dependent activation of SK and BK channels in regulating SPN activity has yet to be determined.
In this study, using mouse striatal slices, we show that the mGluR/mAChR → IP3R → SK/BK channel pathway produces powerful suppression of depolarization-evoked SPN firing, while the AP → RyR → SK channel pathway dominates the slow component of AHPs. Furthermore, our data showing the absence of IP3-induced Ca2+ release in striatal interneurons imply that IP3 signaling plays a privileged role in glutamatergic and cholinergic input-dependent inhibitory regulation of striatal output neurons.
Materials and Methods
Animals.
Male C57BL/6J mice (3–4 weeks of age) were obtained from The Jackson Laboratory and were housed under a 12 h light/dark cycle. Food and water were available ad libitum. All animal procedures were approved by the University of Texas Institutional Animal Care and Use Committee.
Electrophysiology.
Mice were killed by cervical dislocation under isoflurane anesthesia, and oblique horizontal slices (∼20–30°, 200–250 μm) containing the dorsal striatum were cut in an ice-cold solution containing the following (in mm): 205 sucrose, 2.5 KCl, 1.25 NaH2PO4, 7.5 MgCl2, 0.5 CaCl2, 10 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2 (∼300 mOsm/kg) and incubated >1 h at 35°C in a solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2, pH 7.4, ∼295 mOsm/kg. Recordings were made at 34–35°C in the same solution perfused at ∼2.5 ml/min.
Cells were visualized using an upright microscope (Olympus) with infrared/oblique illumination optics. Whole-cell recordings were made with borosilicate glass pipettes (2.2–2.8 MΩ) filled with internal solution containing the following (in mm): 115 K-gluconate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2-phosphocreatine, pH 7.2, ∼285 mOsm/kg, unless indicated otherwise. Series resistance was continuously monitored but left uncompensated during voltage-clamp recording experiments. Recordings were discarded if the series resistance increased beyond 20 MΩ. Bridge balance was adjusted periodically (approximately every 2–3 min) during current-clamp recording experiments. The membrane potential was corrected for a liquid junction potential of −7 mV. A Multiclamp 700B amplifier (Molecular Devices) and AxoGraph X software (AxoGraph Scientific) were used to record and collect data (filtered at 0.5–2 kHz and digitized at 1–5 kHz for voltage-clamp recordings; filtered at 10 kHz and digitized at 20 kHz for current-clamp recordings). The majority of experiments were performed in the presence of picrotoxin (100 μm) to block GABAA inputs.
For pressure ejection, patch pipettes (∼2 μm tip diameter) were filled with (S)-3,5-dihydroxyphenylglycine (DHPG) (100 μm) or muscarine chloride (100 μm), and pressure of 20 psi was applied using a TooheySpritzer Pressure System IIe to eject the drug. Synaptic responses were evoked with a bipolar tungsten electrode in the presence of DNQX (20 μm) and picrotoxin (100 μm) to block AMPA and GABAA receptors and slices were preincubated with MK-801 (10 μm) to block NMDA receptors.
Flash photolysis.
Cells were dialyzed with caged IP3 (400 μm) through the whole-cell pipette for 15–20 min after break-in. A brief UV flash (∼1 ms) was applied with a xenon arc lamp driven by a photolysis system (Cairn Research) to rapidly photolyze caged IP3 (∼3 ms) (Walker et al., 1989). The UV flash was focused through a 60× objective onto a 350 μm area surrounding the recorded cell. The Cairn system has the capacity to vary the intensity of UV flash, which was measured at the tip of the objective (expressed in microjoules). The degree of photolysis of caged compounds is known to be proportional to the UV flash intensity (McCray et al., 1980). The maximal UV flash intensity was ∼180–250 μJ with our system, depending on the age of the xenon arc lamp at the time of experiments.
Ca2+ imaging.
Fluorescence imaging of [Ca2+]i was mostly done using Fluo-4FF (Invitrogen) as Ca2+ indicators. Some of the imaging experiments were performed with Fluo-4 or Fluo-5F. These Ca2+ indicators were loaded into the cell via the whole-cell pipette. Images were taken at 15 Hz using the Olympus Disk Spinning Unit Imaging System. After raw fluorescent signals from selected regions of interest (ROIs) were background subtracted at each time point, Ca2+ transients were expressed as ΔF/F0 = (F(t) − F0)/F0, where F0 was determined as an average of F(t) over a 1 s baseline period. Brief artifacts accompanying UV flashes are omitted from the fluorescence traces in flash photolysis experiments.
Drugs.
Apamin, iberiotoxin, cyclopiazonic acid, DHPG, (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385), MPEP, [S-(R*,R*)]-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](cyclohexylmethyl)phosphinic acid (CGP54626), DNQX, and picrotoxin were obtained from Tocris Biosciences. TTX was obtained from Alomone Labs. Fluo-4, Fluo-5F, and Fluo-4FF were purchased from Invitrogen. Caffeine, heparin sodium salt (from porcine intestinal mucosa), and muscarine chloride were obtained from Sigma RBI. Caged IP3 was a generous gift from Dr. Kamran Khodakhah (Albert Einstein College of Medicine, Bronx, NY).
Data analysis.
AP parameters were obtained from the first APs evoked during depolarizing current injections. AP threshold was determined as the point where the first derivative of the membrane potential (dv/dt) exceeded 20 V/s. AP half-width was measured at the potential halfway between AP threshold and peak. AHP amplitude was determined as the amount of hyperpolarization from AP threshold.
Data are expressed as mean ± SEM. Statistical significance was determined by Student's t test or ANOVA followed by Bonferroni post hoc test. The difference was considered significant at p < 0.05.
Results
Transient mGluR and mAChR stimulation inhibits SPN activity and activates SK and BK channels
To examine the effects of transient mGluR and mAChR activation on the firing of SPNs, whole-cell current-clamp recordings of these neurons were made in the dorsal striatum and AP firing was evoked by depolarizing current injections (200–350 pA; adjusted to produce ∼5–7 Hz firing). Local pressure ejection (500 ms) of the group I mGluR (mGluR1/5) agonist DHPG (100 μm in a pipette placed ∼50–80 μm from the recorded cell) produced a transient hyperpolarization and a pause in firing lasting 1.85 ± 0.11 s, followed by a delayed increase in the firing frequency, in eight cells tested (Fig. 1A). When DHPG application was made at the resting membrane potential (approximately −80 to −95 mV) in six of these eight cells, no measurable response was observed. Pressure ejection of muscarine (100 μm; 500 ms), a broad-spectrum mAChR agonist, also caused membrane hyperpolarization and a pause in SPN firing (1.38 ± 0.15 s; n = 5), followed by gradual acceleration of firing (Fig. 1B).
Figure 1.

Transient mGluR and mAChR activation produces pauses in SPN firing and activates SK and BK channels. A, B, Example traces depicting DHPG- and muscarine-induced pauses in SPN firing evoked by depolarizing current injections. Local pressure ejection (500 ms) of DHPG (100 μm) (A) or muscarine (100 μm) (B) was made at the time indicated. C, Example traces and summary graph showing the effects of LY367385 (75 nm) and MPEP (50 nm) on IDHPG (Vh = −57 mV). D, Example traces illustrating the effect of atropine (2 μm) on muscarine-evoked currents. E, Representative traces and summary graph showing that IDHPG was suppressed by hyperpolarization to −87 mV (t(8) = 9.24, ***p < 0.0001; paired t test). F, Example traces and summary graph depicting the effects of apamin (100 nm) and IbTX (100 nm) on IDHPG. G, Left, Traces of the apamin-sensitive SK-dependent component and the IbTX-sensitive BK-dependent component of IDHPG shown in F. These traces were obtained by subtracting traces before and after apamin/IbTX applications. Right, Summary graphs plotting the fraction of charge carried by SK- and BK-dependent components (t(4) = 9.68, ***p < 0.001; paired t test) and the latency to peak after the onset of DHPG pressure ejection. Error bars indicate SEM.
In voltage clamp (Vh = −57 mV), pressure ejection of DHPG (100 μm, 500 ms, once a minute) produced transient outward currents (∼1–2 s), which were followed by prolonged inward currents (∼20–50 s) (Fig. 1C). Bath perfusion of the mGluR1 antagonist LY367385 (75 nm) and the mGluR5 antagonist MPEP (50 nm) suppressed DHPG-induced outward currents (termed IDHPG) by 47 ± 9 and 40 ± 8%, respectively, in six cells where these two antagonists were sequentially applied (three cells: LY367385 → MPEP + LY367385; three cells: MPEP → LY367385 + MPEP), indicating that these two mGluR subtypes make relatively equal contributions. LY367385 and MPEP similarly inhibited DHPG-induced delayed inward currents by 44 ± 7 and 51 ± 6%, respectively. Pressure ejection of muscarine also produced transient outward currents followed by inward currents, which were abolished by the general mAChR antagonist atropine (2 μm) (n = 3; Fig. 1D).
Group I mGluRs and mAChRs are coupled to Ca2+-activated SK channels in many different types of neurons in other brain areas (Fiorillo and Williams, 2000; Morikawa et al., 2003; Gulledge and Stuart, 2005; Hagenston et al., 2008; Power and Sah, 2008; El-Hassar et al., 2011) and to BK channels in cerebellar Purkinje neurons (Canepari and Ogden, 2006). Indeed, IDHPG amplitude was dramatically reduced when the holding potential was hyperpolarized to −87 mV (Fig. 1E), consistent with the involvement of K+ conductance. Thus, we examined the effects of the SK channel blocker apamin (100 nm) and the BK channel blocker iberiotoxin (IbTX) (100 nm) on IDHPG. Apamin and IbTX reduced the peak amplitude of IDHPG by 75 ± 4 and 26 ± 3%, respectively, and combined application of apamin and IbTX eliminated IDHPG without affecting delayed inward currents (n = 5; three cells: apamin → IbTX + apamin; two cells: IbTX → apamin + IbTX) (Fig. 1F). Accordingly, the apamin-sensitive SK-dependent component accounted for the majority (80 ± 4%) of charge transfer mediating IDHPG (Fig. 1G). SK- and BK-dependent components reached their peak amplitude with similar latency after DHPG application, suggesting that both types of Ca2+-gated channels may be activated by the same Ca2+ signals. Therefore, transient activation of mGluR1/5 and mAChRs activates Ca2+-sensitive SK and BK channels to regulate SPN excitability. DHPG- and muscarine-induced delayed inward currents were also depressed by membrane hyperpolarization (Fig. 1E), suggesting that they result from closure of K+ conductance.
IP3R-dependent Ca2+ release from intracellular stores mediates SPN inhibition
When intracellular Ca2+ ([Ca2+]i) was monitored at the soma with the low-affinity (Kd = 9.7 μm) indicator Fluo-4FF (100 μm), local pressure ejection of DHPG produced a rise in [Ca2+]i that coincided with IDHPG (n = 7) (Fig. 2A). We next made rapid application of IP3 directly into the cytosol using UV photolysis of caged IP3 (400 μm; dialyzed into the cytosol through the whole-cell pipette). Photolytic IP3 application produced a transient rise in [Ca2+]i and an outward current (termed IIP3) in a concentration-dependent manner when three different UV flash intensities (50, 100, and 200 μJ) were used to vary the amount of IP3 released (n = 3) (Fig. 2B). Furthermore, intracellular application of heparin (1 mg/ml; whole-cell dialysis), an IP3R antagonist (Ghosh et al., 1988), or bath application of cyclopiazonic acid (CPA) (20 μm), which depletes intracellular Ca2+ stores (Seidler et al., 1989), completely abolished both IDHPG and IIP3 without affecting DHPG-induced delayed inward currents (Fig. 2C–F; recordings made with standard internal solution containing 25 μm EGTA). CPA also eliminated DHPG- and muscarine-induced suppression of SPN firing (n = 4 and 3, respectively) (Fig. 2G).
Figure 2.
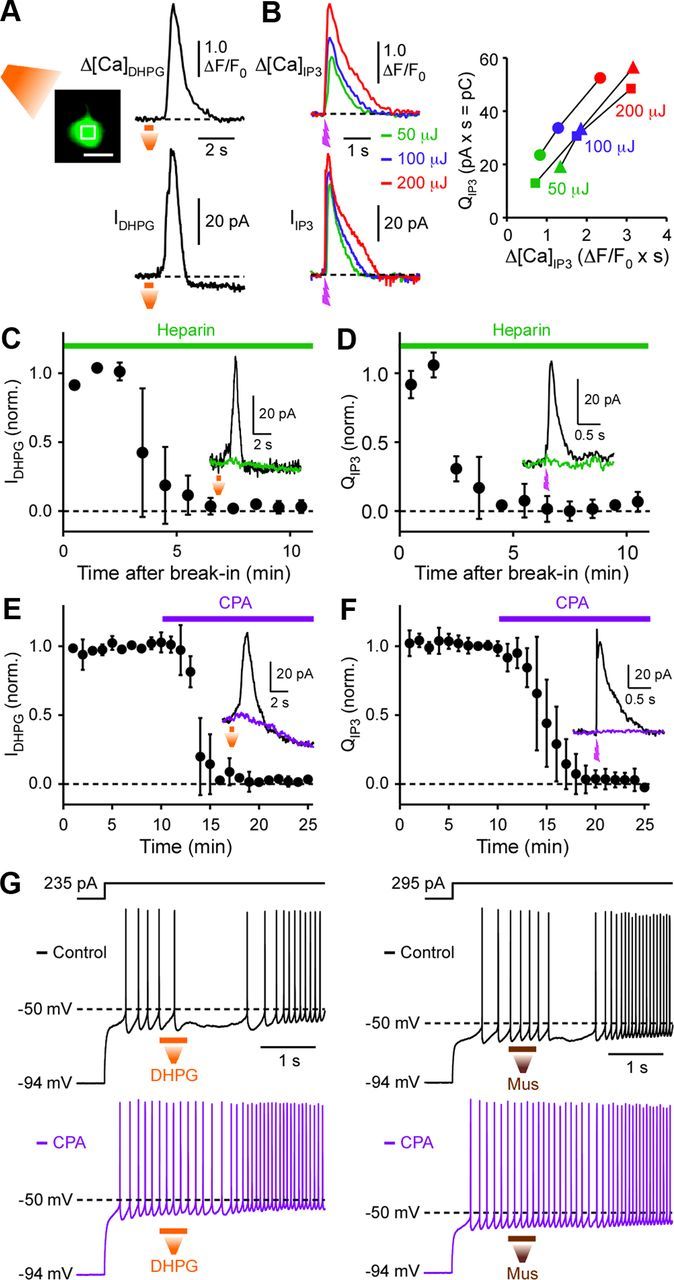
IP3R-dependent Ca2+ release from intracellular stores mediates mGluR/mAChR-induced SPN inhibition. A, Representative experiment imaging DHPG-evoked Ca2+ transient. Fluorescence change, monitored at the ROI indicated on the left (scale bar: 10 μm), and an outward current evoked by pressure ejection of DHPG (100 μm; in a pipette placed ∼60 μm from the recorded cell) are shown in an SPN loaded with Fluo-4FF (100 μm). Note that DHPG-evoked delayed inward current, which is independent of Ca2+ rise, outlasts the fluorescence change. B, Example experiment monitoring Ca2+ transients and outward currents elicited by flash photolysis of caged IP3 (400 μm) using three different UV flash intensities (50, 100, and 200 μJ). UV flashes were applied at the time indicated. Fluorescence changes were measured at the ROI placed in the soma as in A (cell image not shown). Right, The total charge transfer of IIP3 (termed QIP3) exhibited approximately linear relationship with the total fluorescence change (integrated over time) in three cells tested for different UV intensities. Fluo-4FF (100 μm) was used to monitor [Ca2+]i in these three cells. C–F, Summary time graphs showing that cytosolic application of heparin (1 mg/ml) or bath application of CPA (20 μm) abolished outward currents elicited by DHPG (C, E) and IP3 (D, F). Traces from a sample experiment are shown as an inset in each panel. G, Example traces depicting that CPA suppressed the pauses in SPN firing produced by pressure ejection of DHPG (left) and muscarine (right). Note that DHPG and muscarine still caused delayed acceleration in firing after CPA treatment.
We further made detailed analyses of IIP3, since flash photolysis of caged IP3 enables much more quantitative and temporally precise application of IP3 compared with local pressure ejection of DHPG or muscarine. IIP3 displayed concentration-dependent (i.e., UV flash intensity-dependent) increases in total charge transfer (termed QIP3) and half-width and a reduction in onset latency, accompanied by the emergence of an initial transient component with higher UV flash intensities (100–200 μJ) (n = 5) (Fig. 3A–C). We next replaced EGTA in the internal solution with the same concentration (25 μm) of BAPTA (n = 5). Although these two Ca2+ buffers have similar steady-state affinities for Ca2+ (Kd = ∼0.2 μm), thus similarly affecting baseline [Ca2+]i, BAPTA is much more effective in disrupting transient localized Ca2+ signaling processes due to its faster Ca2+ binding rate (∼40–150 times) compared with EGTA (Fakler and Adelman, 2008; Eggermann et al., 2011). BAPTA (25 μm) reduced QIP3 and half-width of IIP3 (measured at half the peak amplitude of the slow component) evoked by the highest UV intensity (200 μJ); however, it had no significant effect on the onset latency or the peak amplitudes of the initial or slow components. Increasing the BAPTA concentration to 1 mm completely eliminated IIP3 (n = 4), whereas measurable IIP3 was still observed with 1 mm EGTA (n = 4) (Fig. 3D). These data suggest that localized Ca2+ signaling is involved in the generation of IIP3, presumably at the level of CICR among neighboring IP3Rs that acts to sustain IP3-induced Ca2+ rises.
Figure 3.

IP3 triggers BAPTA-sensitive localized Ca2+ signaling. A, Representative traces of IIP3 using three different UV intensities (50, 100, and 200 μJ) in cells filled with EGTA (25 μm) and BAPTA (25 μm). UV flashes were applied at the time indicated. B, Summary plots showing the effects of BAPTA on QIP3 (Ca2+ buffer type: F(1,16) = 6.22, p < 0.05; Ca2+ buffer concentration: F(2,16) = 36.2, p < 0.0001; Ca2+ buffer type by concentration: F(2,16) = 10.1, p < 0.01) and IIP3 half-width (Ca2+ buffer concentration: F(2,16) = 37.8, p < 0.0001; Ca2+ buffer type by concentration: F(2,16) = 18.9, p < 0.0001; mixed two-way ANOVA). ***p < 0.001 versus EGTA (Bonferroni's post hoc test). C, Summary graphs plotting three parameters of IIP3 (onset latency, peak amplitude of the initial component, and peak amplitude of the late component) using three different UV flash intensities in cells filled with EGTA (25 μm) or BAPTA (25 μm). These parameters were not significantly affected by BAPTA. D, Summary bar graph showing the effects of two different concentrations (25 μm and 1 mm) of EGTA and BAPTA on QIP3 (200 μJ UV intensity) (Ca2+ buffer type: F(1,14) = 11.7, p < 0.01; Ca2+ buffer concentration: F(1,14) = 32.0, p < 0.0001; two-way ANOVA). **p < 0.01 versus EGTA (Bonferroni's post hoc test). Error bars indicate SEM.
When apamin and IbTX were tested on IIP3 (200 μJ UV intensity), SK- and BK-dependent components accounted for 78 ± 7 and 22 ± 7% of QIP3, respectively (n = 5; three cells: apamin → IbTX + apamin; two cells: IbTX → apamin + IbTX) (Fig. 4A). The initial fast component was mostly BK-mediated, suggesting that certain population of BK channels may have more direct access to IP3R-mediated Ca2+ release. Small IP3-induced inward currents were observed in the presence of apamin and IbTX. These inward currents became larger with membrane hyperpolarization to −87 mV (12.9 ± 1.6 pA at −57 mV vs 26.4 ± 2.0 pA at −87 mV, n = 7; t(6) = 7.60, p < 0.001, paired t test), suggesting the involvement of a Ca2+-activated cationic conductance (Fleig and Penner, 2004). Stable IP3-induced currents were observed with repeated IP3 applications (once a minute) even when recorded cells were constantly held at −87 mV (n = 5), indicating that Ca2+ stores can be replenished without VGCC-mediated Ca2+ influx (i.e., priming) in SPNs (Hong and Ross, 2007; Wu et al., 2011).
Figure 4.
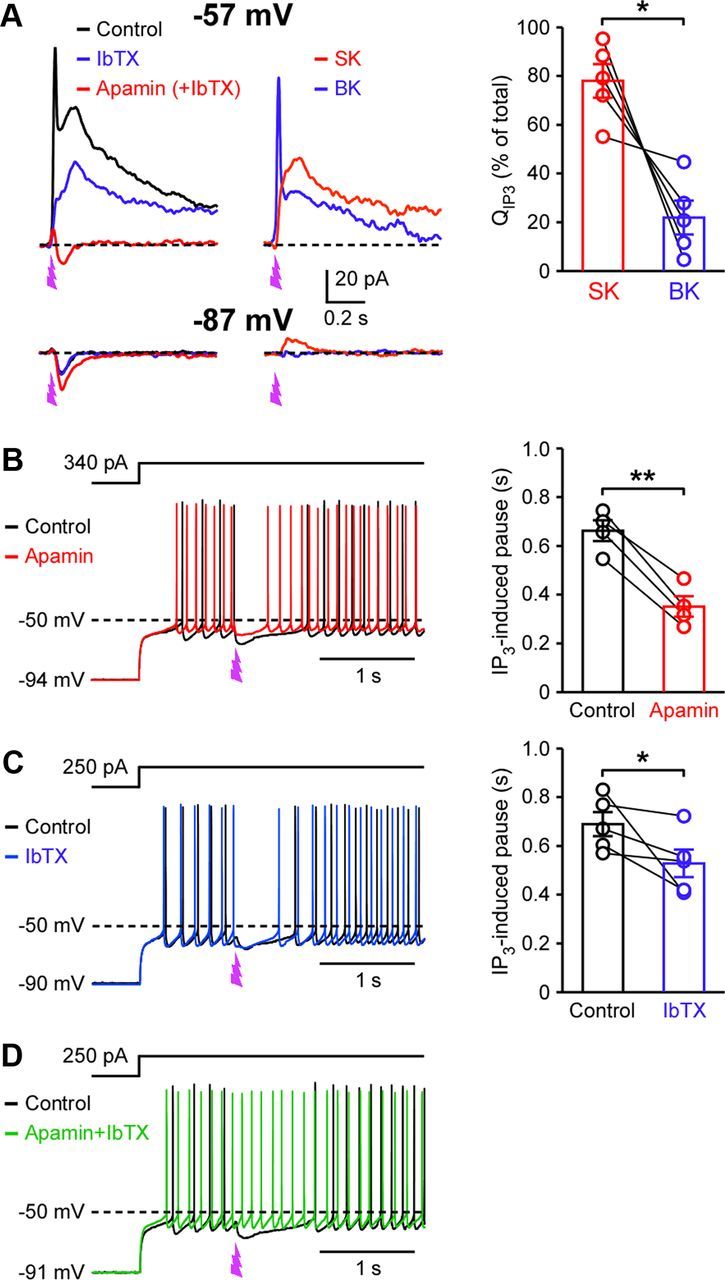
IP3-induced activation of SK and BK channels causes SPN inhibition. A, Example traces showing the effects of apamin (100 nm) and IbTX (100 nm) on IIP3 (200 μJ UV intensity) measured at −57 and −87 mV. Traces of SK-and BK-dependent components were obtained by subtracting traces before and after apamin/IbTX applications. Note the absence of BK component at −87 mV, reflecting the voltage dependence of BK activation (Fakler and Adelman, 2008). Right, Summary graph plotting the fraction of charge carried by SK- and BK-dependent components at −57 mV (t(4) = 4.04, p < 0.05; paired t test). B, C, Example traces depicting the effects of apamin (B) and IbTX (C) on IP3-induced pause in SPN firing. The duration of IP3-induced pause before and after application of apamin (t(3) = 9.06; p < 0.01) or IbTX (t(4) = 2.27, p < 0.05; paired t test) is plotted on the right. D, Example traces showing that combined application of apamin and IbTX eliminated the IP3-induced pause. *p < 0.05, **p < 0.01. Error bars indicate SEM.
In current clamp, photolytic IP3 application produced a pause in SPN firing (0.68 ± 0.03 s; n = 9). Apamin and IbTX reduced the duration of this IP3-induced pause by 47 ± 5% (n = 4) and 22 ± 9% (n = 5), respectively, while combined application of apamin and IbTX eliminated the pause (n = 3) (Fig. 4B–D). IP3 application produced small depolarizations at the resting membrane potential (1.8 ± 0.1 mV; n = 10), consistent with the cationic conductance described above.
Together, these data indicate that IP3R-mediated Ca2+ release from intracellular stores, causing activation of SK and BK channels, is responsible for mGluR- and mAChR-induced inhibition of SPNs.
Synaptic stimulation triggers mGluR- and mAChR-mediated IPSCs
We next examined synaptic responses (AMPA, NMDA, and GABAA receptors blocked) elicited by intrastriatal stimulation using a bipolar stimulating electrode (∼600 μm tip separation) placed caudal to the recorded cell (∼500–600 μm away) (Fig. 5A). This likely causes stimulation of a large number of inputs arising from all available sources, both intrinsic fibers from striatal neurons and extrinsic inputs from other structures, including those from the cortex via antidromic stimulation (Cowan and Wilson, 1994). Although single stimuli (500 μs; ∼1–2 mA) failed to produce measurable responses, trains of 2–10 stimuli at 50 Hz evoked slow IPSC-like outward currents (Vh = −57 mV) (Fig. 5B). A train of 5 stimuli produced a near-saturating response in four cells tested for various train lengths (1–10 stimuli), and hence were used in the following experiments. IPSC-like outward currents thus evoked, which peaked at 0.47 ± 0.03 s after the onset of stimulus train and had a half-width of 0.58 ± 0.03 s (n = 14), were largely suppressed by CPA (20 μm; n = 4) and also by combined application of LY367385 (mGluR1 antagonist; 75 nm), MPEP (mGluR5 antagonist; 50 nm), and atropine (mAChR antagonist; 2 μm) (n = 10) (Fig. 5C,D), indicating that they mostly represent Ca2+ store-dependent mGluR/mAChR IPSCs. Small outward currents (∼10 pA or less) routinely remained after application of CPA or the mGluR/mAChR antagonist mixture. The outward currents in the presence of antagonist mixture were not suppressed by subsequent application of CPA (n = 3) or the GABAB receptor antagonist CGP54626 (1 μm; n = 3), and thus their identity is currently unknown. On average, mGluR- and mAChR-mediated components made equal contributions to IPSCs, while mGluR1 accounted for the vast majority of the mGluR-dependent component. All 10 cells examined had both mGluR- and mAChR-mediated components, although the relative size varied from cell to cell. These results demonstrate that synaptic stimulation of glutamatergic and cholinergic inputs produces Ca2+ store-dependent inhibition of SPNs. Delayed EPSCs (∼10 pA) sensitive to atropine were observed in 3 of 14 cells.
Figure 5.

Intrastriatal stimulation elicits Ca2+ store-dependent mGluR1/mAChR IPSCs. A, Image of an oblique horizontal striatal slice depicting the placement of a bipolar stimulating electrode (tips inside large circles) with respect to the recorded cell (inside the small circle). Ctx, Cortex; CC, corpus callosum. B, Example traces illustrating synaptic responses produced by trains of 1–10 intrastriatal stimuli (50 Hz). The arrow indicates the onset of stimulus trains. Stimulus artifacts are omitted from these traces for clarity. Right, Summary graph plotting the IPSC amplitude evoked by trains of 1–10 stimuli in four cells. C, Representative traces showing the effect of CPA (20 μm). D, Example traces and summary graph showing the effects of atropine (2 μm), LY367385 (75 nm), and MPEP (50 nm) on IPSCs. Percentage inhibition was calculated after removing the current insensitive to the combined application of these antagonists in each cell. Of 10 cells examined for all three antagonists, LY367385 and MPEP were applied together at the same time in 5 cells and sequentially in the remaining 5 cells. The order of drug application was varied in these experiments. Error bars indicate SEM.
RyR-dependent, but IP3R-independent, Ca2+ release contributes to slow AHPs
It has been shown that AP-evoked Ca2+ influx activates BK and SK channels, thereby producing fast and slow AHPs (fAHPs and sAHPs) in SPNs (Pineda et al., 1992; Hopf et al., 2010). In line with these studies, IbTX increased the AP half-width and suppressed fAHPs (peaked at ∼2–3 ms after AP onset) without affecting the interspike interval (n = 6), indicating the role of BK channels in AP repolarization, whereas apamin depressed sAHPs (peaked at ∼20–40 ms after AP onset) and decreased the interspike interval (n = 6) (Fig. 6A,B; Table 1). CICR from intracellular stores triggered by AP-evoked Ca2+ influx may contribute to sAHPs (Berridge, 1998). We found that Ca2+ store depletion with CPA dramatically suppressed sAHPs and decreased the interspike interval (n = 8), an effect that was occluded by previous application of apamin (n = 4) (Fig. 6C, Table 1). Therefore, CICR contributes to SK-dependent sAHPs in SPNs. Apamin and CPA also accelerated the initial ramp-like depolarizations preceding the first AP during depolarizing current injections, demonstrating that CICR-dependent SK channel activation, in addition to activation of voltage-gated K+ conductances (Nisenbaum and Wilson, 1995), is capable of delaying the firing response to depolarizing inputs.
Figure 6.

Ca2+ release from intracellular stores regulates SK-dependent slow AHPs. Example traces showing the effects of IbTX (100 nm; A), apamin (100 nm; B), and CPA (20 μm; C) on SPN firing. Right, First APs evoked by depolarizing current injections are shown on expanded timescales, with the AP onset aligned, to illustrate the effects of these drugs on the AP shape and two AHP components.
Table 1.
Effects of IbTX, apamin, and CPA on electrophysiological properties of SPNs
| IbTX (n = 6) |
Apamin (n = 6) |
CPA (n = 8) |
CPA in apamin (n = 4) |
|||||
|---|---|---|---|---|---|---|---|---|
| Control | IbTX | Control | Apamin | Control | CPA | Apamin | CPA | |
| AP threshold (mV) | −46.6 ± 1.5 | −46.8 ± 1.7 | −47.6 ± 1.4 | −48.2 ± 1.5 | −47.3 ± 1.6 | −47.7 ± 1.6 | −48.2 ± 1.2 | −49.5 ± 1.1 |
| AP half-width (ms) | 0.80 ± 0.02 | 1.05 ± 0.05** | 0.72 ± 0.02 | 0.73 ± 0.02 | 0.75 ± 0.02 | 0.76 ± 0.02 | 0.74 ± 0.02 | 0.75 ± 0.02 |
| AP decay rate (ms) | 0.38 ± 0.02 | 0.59 ± 0.03** | 0.33 ± 0.01 | 0.33 ± 0.01 | 0.35 ± 0.01 | 0.36 ± 0.01 | 0.34 ± 0.01 | 0.34 ± 0.00 |
| Latency to first AP (ms) | 567 ± 34 | 510 ± 23 | 560 ± 25 | 410 ± 21** | 551 ± 37 | 401 ± 51** | 437 ± 17 | 458 ± 22 |
| Interspike interval (ms) | 175 ± 6 | 174 ± 10 | 171 ± 8 | 101 ± 2*** | 159 ± 7 | 116 ± 11** | 104 ± 1 | 108 ± 5 |
| fAHP amplitude (mV) | 9.7 ± 0.6 | 4.0 ± 0.8*** | 10.2 ± 1.1 | 9.8 ± 1.1 | 11.5 ± 0.4 | 10.9 ± 0.4 | 11.1 ± 1.0 | 11.1 ± 1.1 |
| sAHP amplitude (mV) | 14.5 ± 0.5 | 14.2 ± 0.4 | 14.5 ± 0.4 | 11.3 ± 0.6*** | 14.4 ± 0.5 | 12.9 ± 0.4** | 12.0 ± 0.3 | 11.9 ± 0.4 |
| RMP (mV) | −87.1 ± 1.6 | −87.6 ± 1.3 | −89.8 ± 1.1 | −90.7 ± 1.3 | −89.4 ± 1.7 | −90.1 ± 2.0 | −91.3 ± 0.9 | −91.8 ± 1.0 |
| Input resistance (MΩ) | 61.0 ± 9.6 | 65.6 ± 11.1 | 53.2 ± 5.2 | 54.5 ± 5.4 | 56.1 ± 5.7 | 56.8 ± 5.5 | 53.3 ± 8.4 | 53.6 ± 8.5 |
*p < 0.05;
**p < 0.01;
***p < 0.001 (paired t test).
We next measured the currents underlying sAHPs in voltage clamp, as has been done previously in midbrain dopamine neurons and striatal cholinergic interneurons (Goldberg and Wilson, 2005; Cui et al., 2007). Here, 2 ms depolarizing pulses to −7 mV from a holding potential of −57 mV were applied to evoke unclamped APs, which resulted in tail outward currents (IsAHP) lasting ∼200 ms (Fig. 7A). We calculated the time integral of IsAHP (i.e., the total charge transfer) after removing a 10 ms window following the 2 ms depolarizing pulse. QsAHP thus obtained was largely suppressed (87 ± 2%; n = 4) by TTX (1 μm), consistent with the role of unclamped APs in generating IsAHP. Apamin and IbTX reduced QsAHP by 76 ± 5 and 10 ± 3%, respectively (n = 5; three cells: apamin → IbTX + apamin; two cells: IbTX → apamin + IbTX) (Fig. 7B,C). Ca2+ store depletion with CPA depressed QAHP by 56 ± 7% (n = 5) (Fig. 7D,E). Furthermore, prior application of CPA dramatically reduced the SK-dependent component of QsAHP and eliminated the small BK-dependent component (n = 4) (Fig. 7C,E). Thus, CICR-dependent activation of SK channels largely mediates IsAHP evoked by unclamped APs, consistent with its dominant role in controlling sAHPs and interspike interval during depolarization-induced firing described above (Fig. 6, Table 1).
Figure 7.

Ca2+ store-dependent regulation of slow AHP currents is not dependent on IP3Rs. A, Example traces showing that TTX (1 mm) largely suppressed IsAHP. Inset, Traces of currents during the 2 ms depolarization are shown on an expanded scale (2 Hz sampling frequency). Note that TTX abolished the inward action current (asterisk). B, Example traces depicting the effects of apamin (100 nm) and IbTX (100 nm) on IsAHP elicited by unclamped APs. Currents during unclamped APs (at the time indicated by the arrow) are omitted for clarity. Traces of SK- and BK-dependent components of IsAHP are shown on the right. The small apamin/IbTX-insensitive component likely reflects activation of voltage-gated K+ conductance. C, Summary bar graph showing the fraction of QsAHP carried by SK- and BK-dependent components in control solution and after application of CPA (20 μm). Note that the data in CPA represent percentage of total QsAHP before CPA application in each cell [external solution (control vs CPA): F(1,14) = 77.7, p < 0.0001; current type (SK vs BK): F(1,14) = 158, p < 0.0001; external solution by current type: F(1,14) = 43.9, p < 0.0001; two-way ANOVA]. ***p < 0.001 versus control solution (Bonferroni's post hoc test). D, Summary time graph illustrating CPA-induced suppression of QsAHP. E, Example traces of IsAHP depicting the effects of CPA, apamin, and IbTX applied sequentially. F, Summary time graph demonstrating that heparin failed to affect CPA-induced suppression of QsAHP. Error bars indicate SEM.
AP-induced CICR that mediates sAHPs frequently occurs via RyRs (Sah and McLachlan, 1991; Kakizawa et al., 2007), although IP3Rs may also play a role (Cui et al., 2007). We next recorded IsAHP with heparin (1 mg/ml) in the recording pipette to test the involvement of IP3Rs. Heparin, which abolished IDHPG and IIP3 in ∼5 min (Fig. 2C,D), had no effect on QsAHP (n = 5) (Fig. 7F). Furthermore, it failed to suppress CPA-induced depression of QsAHP (55 ± 8%, n = 5; p = 0.94 vs CPA effect with control internal solution, unpaired t test). Therefore, IP3Rs are not involved in the CICR-dependent component of sAHPs.
To examine the involvement of RyRs, we used caffeine, which increases the Ca2+ sensitivity of RyRs while at the same time blocks IP3Rs (Ehrlich et al., 1994). Bath application of caffeine (1 and 2 mm) reversibly increased QsAHP but depressed IIP3 and IDHPG in a concentration-dependent manner (Fig. 8A,B). We further tested the effect of ryanodine (20 μm), which locks RyR channels in a subconductance open state and thus depletes Ca2+ stores expressing RyRs (Zucchi and Ronca-Testoni, 1997). Ryanodine inhibited QsAHP by 56 ± 8% (n = 5), an inhibition virtually identical with that produced by CPA, and completely abolished IIP3 (Fig. 8C). These data demonstrate that, although IP3Rs and RyRs are coexpressed on the same Ca2+ stores, these two Ca2+ release channels are functionally segregated, where IP3 selectively elicits IP3R-mediated Ca2+ release, without invoking CICR via RyRs, to mediate mGluR/mAChR-induced inhibition of SPN activity, whereas AP-induced Ca2+ influx through VGCCs selectively recruits RyR-mediated Ca2+ release to generate sAHPs (Fig. 8D). Ca2+ influx through certain types of VGCCs at depolarized potentials may enlarge the size of Ca2+ stores (Power and Sah, 2005) or enhance the activity of PLC (Nevian and Sakmann, 2006); however, we found that bath application of the general VGCC blocker Cd2+ (100 μm), which suppressed QsAHP by 78 ± 1% (n = 3), had no significant effect on IDHPG measured at −57 mV (n = 4) (Fig. 8E), further confirming that Ca2+ stores can be maintained without VGCC-mediated Ca2+ influx in SPNs.
Figure 8.
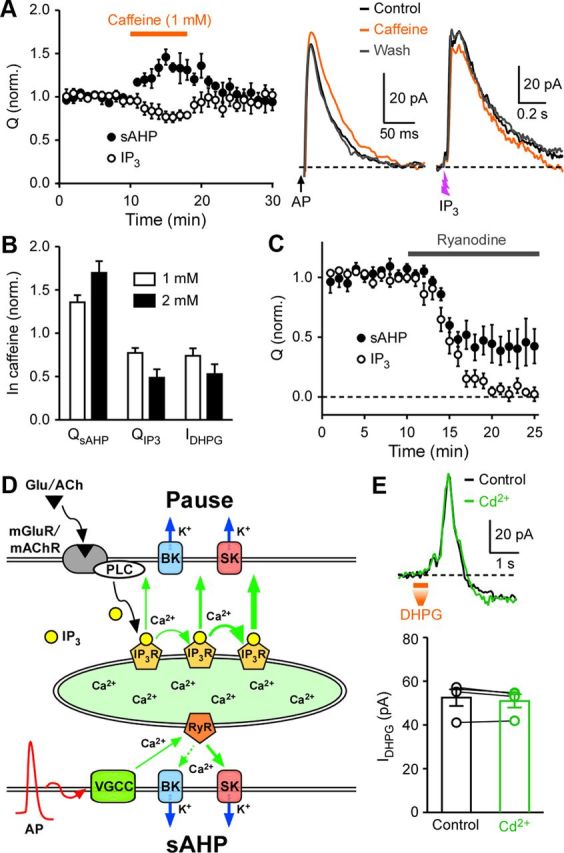
Functional segregation of IP3Rs and RyRs. A, Summary time graph illustrating the effects of caffeine (1 mm) on QsAHP and QIP3. In these experiments, both IsAHP and IIP3 were evoked once a minute (7 s apart) in each cell. Traces of IsAHP and IIP3 from a sample experiment are shown on the right. B, Summary bar graph showing the effects of caffeine (1 and 2 mm) on QsAHP, QIP3, and IDHPG. C, Summary time graph depicting ryanodine (20 μm)-induced depression of QsAHP and QIP3. D, Schematic diagram illustrating the functional segregation two Ca2+ signaling pathways mediating Ca2+ store-dependent activation of SK and BK channels: (1) mGluR/mAChR → IP3 → IP3R pathway and (2) AP → VGCC → RyR pathway. This diagram does not include the direct coupling between VGCC-mediated Ca2+ influx and SK/BK channels (Fakler and Adelman, 2008). E, Representative traces (top) and summary graph (bottom) showing that Cd2+ (100 μm) had no significant effect on IDHPG. Error bars indicate SEM.
We next tested EGTA/BAPTA sensitivity of SK- and BK-dependent components of IsAHP (IsAHP-SK and IsAHP-BK) (Fig. 9). Replacing 25 μm EGTA with 25 μm BAPTA depressed IsAHP-SK, without changing its half-width, and abolished IsAHP-BK. Increasing the BAPTA concentration to 1 mm virtually eliminated IsAHP-SK, while measurable IsAHP-SK was observed with 1 mm EGTA, although reduced in size compared with that in 25 μm EGTA. Thus, both components were more sensitive to BAPTA than to EGTA, implying the involvement of localized Ca2+ signaling processes. The higher sensitivity of IsAHP-BK to these Ca2+ buffers compared with IsAHP-SK likely reflects different Ca2+ sensitivity of BK versus SK channels.
Figure 9.

BAPTA-sensitive localized Ca2+ signaling is involved in the generation of IsAHP. A, Summary bar graphs plotting SK- and BK-dependent components of QsAHP in cells filled with two different concentrations (25 μm and 1 mm) of EGTA or BAPTA (SK-dependent component: Ca2+ buffer type, F(1,11) = 7.90, p < 0.05; Ca2+ buffer concentration, F(1,11) = 29.7, p < 0.001; BK-dependent component: Ca2+ buffer type, F(1,11) = 6.08, p < 0.05; Ca2+ buffer concentration, F(1,11) = 5.50, p < 0.05, Ca2+ buffer type by concentration, F(1,11) = 5.15, p < 0.05; two-way ANOVA). B, Summary bar graph showing that BAPTA (25 μm) did not affect the half-width of the SK-dependent component of IsAHP (t(7) = 0.43, p = 0.68; unpaired t test). Error bars indicate SEM.
IP3-induced Ca2+ signaling is absent in striatal interneurons
A previous immunohistochemical study has shown that expression of neuronal type 1 IP3Rs is restricted to SPNs in the striatum, with little or no IP3R expression in other aspiny neurons, particularly in cholinergic neurons and parvalbumin-containing neurons, whereas RyRs are expressed in virtually all striatal neurons (Martone et al., 1997). In agreement with this study, flash photolysis of caged IP3 (400 μm; ∼200 μJ UV intensity), as well as trains of 5–10 unclamped APs at 20 Hz, invariably produced robust Ca2+ transients in all SPNs tested [nine cells with 100 μm Fluo-4FF (Kd = 9.7 μm), two cells with 100 μm Fluo-5F (Kd = 2.3 μm), one cell with 200 μm Fluo-4 (Kd = 0.35 μm)] (Fig. 10A). In contrast, the same photolytic application of IP3 failed to evoke detectable Ca2+ rises in electrophysiologically identified cholinergic interneurons (ChIs) (n = 4), fast-spiking interneurons (FSIs) (n = 3), and low-threshold spike (LTS) interneurons (n = 3) even when [Ca2+]i was monitored with the high-affinity indicator Fluo-4 (200 μm), while trains of unclamped APs (ChIs and LTS neurons: 5 APs at 10 Hz; FSIs: 5–10 APs at 20 Hz) were capable of producing Ca2+ transients (Fig. 10B–D). When recordings were made with our routine internal solution containing 25 μm EGTA (without Ca2+ indicators), IP3 application elicited no measurable currents, while unclamped APs evoked apamin-sensitive IsAHP, in ChIs (n = 4) and LTS neurons (n = 3; IP3 tested in two of these three cells) (Fig. 10E). No IP3-evoked currents or IsAHP were observed in FSIs (n = 5) (data not shown). These data strongly suggest that functional IP3Rs are selectively expressed in SPNs among different types of striatal neurons.
Figure 10.
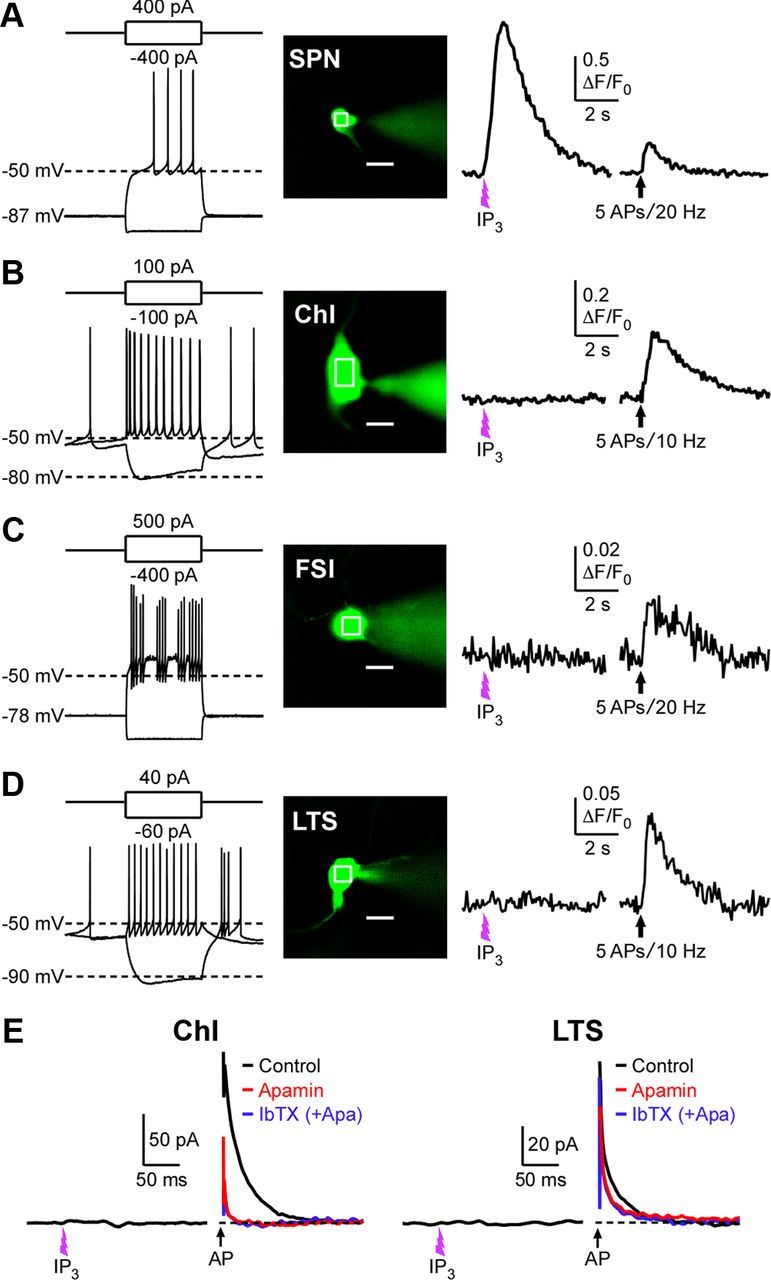
Photolytic application of IP3 produces Ca2+ transients only in SPNs among striatal neurons. A–D, Representative traces of Ca2+ transients evoked by flash photolysis of caged IP3 (400 μm) or trains of APs (5 APs at 10–20 Hz; train onset at the arrow) in an SPN (A), a ChI (B), an FSI (C), and an LTS interneuron (D). Neuron types were identified by characteristic responses to depolarizing and hyperpolarizing current injections (500 ms) (Kawaguchi, 1993). Fluorescence changes were monitored at the ROIs indicated in the images of cells filled with Fluo-4 (200 μm). Scale bar: 10 μm. Cells were voltage clamped at −57 mV in these experiments. E, Example traces illustrating that photolytic application of IP3 failed to elicit outward currents in a ChI and an LTS interneuron, although these neurons exhibited apamin-sensitive (i.e., SK-dependent) IsAHP in response to unclamped APs (Vh = −57 mV). Error bars indicate SEM.
Discussion
Inhibitory regulation of SPN activity is thought to be achieved either through feedback inhibition among SPNs or via feedforward inhibition by GABAergic interneurons, recruited by extrinsic glutamatergic and local cholinergic inputs (Ponzi and Wickens, 2010; English et al., 2012; Gittis and Kreitzer, 2012). Here, we found that glutamatergic and cholinergic inputs can directly inhibit SPNs via mGluR/mAChR-induced intracellular Ca2+ release and subsequent activation of SK and BK channels. IP3Rs are selectively involved in mGluR/mAChR-induced Ca2+ release without playing a role in CICR triggered by AP-evoked Ca2+ influx. Intriguingly, IP3-induced Ca2+ release is not observed in striatal interneurons. Therefore, IP3 signaling is uniquely involved in glutamatergic and cholinergic inhibition of striatal output neuron activity.
Mechanisms of mGluR- and mAChR-induced inhibition
Both local pressure ejection of agonists and intrastriatal stimulation of glutamatergic and cholinergic inputs produce Ca2+ store-dependent inhibitory responses in SPNs. Experiments were mostly performed in the presence of the GABAA blocker picrotoxin, ruling out the contribution of local GABAergic network. Curiously, the mGluR-dependent component of IPSCs is almost exclusively mediated by the mGluR1 subtype, while pressure ejection of DHPG, which would reach an area surrounding the soma, proximal dendrites, and maybe part of more distal dendrites equipped with spines, equally engages both mGluR1 and mGluR5. Previous ultrastructural studies in SPNs have shown that both mGluR1 and mGluR5 are equally distributed on dendritic spines and shafts, mostly at extrasynaptic sites, although mGluR5 is more frequently detected than mGluR1 at the soma (Paquet and Smith, 2003; Mitrano and Smith, 2007). Pharmacologically, these two mGluR subtypes have similar affinities to glutamate as well as to DHPG (Schoepp et al., 1999). Therefore, it is not clear why mGluR5-dependent IPSCs were not detected. Of note, distinct roles of mGluR1 versus mGluR5 on AMPA/NMDA-mediated glutamatergic transmission and plasticity have been reported in SPNs (Gubellini et al., 2004). It remains to be determined how these two mGluR subtypes differentially sense synaptically released glutamate and signal its consequence.
mAChR-dependent inhibitory responses are most likely mediated by the PLC-coupled M1 subtype, which is expressed in virtually all SPNs (Yan et al., 2001). The majority of cholinergic terminals in the striatum, which arise from ChIs, do not make synaptic contacts onto other neurons (Contant et al., 1996). This, together with the extrasynaptic localization of mGluRs described above, may account for the requirement of repetitive, high-frequency stimulation to elicit IPSCs via extrasynaptic diffusion of glutamate/ACh (Brasnjo and Otis, 2001). mAChR-dependent inhibition may contribute, in part, to the suppression of SPN firing caused by selective excitation of ChIs in vivo (Witten et al., 2010; English et al., 2012).
SK channels and, to a smaller degree, BK channels both contribute to mGluR- and IP3-induced outward currents. mGluR/mAChR/IP3-induced outward currents/hyperpolarizations are entirely mediated by SK channels in cortical pyramidal neurons (Gulledge and Stuart, 2005; Hagenston et al., 2008), hippocampal CA1 pyramidal neurons (El-Hassar et al., 2011), basolateral amygdala (BLA) neurons (Power and Sah, 2008), and midbrain dopamine neurons (Fiorillo and Williams, 2000; Morikawa et al., 2000, 2003). In contrast, mGluR/IP3-induced Ca2+ signaling is exclusively coupled to BK channels in cerebellar Purkinje neurons (Khodakhah and Ogden, 1995; Canepari and Ogden, 2006), even though these neurons express SK channels (Womack and Khodakhah, 2003). SK channels are solely gated by Ca2+ with submicromolar affinity, while BK channels are gated by voltage and Ca2+ (Fakler and Adelman, 2008). Both strong depolarization (≥0 mV) and very high [Ca2+]i (≥10 μm) are generally necessary to activate BK channels. This large difference in Ca2+ sensitivity readily accounts for the major contribution of SK channels at rather hyperpolarized potentials (below −50 mV) in many neurons described above. However, recent evidence indicates that BK channel gating is dramatically enhanced by auxiliary proteins, termed LRRC (leucine-rich repeat-containing) proteins, which allow BK channel activation by submicromolar to micromolar [Ca2+]i even at hyperpolarized potentials (Yan and Aldrich, 2010, 2012). mRNAs of these proteins are found in the brain, raising the possibility that certain BK channels might have high Ca2+ sensitivity in neurons. Indeed, BK- and SK-dependent components of IDHPG and IIP3 had similar kinetics (except for the initial BK component of IIP3), suggesting that these two Ca2+-activated channels may be sensing the same Ca2+ signals in SPNs.
As in cortical and hippocampal pyramidal neurons and BLA neurons (Power and Sah, 2002, 2005; Gulledge and Stuart, 2005; Hagenston et al., 2008), mGluR-induced, and likely mAChR-induced, Ca2+ signaling in SPNs is entirely dependent on IP3-mediated Ca2+ release, boosted by CICR among neighboring IP3Rs without recruiting CICR via RyRs. Both IP3Rs and RyRs are involved in mGluR-induced Ca2+ release in dopamine neurons; however, these two Ca2+ release mechanisms occur independently, being mediated by distinct intracellular second messengers, IP3 for IP3Rs and cyclic ADP-ribose for RyRs (Morikawa et al., 2003). Therefore, IP3Rs and RyRs generally exhibit functional segregation, although these two Ca2+ release channels have been shown to be coexpressed on the same pool of Ca2+ stores in all of these neurons, consistent with the idea that endoplasmic reticulum Ca2+ stores form a single continuous network in individual cells (Berridge, 1998). Functional segregation of IP3Rs and RyRs may, at least partially, be accounted for by their differential subcellular localization (Sharp et al., 1993).
mGluR/mAChR activation also produces an inward current independent of Ca2+ release in SPNs. This most likely results from closure of KCNQ K+ channels as a consequence of PLC-dependent depletion of a membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (Shen et al., 2005). In contrast, photolytic application of IP3 causes a transient inward current, which is likely due to a TRPC-like cationic conductance activated by Ca2+ (Zhu, 2005). mGluR/mAChR-induced excitatory responses observed in many different neurons generally depend on either, or both, of these two ionic mechanisms (Fiorillo and Williams, 2000; Gulledge and Stuart, 2005; Canepari and Ogden, 2006; Hagenston et al., 2008; El-Hassar et al., 2011). The amount and spatiotemporal profile of IP3 achieved by mGluR/mAChR activation appears to be insufficient to activate TRPC-like channels in SPNs.
Ca2+ store dependence of slow AHPs
BK and SK channels make distinct contributions to different phases of AHPs (Fakler and Adelman, 2008). Indeed, BK and SK channels contribute to fAHPs and sAHPs, respectively, in SPNs. Our data further demonstrate that sAHPs are largely dependent on intracellular Ca2+ release. IsAHP evoked by unclamped APs was augmented by caffeine, which increases Ca2+ sensitivity of RyRs, but unaffected by heparin, an IP3R antagonist. Thus, RyR-dependent CICR triggered by AP-induced Ca2+ influx accounts for the Ca2+ store dependence of sAHPs, as in other neurons (Sah and McLachlan, 1991; Berridge, 1998; Kakizawa et al., 2007). This is also in line with the preferential localization of RyRs at the soma and proximal dendrites in SPNs (Martone et al., 1997), which display limited backpropagation of APs into distal dendrites (Day et al., 2008). However, AP-induced Ca2+ influx can trigger IP3R-dependent CICR when IP3 levels are elevated by activation of mGluRs and other PLC-coupled receptors in cortical and hippocampal pyramidal neurons (Nakamura et al., 2000; Stutzmann et al., 2003), BLA neurons (Power and Sah, 2008), and dopamine neurons (Cui et al., 2007). It remains to be determined whether similar “cross talk” of AP-induced Ca2+ influx and IP3 signaling occurs in SPNs.
IsAHP had a small BK component, while the BK channel blocker IbTX failed to affect sAHPs. A small reduction in BK-dependent sAHPs might be offset by IbTX-induced AP broadening, which would augment AP-evoked Ca2+ influx and subsequent SK channel activation. Based on differential sensitivity to EGTA and BAPTA, localized Ca2+ signaling around VGCCs is thought to drive BK channel activation that mediates AP repolarization and fAHPs (Fakler and Adelman, 2008). Our data suggest that the Ca2+ store-dependent component of sAHPs also involves highly BAPTA-sensitive, localized Ca2+ signaling, from RyRs to SK/BK channels and/or from VGCCs to RyRs, in SPNs (Fig. 7D).
IP3-induced Ca2+ signaling selectively regulates SPNs in the striatum
All SPNs showed robust responses to DHPG, muscarine, or IP3 applications. In contrast, striatal interneurons failed to respond to photolytic IP3 application using maximal UV intensity used in the present study, in agreement with an immunohistochemical study reporting selective expression of IP3Rs in SPNs among striatal neurons (Martone et al., 1997). To our knowledge, this is the first indication that functional IP3 signaling is specific to projection neurons in a given brain area, although we cannot completely rule out the possibility that striatal interneurons might express IP3Rs with extremely low sensitivity.
SPNs fire clusters of APs interrupted by periods of pauses in vivo, sometimes without transition to a full DOWN state (Wilson and Kawaguchi, 1996; Stern et al., 1998). Firing pauses in distinct populations of SPNs are thought to be important for action selection and deselection (Jin and Costa, 2010; Krause et al., 2010). Working in concert with feedback and feedforward GABAergic mechanisms, robust IP3-mediated inhibition driven by glutamatergic and cholinergic inputs onto SPNs themselves might contribute to these pauses observed in behaving animals.
Footnotes
This work was supported by NIH Grants DA015687 and AA015521. We thank Dr. Kamran Khodakhah for the generous gift of caged IP3 made in his laboratory. We also thank Dr. Mark Harnett for critical comments on this manuscript.
References
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/s0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Canepari M, Ogden D. Kinetic, pharmacological and activity-dependent separation of two Ca2+ signalling pathways mediated by type 1 metabotropic glutamate receptors in rat Purkinje neurones. J Physiol. 2006;573:65–82. doi: 10.1113/jphysiol.2005.103770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contant C, Umbriaco D, Garcia S, Watkins KC, Descarries L. Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience. 1996;71:937–947. doi: 10.1016/0306-4522(95)00507-2. [DOI] [PubMed] [Google Scholar]
- Cowan RL, Wilson CJ. Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J Neurophysiol. 1994;71:17–32. doi: 10.1152/jn.1994.71.1.17. [DOI] [PubMed] [Google Scholar]
- Cui G, Bernier BE, Harnett MT, Morikawa H. Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci. 2007;27:4776–4785. doi: 10.1523/JNEUROSCI.0139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- El-Hassar L, Hagenston AM, D'Angelo LB, Yeckel MF. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca2+ wave-dependent activation of SK and TRPC channels. J Physiol. 2011;589:3211–3229. doi: 10.1113/jphysiol.2011.209783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsáki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci. 2000;20:7855–7860. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig A, Penner R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci. 2004;25:633–639. doi: 10.1016/j.tips.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35:557–564. doi: 10.1016/j.tins.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Pisani A, Centonze D, Bernardi G, Calabresi P. Metabotropic glutamate receptors and striatal synaptic plasticity: implications for neurological diseases. Prog Neurobiol. 2004;74:271–300. doi: 10.1016/j.pneurobio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18:407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka O, Sakamoto M, Usui S. Functional properties of monkey caudate neurons. III. Activities related to expectation of target and reward. J Neurophysiol. 1989;61:814–832. doi: 10.1152/jn.1989.61.4.814. [DOI] [PubMed] [Google Scholar]
- Hong M, Ross WN. Priming of intracellular calcium stores in rat CA1 pyramidal neurons. J Physiol. 2007;584:75–87. doi: 10.1113/jphysiol.2007.137661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Seif T, Mohamedi ML, Chen BT, Bonci A. The small-conductance calcium-activated potassium channel is a key modulator of firing and long-term depression in the dorsal striatum. Eur J Neurosci. 2010;31:1946–1959. doi: 10.1111/j.1460-9568.2010.07231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature. 2010;466:457–462. doi: 10.1038/nature09263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, Shimizu H, Fukaya M, Nishi M, Sakagami H, Ikeda A, Kondo H, Kano M, Watanabe M, Iino M, Takeshima H. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 2007;26:1924–1933. doi: 10.1038/sj.emboj.7601639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodakhah K, Ogden D. Fast activation and inactivation of inositol trisphosphate-evoked Ca2+ release in rat cerebellar Purkinje neurones. J Physiol. 1995;487:343–358. doi: 10.1113/jphysiol.1995.sp020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, German PW, Taha SA, Fields HL. A pause in nucleus accumbens neuron firing is required to initiate and maintain feeding. J Neurosci. 2010;30:4746–4756. doi: 10.1523/JNEUROSCI.0197-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone ME, Alba SA, Edelman VM, Airey JA, Ellisman MH. Distribution of inositol-1,4,5-trisphosphate and ryanodine receptors in rat neostriatum. Brain Res. 1997;756:9–21. doi: 10.1016/s0006-8993(96)01430-8. [DOI] [PubMed] [Google Scholar]
- McCray JA, Herbette L, Kihara T, Trentham DR. A new approach to time-resolved studies of ATP-requiring biological systems; laser flash photolysis of caged ATP. Proc Natl Acad Sci U S A. 1980;77:7237–7241. doi: 10.1073/pnas.77.12.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrano DA, Smith Y. Comparative analysis of the subcellular and subsynaptic localization of mGluR1a and mGluR5 metabotropic glutamate receptors in the shell and core of the nucleus accumbens in rat and monkey. J Comp Neurol. 2007;500:788–806. doi: 10.1002/cne.21214. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20 doi: 10.1523/JNEUROSCI.20-20-j0003.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Wilson CJ. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci. 1995;15:4449–4463. doi: 10.1523/JNEUROSCI.15-06-04449.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet M, Smith Y. Group I metabotropic glutamate receptors in the monkey striatum: subsynaptic association with glutamatergic and dopaminergic afferents. J Neurosci. 2003;23:7659–7669. doi: 10.1523/JNEUROSCI.23-20-07659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JC, Galarraga E, Bargas J, Cristancho M, Aceves J. Charybdotoxin and apamin sensitivity of the calcium-dependent repolarization and the afterhyperpolarization in neostriatal neurons. J Neurophysiol. 1992;68:287–294. doi: 10.1152/jn.1992.68.1.287. [DOI] [PubMed] [Google Scholar]
- Ponzi A, Wickens J. Sequentially switching cell assemblies in random inhibitory networks of spiking neurons in the striatum. J Neurosci. 2010;30:5894–5911. doi: 10.1523/JNEUROSCI.5540-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Intracellular calcium store filling by an L-type calcium current in the basolateral amygdala at subthreshold membrane potentials. J Physiol. 2005;562:439–453. doi: 10.1113/jphysiol.2004.076711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Competition between calcium-activated K+ channels determines cholinergic action on firing properties of basolateral amygdala projection neurons. J Neurosci. 2008;28:3209–3220. doi: 10.1523/JNEUROSCI.4310-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Ca2+-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca2+-activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-z. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Jaeger D, Wilson CJ. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature. 1998;394:475–478. doi: 10.1038/28848. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, LaFerla FM, Parker I. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Wilson CJ, Koós T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008;58:272–281. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JW, Feeney J, Trentham DR. Photolabile precursors of inositol phosphates. Preparation and properties of 1-(2-nitrophenyl)ethyl esters of myo-inositol 1,4,5-trisphosphate. Biochemistry. 1989;28:3272–3280. doi: 10.1021/bi00434a023. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–2410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci. 2003;23:2600–2607. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem Biol. 2011;18:777–793. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature. 2010;466:513–516. doi: 10.1038/nature09162. [DOI] [PubMed] [Google Scholar]
- Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci U S A. 2012;109:7917–7922. doi: 10.1073/pnas.1205435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Flores-Hernandez J, Surmeier DJ. Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience. 2001;103:1017–1024. doi: 10.1016/s0306-4522(01)00039-2. [DOI] [PubMed] [Google Scholar]
- Zhu MX. Multiple roles of calmodulin and other Ca2+-binding proteins in the functional regulation of TRP channels. Pflugers Arch. 2005;451:105–115. doi: 10.1007/s00424-005-1427-1. [DOI] [PubMed] [Google Scholar]
- Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51. [PubMed] [Google Scholar]