

Abstract
Autophagy is a critical cellular process orchestrating the lysosomal degradation of cellular components in order to maintain cellular homeostasis and respond to cellular stress. A growing research effort over the last decade has proven autophagy to be essential for constitutive protein and organelle turnover, for embryonic/neonatal survival, and for cell survival during conditions of environmental stress. Emphasizing its biological importance, dysfunctional autophagy contributes to a diverse set of human diseases. Cellular stress induced by xenobiotic exposure typifies environmental stress, and can result in the induction of autophagy as a cytoprotective mechanism. An increasing number of xenobiotics are notable for their ability to modulate the induction or the rate of autophagy. The role of autophagy in normal cellular homeostasis, the intricate relationship between cellular stress and the induction of autophagy, and the identification of specific xenobiotics capable of modulating autophagy, point to the importance of the autophagic process in toxicology. This review will summarize the importance of autophagy and its role in cellular response to stress, including examples in which consideration of autophagy has contributed to a more complete understanding of toxicant-perturbed systems.
2. Introduction
Autophagy, of Greek derivation meaning self-eating, was first characterized by Christian de Duve nearly five decades ago. The term was coined to describe observations from electron microscopy studies that demonstrated novel single- or double-membrane vesicles containing parts of the cytoplasm, including organelles, in various degrees of disintegration (Ashford and Porter 1962; Clark 1957; De Duve and Wattiaux 1966). Today autophagy is recognized as a fundamental cellular process that orchestrates the constitutive degradation of proteins and organelles, as well as the removal of damaged cellular constituents through lysosomal degradation. The exponential increase in autophagy publications over roughly the past decade has contributed to a rapidly evolving understanding of autophagy as a pivotal cellular process that is necessary for constitutive protein turnover, embryonic development, neonatal survival, and cellular adaptation to environmental stress. Perturbation of autophagy can lead to malignant transformation and can enhance tumor survival under environmental stress (Liang et al., 1999; Liu et al., 2010). Autophagy can be an adaptive response to cellular stress, including stress induced by xenobiotics or their intermediary effects (Lim et al., 2010; Shen et al., 2011). Under specific conditions autophagy may be capable of executing a type of programmed cell death. Several xenobiotics are well-characterized for their effect in inhibiting or in modulating autophagy. Taken together, the importance of autophagy in homeostasis, the close relationship between cell stress and autophagy, and the identification of diverse environmental stressors and xenobiotics that are capable of modulating autophagy, all underscore the importance of this cellular process to the field of toxicology. The intent of this review is to advance the appreciation of this important cellular stress response among toxicologists, a research community focused on stressed biological systems.
3. The process of autophagy
The unifying feature of all types of autophagy is the delivery of cellular constituents to the lysosome, where they are degraded. As illustrated in Figure 1, three substantially different mechanisms have been defined that can carry out this process: macroautophagy, chaperone-mediated autophagy, and microautophagy. The processes are not exclusive and may occur simultaneously.
Figure 1.
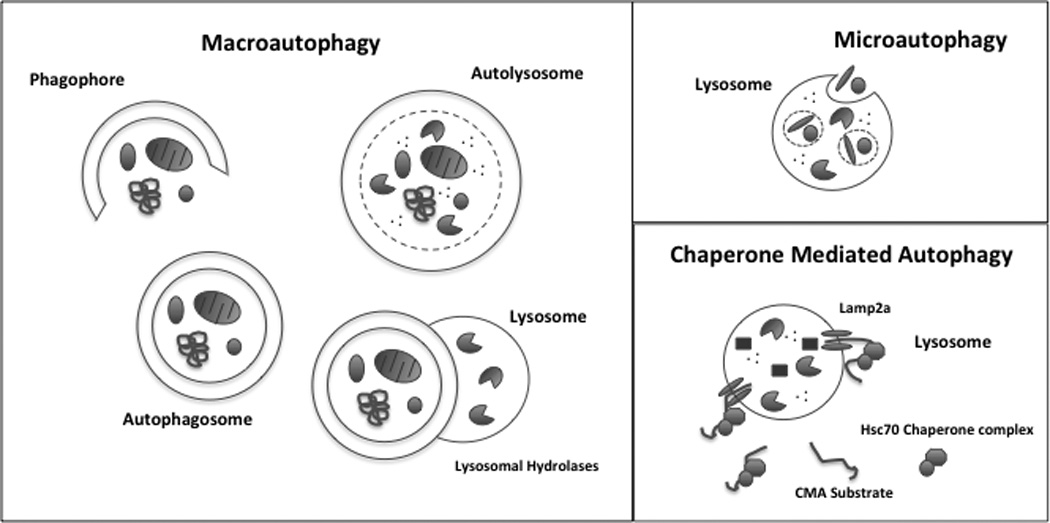
Primary autophagic pathways in mammalian cells. Macroautophagy involves the sequestration of cytoplasmic components by the generation of a denovo double membrane structure (phagophore) from within the cell to form an autophagosome. The newly formed autophagosome then fuses with a lysosome, forming an autolysosome, where lysosomal hydrolases degrade the cellular components. Microautophagy mediates the sequestration of cellular cargo, proteins and organelles through the invagination of the lysosomal membrane. Once cellular components are localized in the lysosome, lysosomal hydrolases degrade the components. Chaperone Mediated Autophagy (CMA) involves the specific targeting and shuttling of individual proteins to the lysosome for degradation. Internalization of substrate proteins into the lysosome is mediated through coordinated interaction of chaperone proteins and the lysosomal receptor Lamp2a on the membrane of the lysosome.
3.1: Macroautophagy
Macroautophagy is initiated through the formation of a de novo double membrane structure or phagophore from within the cell. The growing phagophore gradually surrounds cellular components, then fuses at its ends, forming a unique double-membrane vesicle called an autophagosome (Mizushima et al., 2001). This sequestration of cellular components by the autophagosome can either be a non-selective process resulting in the engulfment of bulk cytoplasm, or alternatively can involve the selective targeting of specific cellular components for degradation (Nakagawa et al., 2004; Rodriguez-Enriquez et al., 2006). The autophagosome is shuttled to a lysosome, with which it fuses by merging its outer membrane with the lysosomal membrane, introducing a now single-membrane autophagosome into the lysosome. The newly formed combined organelle is referred to as an autolysosome. In the autolysosome, lysosomal hydrolases degrade the introduced cellular components, eventually resulting in a vacuole.
Macroautophagy is an evolutionarily conserved process, with much of the molecular characterization of pathways and pathway members resulting from studies of yeast (Barth and Thumm 2001). To date over 30 genes involved in the execution and regulation of macroautophagy, referred to as autophagy-related genes (Atg), have been identified in yeast (Review in (Klionsky et al., 2003)). Many mammalian homologues of the yeast Atg have been identified and several reviews have provided a detailed description of their role in the initiation, machinery, and regulation of the autophagic process (Eskelinen 2008a; He and Klionsky 2009; Kroemer et al., 2010; Kundu and Thompson 2008; Yorimitsu and Klionsky 2005).
In mammalian systems the central inhibitor of macroautophagy is the nutrient sensing serine/threonine kinase, mammalian target of rapamycin (mTOR) (Reviewed in (Pattingre et al., 2008)). mTOR forms an inhibitory complex, mTORC1, with its substrate binding subunit, raptor, and the polypeptide Lst8/GβL (Kim et al., 2002; Kim et al., 2003). Under nutrient rich conditions in eukaryotes, mTORC1 is activated through either the PI3K-1/AKT or MAPK/Erk1/2 signaling pathways, to enable cell growth and to inhibit autophagy by negatively regulating the autophagy genes, Unc-51-like kinase 1 (ULK1) and - 2 (ULK2) (yeast homolog, Atg1). Environmental stress conditions or exposure to certain xenobiotics (e.g. rapamycin) cause inhibition of the mTORC1 complex, resulting in downstream activation of ULKs. Activation of ULKs promotes recruitment of the focal adhesion kinase family-interacting protein of 200kD (FIP200) (yeast homolog, Atg17) and Atg13 to form the ULKs-Atg13-FIP200 initiation scaffold as well as recruitment of other Atg proteins to the phagophore assembly site (PAS) to initiate autophagosome formation. The nucleation and assembly of the initial phagophore membrane requires the class III phophatidylinositol 3-kinase (PtdIns3K) complex, which is composed of PtdIns3K, vacuolar protein sorting 34 (Vps34), p150 (yeast homolog, Vps15), Barkor or mammalian Atg14 (mAtg14) (yeast homolog, Atg14), and Beclin 1 (yeast homolog, Atg6/Vps30). In addition, two positive regulators of autophagy, activating molecule in beclin-1-regulated autophagy (Ambra1) and ultraviolet irradiation resistance-associated gene (UVRAG), can associate with the PtdIns3K complex, driving autophagosome maturation (Fimia et al., 2007; Liang et al., 2008). The PtdIns3K complex along with other Atg proteins further recruits two interrelated ubiquitin-like conjugation systems Atg12-Atg5-Atg16 and Atg8-phosphatidylethanolamine (Atg8-PE) (mammalian homolog, microtubule-associated protein 1 light chain 3 (LC3)) to the phagophore, which play an integral role in regulating the membrane elongation and expansion of the forming autophagosome (Reviewed in (He and Klionsky 2009)). Atg12 and Atg8 are considered ubiquitin-like proteins because they undergo a conjugation process in a similar manner as ubiquitin. Atg12 is activated by Atg7 (E1 activating enzyme), transferred to Atg10 (E2 conjugating enzyme) and attached to an internal lysine of the substrate protein Atg5. The Atg12-Atg5 conjugate further interacts with Atg16, which links the Atg12-Atg5-Atg16 complex into a tetramer and attaches it to the phagophore (Mizushima et al., 2003; Mizushima et al., 1999). In the Atg8 conjugation system, Atg8 is first processed by Atg4 to expose a C-terminal glycine residue. Atg8 is activated by the same E1 enzyme, Atg7, and then transferred to Atg3 (E2 conjugating enzyme). Atg8 is conjugated to the target lipid PE, facilitated by the E3-like Atg12-Atg5 conjugate, and then attaches to both the inner and outer membranes of the phagophore (Hanada et al., 2007).
Autophagy genes can be regulated at the transcriptional level in response to stress stimuli. For example, under nutrient starved conditions, gene expression of the autophagy gene Atg8/LC3 is rapidly induced in yeast and certain mammalian cells (Kirisako et al., 1999; Mizushima et al., 2004). However, the underlying transcriptional signaling that regulates autophagy gene expression is poorly understood. The transcription factor, FoxO3 (forkhead box transcription factor class O 3) induces the expression of multiple autophagy genes, including LC3B, GABA(A) receptor-associated protein like 1 (GabarapL1), Atg12, Atg4B, Vps34, ULK2, Beclin 1, BCL2/ adenovirus E1B 19kDa interacting protein 3 (Bnip3), and Bnip3L in atrophied skeletal muscle cells (Mammucari et al., 2007; Zhao et al., 2008). In addition, signaling pathways that induce translational arrest such as the eukaryotic translation initiation factor 2-alpha kinase (eIF2α) mediated signaling pathway positively regulate autophagy. Phosphorylation of eIF2α at serine 51 is essential for global translational arrest and selective translational induction of specific transcriptional activators such as GCN4, which stimulate transcription of starvation-induced genes (Talloczy et al., 2002). Recently, transcription factor EB (TFEB), which regulates lysosome biogenesis, has also been identified as a master regulator of autophagy through the induction of both lysosomal and autophagy genes (Settembre et al., 2011).
Macroautophagy can be a non-selective bulk degradation process or, conversely, can selectively target a particular organelle for degradation in order to regulate organelle number or maintain organelle quality control. Alternative terms describing macroautophagy have been coined to identify particular subtypes of macroautophagy that selectively target a particular superfluous or damaged organelle for degradation. For example, mitophagy is the selective removal of mitochondria from the cell by autophagic degradation (Lemasters 2005). Mitophagy is critical for the removal of mitochondria from immature red blood cells in mammals during maturation (Kundu et al., 2008). In addition, damaged mitochondria can be tagged for autophagic removal (Kim et al., 2007). Pexophagy is the selective removal of peroxisomes. In methylotrophic yeast species a shift in the main carbon source from glucose to methanol results in an increased proliferation of peroxisomes, which are removed through pexophagy once normal nutrient conditions are restored (Oku and Sakai 2010). Additional examples of cargo-specific autophagy include ribophagy (ribosomes) and xenophagy (intracellular pathogens) (Gutierrez et al., 2004; Kraft et al., 2008).
3.1.1: Experimental Measurement of Macroautophagy
There is currently no single experimental measurement that unequivocally establishes the presence or rate of macroautophagy. Many investigators consider the electron microscope-documented presence of cellular structures consistent with autophagosomes and autolysosomes to be a gold standard for establishing autophagy (Eskelinen 2008b; Klionsky et al., 2008).
Autophagy can also be monitored with fluorescent dyes that accumulate in acidic vesicles, allowing measurement of the number and size of autolysosomes. Commonly used acidophilic dyes that can be measured microscopically or by flow cytometry include lysotracker red, acridine orange, and monodansylcadaverine (Bolt et al., 2010b; Contento et al., 2005; Paglin et al., 2001).
Immunoblot analysis of cellular protein levels of LC3-II (an alias for Atg8-PE, mentioned previously) is commonly used to monitor autophagy. Upon the induction of autophagy, the cytosolic protein LC3 is cleaved at its C terminus by Atg4 to form LC3-I. LC3-I is then covalently conjugated to phosphatidylethanolamine via an ubiquitylation-like system through Atg7 and Atg3 to form LC3-II, which is specifically located on both the inner and outer membranes of autophagosomes. Thus, there are two pools of LC3-II in autophagosomes. The LC3-II bound to the outer membrane can be removed by Atg4-mediated delipidation during the maturation of the autophagosome, potentially recycling LC3-I back to the cytoplasm. Upon fusion of the autophagosome with the lysosome the inner autophagosome membrane is inserted into the lysosomal lumen. This results in the degradation of LC3-II present on that membrane (Kabeya et al., 2004; Kabeya et al., 2000). Thus, the quantity of LC3-II protein correlates with the number of autophagosomes, but this is complicated by the fact that LC3-II is in a state of flux during autophagy, with production by Atg genes that post-translationally modify LC3, and with degradation by the lysosomal introduction of the inner autophagosomal membrane as autophagy proceeds. As a result, measuring LC3-II flux through the autolysosome over time is an essential element in quantifying autophagy. Simply observing a decrease in steady-state LC3-II levels in experimental groups, compared to controls, could result from decreased autophagy and fewer autophagosomes, or it could result from increased autophagy that consumes LC3-II more rapidly than it is being produced. In practice, teasing this apart is accomplished through the use of inhibitors of lysosomal degradation. One commonly used inhibitor is bafilomycin A1 (BafA1), a proton pump inhibitor that increases lysosomal pH, inactivating lysosomal proteases (Yamamoto et al., 1998). For example, cultured cells, in the presence or absence of an experimental treatment, would be subjected to exposure to BafA1 for 0 hrs, 4 hrs, and 8 hrs. The 0 hr time-point reflects the steady-state LC3-II level, and the delta of the band intensities between the subsequent time-points and the 0 time-point reflects the amount of LC3-II that would have been degraded by autophagy during that time without BafA1 present.
3.2 Chaperone-Mediated Autophagy
In contrast to macroautophagy, CMA is a selective process that targets individual proteins and directly shuttles them from the cytosol across the lysosomal membrane into the lysosomal lumen via specific receptors, leading to their degradation by lysosomal hydrolases. Unlike either macro or micro-autophagy, CMA has only been described in mammals. Currently the signal transduction pathways that regulate CMA remain elusive. Inhibition of p38 MAPK can partially prevent activation of CMA, implicating this pathway in the activation of CMA (Finn et al., 2005). In addition, protein levels of CMA components can regulate CMA activity. The quantity of lysosomal-associated membrane protein 2a (Lamp2a), the lysosomal membrane protein that interacts with CMA chaperones and internalizes CMA substrate proteins to the lysosome, directly correlates with CMA activity. Under nutrient-starved conditions, the half-life of Lamp2a in mouse fibroblasts is increased, resulting in more Lamp2a protein in the lysosomal membrane and an elevation in CMA-mediated proteolytic activity (Cuervo and Dice 2000). In addition, researchers have determined that, under conditions of starvation or oxidative stress, protein levels of the CMA chaperone, Hsc70, in the lysosome, as well as the number of Hsc70-positive lysosomes gradually increased with increased CMA activity (Chiang et al., 1989; Kiffin et al., 2004).
CMA incorporates substrate selectivity and direct translocation of substrates across the lysosomal lumen, which distinguishes CMA from the other two autophagic processes. CMA substrate selectivity and direct shuttling to the lysosome allow for the selective removal of non-essential proteins for degradation to generate new pools of free amino acids required for protein synthesis during starvation. This system also allows for the removal of specific, damaged or misfolded proteins during cellular stress without disturbing undamaged proteins or functionally critical proteins (Cuervo et al., 1995; Kiffin et al., 2004). CMA is also involved in specialized cellular functions such as the processing and major histocompatibility complex (MHC) - loading of endogenous and exogenous antigens during antigen presentation (Zhou et al., 2005).
The CMA process centers around three components: chaperone proteins, receptor proteins and substrate proteins. The main chaperone protein is Hsc70, which is a constitutively expressed member of the heat shock protein 70 family of chaperones. Chaperone proteins are involved in the recognition and shuttling of individual proteins to the lysosome. In order for the chaperones to recognize proteins that are substrates of CMA, the proteins must contain a specific motif homologous to the consensus amino acid sequence KFERQ (Cuervo et al., 1994; Salvador et al., 2000). During CMA, cytosolic-Hsc70 interacts with the KFERQ motif in substrate proteins, after which the protein pair is shuttled to the lysosome. The docking partner present on the lysosomal membrane is Lamp2a. CMA substrate proteins directly bind to the short cytosolic tail of Lamp2a, which is considered the rate-limiting step in the CMA process (Majeski and Dice 2004). The positively charged amino acid residues in the cytosolic tail of Lamp2a are important for this binding. Upon formation of the 3-protein complex, the substrate protein unfolds, Lamp2a forms a multimeric channel, and the substrate protein is translocated into the lysosomal lumen through that channel (Cuervo et al., 1994; Salvador et al., 2000). Co-chaperone proteins that interact with Hsc70 and facilitate substrate recognition and transport include heat shock protein 40 kD (Hsp40), heat shock protein 90 kD (Hsp90), and Hsp70-interacting protein (Hip) (Agarraberes and Dice 2001).
Antibodies raised against the CMA recognition motif KFERQ immunoprecipitate roughly one-third of cytosolic proteins in mammalian cells, suggesting that a substantial portion of the proteome can be degraded through CMA (Dice 2007). Of the cellular proteins that contain the recognition motif, about 80% of the motifs are recognizable by KFERQ specific antibodies without denaturing the protein, evidence that the motif is accessible under normal protein folding (Chiang and Dice 1988). These proteins could represent a pool subject to starvation-induced CMA degradation. On the other hand, some substrate recognition motifs are only accessible after protein unfolding. This suggests a mechanism for the selective removal of improperly folded proteins by the KFERQ becoming accessible to the chaperone proteins when the protein is misfolded, but not in the native folding state (Kiffin et al., 2004).
3.2.1: Experimental Measurement of CMA
Methods used to monitor CMA include measuring protein degradation rates of CMA degraded proteins, or measuring protein levels of key CMA pathway members such as Hsc70 or Lamp2a by immunoblot analysis. An in vitro measurement of CMA activity can be performed by monitoring the direct translocation of a known CMA substrate, which is radio-labeled, into lysosomes isolated from a particular tissues or cell type of interest (Kaushik and Cuervo 2009).
3.3: Microautophagy
Microautophagy is characterized by the direct sequestration of cellular components into the lysosome through invagination of the lysosomal membrane. The vesicle containing cytoplasmic contents is then pinched off into the lysosomal lumen and rapidly degraded by lysosomal hydrolyses (Ahlberg and Glaumann 1985). The degradation of both organelles and proteins through microautophagy has been described in yeast (Yuan et al., 1997). As opposed to macroautophagy where there is considerable homology in the Atg genes from yeast to humans, the mammalian homologues of the yeast genes unique to microautophagy have not yet been identified. The lack of molecular characterization of the microautophagy machinery in mammalian systems has limited our understanding of this process in higher organisms. Methods to monitor or to modulate microautophagy are also limited. Currently, the most widely used method of monitoring microautophagy is through visualization of morphological characteristics by transmission electron microscopy (Reviewed in (Klionsky et al., 2007)).
These three types of autophagy share the common function of delivering both bulk, non-selective cytosolic cargo, as well as cargo that has been specifically tagged for degradation, to the lysosome. Because of our limited understanding of microautophagy, this review will focus on macroautophagy and chaperone-mediated autophagy as they relate to toxicology.
4. Autophagy: Lifeboat or Torpedo?
Molecular and morphological evidence of autophagy is commonly observed in dead or dying cells. In part, this has led to the classification of autophagy as an alternative programmed cell death mechanism (Bursch et al., 1996). There has been considerable debate over whether autophagy is actually a programmed cell death mechanism, or conversely, if the presence of autophagy markers in dead or dying cells is a remnant of failed cytoprotection (Reviewed in (Bursch et al., 2008; Debnath et al., 2005)). There are many examples of xenobiotic-induced cellular death that are associated with characteristic markers of autophagy; including environmental toxicants such as arsenic as well as other xenobiotics including dexamethosome, histone deacetylase inhibitors, tamoxifen, etoposide, and staurospaurine (Bolt et al., 2010b; Bursch et al., 1996; Kanzawa et al., 2005; Laane et al., 2009; Lee et al., 2011; Shao et al., 2004; Shimizu et al., 2004; Zhao et al., 2010). Yet, deciphering the precise role autophagy plays in the cell death process has been challenging.
There are, in fact, relatively few examples in which specific inhibition of autophagy through knockout or knockdown of essential autophagy-relevant genes fully inhibits cell death induced by specific stimuli. Shimizu et al. demonstrated that embryonic fibroblasts from Bax/Bak double knockout mice are resistant to apoptosis, but underwent non-apoptotic cell death after treatment with etoposide or staurospaurine. Morphological and molecular characterization identified that cell death was associated with autophagy. Inhibition of autophagy by knockout of autophagy genes Atg5 and Beclin 1 or chemical inhibition with 3-methyladenine (3MA) resulted in a reduction of cell death (Shimizu et al., 2004).In addition, autophagic cell death is required for the selective destruction of salivary gland cells during Drosophila development (Berry and Baehrecke 2007). In this model, Atg loss-of-function genetic mutants are impaired in the developmentally programmed degradation of salivary gland cells. Instead, salivary gland cells were vacuolated and fragments of the cells persisted for up to 24 hours. Further studies have established that the induction of autophagy leads to autophagic cell death of salivary gland cells in a caspase-independent manner. Autophagy has also been shown to be required for the induction of necrotic cell death in C. elegans neuronal cells (Samara et al., 2008). Loss of the gene unc-51, the C. elegans ortholog of Atg1 involved in induction of autophagosome formation, substantially suppressed necrotic cell death induced by a diverse set of genetic neurotoxic insults. Thus, in certain situations, autophagy can execute programmed cell death. These studies however, should be distinguished from studies that simply associate cell death with autophagy markers.
More commonly, autophagy is considered a cytoprotective process. Recently, Shen et al. investigated whether autophagy is capable of mediating cell death in mammalian cells by performing a high-content screen of cytotoxic compounds from the National Cancer Institute (NCI) mechanistic library (Shen et al., 2011). Of the 59 cytotoxic compounds that robustly induced the formation of GFP-LC3 puncta (indicative of autophagy induction) that was enhanced by Baf-A1 exposure (confirming active autophagic flux), no compounds caused cell death that was inhibited by Atg7 knockdown. The authors concluded that, in general, xenobiotic stress does not induce autophagy as a cell death mechanism.
5. Autophagy: A view from the cell
5.1: Adaptation to Nutrient Deprivation
Arguably the most well-understood function of the autophagic process is its role as an adaptive process to replenish the nutrient pools of the cell in response to conditions of amino acid starvation or general nutrient deprivation. Nutrient-deprived cells must reduce the energy intensive process of new protein synthesis due to limited supply of amino acids and energy. Maintaining homeostasis under these conditions requires the cell to set a new balance between protein synthesis and protein degradation rates. Autophagy induction addresses this need through a reduction in new protein synthesis and an increase in protein degradation. Inhibition of protein translation and the induction of autophagy are coordinately regulated through eIF2α- and mTOR- mediated pathways (Harding et al., 2000; Hosokawa et al., 2009). Phosphorylation of eIF2α at serine 51 downregulates cap-dependent RNA translation leading to globally reduced protein synthesis and can also play a role in the induction of autophagy. In mammals there are four eIF2α kinases that respond to different stress stimuli: GCN2, PKR, HRI and PERK. GCN2 is activated under nutrient-deprived conditions through the binding to uncharged tRNAs that accumulate under amino acid-limited conditions to mediate translational control (Sood et al., 2000). eIF2α has also been shown to indirectly induce autophagy by phosphorylating eukaryotic elongation factor 2 (eEF-2) during nutrient deprivation (Py et al., 2009). AMP-activated protein kinase (AMPK) senses the energy status through monitoring the AMP:ATP ratio inside the cell. Under conditions of starvation or low energy (increasing AMP:ATP ratio), AMPK activates autophagy through the inhibition of mTOR, an event mediated by phosphorylation of the tuberous sclerosis complex 2 (TSC2) and the regulatory protein associated with mTOR, Raptor (Gwinn et al., 2008; Inoki et al., 2003).
The rapid induction of autophagic degradation of cellular organelles and proteins serves multiple objectives for the starved cell. First, degraded, superfluous organelles and proteins can contribute recycled components, including amino acids, to pathways that are critical for cell survival (Onodera and Ohsumi 2005; Yang et al., 2006). Second, by targeting some mitochondria for degradation the cell can alter its overall metabolic state to become less energy consumptive (Egan et al., 2011). While macroautophagy is the principal autophagy pathway shown to mitigate starvation stress, CMA also plays a role in the degradation of specific proteins. During starvation macroautophagy is activated very rapidly, often within minutes. The advantages of this are evident, but because macroautophagy is not an entirely selective process, extended induction carries the risk of eventually destroying essential cellular components. The starvation-stressed cell can mitigate this risk is by switching from macroautophagy to CMA, which facilitates targeting of non-essential proteins (proteins with natively-exposed motifs) for degradation to obtain amino acids required to maintain synthesis of essential proteins (Fuertes et al., 2003; Massey et al., 2006).
Onodera and Ohsumi investigated the role of autophagy in nutrient recycling and protein translation rates in wild type or Atg7-deficient strains of yeast (S. cerevisiae) under nitrogen starved conditions for up to 24 hours. Autophagy was strongly induced by nutrient starvation and was characterized by an increase in amino acid synthesis enzymes and vacuolar (analogous to lysosomal) enzymes in wild type cells, but not in Atg7-deficient cells. However, global protein synthesis and total intracellular amino acid pools were significantly decreased in Atg7-deficient cells in comparison to wild type controls, demonstrating that under nutrient deprived conditions, autophagy plays an important role in the recycling of cellular components in order to replenish depleted amino acid pools helping cells survive and maintain normal protein synthesis function (Onodera and Ohsumi 2005).
Autophagy also plays a role in maintaining nutrient homeostasis in mammalian systems. Amino acid-starvation of human HepG2 hepatocytes resulted in the induction of autophagy, characterized by extensive vacuolization, depletion of organelles, increase in acidic vesicles, and accumulation of lipid droplets. The induction of autophagy was associated with a decline in global translation (reduced de novo protein synthesis, decrease in total protein content and dephosphorlyation of p70 S6 kinase) after a 48 hour period (Martinet et al., 2006). In HeLa cells, deprivation of serum and amino acids caused an induction of autophagy (Boya et al., 2005). Knock down of autophagy genes in this model (Beclin 1, Atg5, Atg12, or Atg10) using RNA interference, or chemical inhibition of autophagy, resulted in increased apoptotic cell death of the nutrient deprived cells.
5.2: Adaptation to Diverse Environmental Stressors
Autophagy is also induced in response to diverse cellular stressors, including ER stress, oxidative stress, hypoxia, and DNA damage (Abedin et al., 2007; Bellot et al., 2009; Kiffin et al., 2004; Ogata et al., 2006; Onodera and Ohsumi 2005). In this context, autophagy serves as a cytoprotective mechanism, degrading damaged cellular proteins and organelles that can be toxic to the cell and thus restoring cellular homeostasis (Ogata et al., 2006). Both macroautophagy as well as CMA have been shown to function in the removal of damaged cellular components from the cell in response to environmental stress (Kiffin et al., 2004; Ogata et al., 2006).
5.2.1: Endoplasmic Reticulum Stress
ER stress due to the accumulation of misfolded proteins in the ER lumen activates the unfolded protein response (UPR). There are three major branches of the UPR, mediated by three ER-membrane associated “sensor” proteins: PKR-like eIF2α kinase (PERK), inositol required enzyme 1 (IRE1), and activating transcription factor-6 (ATF6). Xenobiotics such as thapsigargin, cyclosporine, cadmium, and tunicamycin have all been shown experimentally to induce ER stress (Lim et al., 2010; Ogata et al., 2006; Pallet et al., 2008). Macroautophagy is induced in response to UPR activation, mediated through activation of one or more of the ER stress-sensing pathways (PERK, IRE1, and ATF6), or through an elevation in cytosolic calcium levels (Hoyer-Hansen et al., 2007; Kouroku et al., 2007; Ogata et al., 2006). Treatment of either yeast or mammalian cells with ER stress inducing agents including tunicamycin or dithiolthreitol (DTT) effectively triggered the induction of autophagy (Ogata et al., 2006; Yorimitsu et al., 2006). The activation of autophagy in response to ER stress is crucial for the removal of misfolded proteins that could accumulate, inducing further ER stress and damage. An additional role of autophagy during ER stress is in maintaining total cellular ER volume. In yeast, ER stress leads to an expansion of the ER lumen in response to the transcriptional activation of the UPR (Bernales et al., 2006). The autophagic degradation of ER serves to normalize the volume of ER present in cells that have recovered from ER stress.
5.2.2: Hypoxia
Hypoxic (3.0%–0.1% oxygen) or anoxic (<0.1% oxygen) conditions induce mitophagy through two distinct pathways. Enhanced mitophagy during hypoxia is an adaptive response, reducing the levels of reactive oxygen species (ROS) and protecting cell integrity (Rouschop et al., 2009). Hypoxic conditions can rapidly induce autophagy through activation of hypoxia-inducible factor 1 alpha (HIF-1α), a transcription factor that induces genes expression leading to rapid adaptation to reduced oxygen conditions. In mouse embryonic fibroblasts (MEF), selective removal of mitochondria through mitophagy is induced in response to hypoxic conditions via HIF-1α and downstream induction of Bnip3 (Zhang et al., 2008). Bnip3 competes with the autophagy protein Beclin 1 for binding to the anti-apoptotic protein B-cell CLL/lymphoma 2 (BCL2) and thus releases Beclin 1 for participation in mitophagy (Bellot et al., 2009).
Autophagy is also capable of being activated during hypoxia or anoxia in a HIF-1α-independent manner. Often anoxic conditions are accompanied by dramatic glucose and amino acid restriction, leading to autophagy induction during hypoxia/anoxia through the previously described mTOR/AMPK pathways (Mazure and Pouyssegur 2010; Papandreou et al., 2008). In addition, anoxic conditions can lead to ER stress, activating autophagy through up-regulation of the UPR (Koritzinsky et al., 2006).
5.2.3: Oxidative Stress
Autophagy can also mitigate damage from oxidative stress. Mild oxidative damage induces partial protein unfolding and facilitates proteolytic degradation of damaged proteins (Grune et al., 1997; Grune et al., 1995). However, persistent or extensive oxidative damage can lead to protein aggregation when damaged proteins adopt conformations that expose previously “hidden” regions of hydrophobic amino acids. Aggregated proteins become less susceptible to proteolytic cleavage (Davies 2001; Grune et al., 1997). CMA facilitates the selective removal of individual damaged proteins induced by oxidative stress without disrupting other non-damaged protein critical for normal cellular function (Cuervo et al., 1998; Kiffin et al., 2004). In addition, CMA activity increases in vitro in the presence of compounds that induce oxidative stress, further supporting the hypothesis that CMA is involved in the removal of oxidatively damaged proteins (Kiffin et al., 2004). Degradation of damaged proteins in the proteasome or via CMA is efficient only for monomeric proteins. However, when the oxidative damage is extensive and protein aggregates form, macroautophagy is required to remove the protein aggregates and to prevent further damage to the cell.
Mitochondria are both ROS producers and ROS targets, pointing to their central role in the oxidatively stressed cell (Hamanaka and Chandel 2010). Autophagy removes damaged mitochondria selectively (mitophagy) to reduce ROS levels and to maintain a functional mitochondrial pool under conditions of cellular stress (Kim et al., 2007).
5.2.4: DNA Damage
Exogenous and endogenous chemical agents can induce DNA damage. The fate of a DNA-damaged cell can be influenced by autophagy (Abedin et al., 2007; Kang et al., 2009). DNA-damaging agents camptothecin, etoposide, tomozolomide, p-anilioaniline, doxorubicin, and ionizing radiation have all been shown to induce autophagy (Abedin et al., 2007; Elliott and Reiners 2008; Katayama et al., 2007; Munoz-Gamez et al., 2009; Rieber and Rieber 2008). Yet, the role that autophagy serves in the response to DNA damage is still unclear. Autophagy delays apoptotic cell death induced by the DNA damaging agent camptothecin through activation of mitophagy (Abedin et al., 2007). In 3T3 fibroblasts, doxorubicin induced DNA-damage is associated with characteristics of autophagy, which are dependent on activation of the DNA repair enzyme poly (ADP-ribose) polymerase 1 (PARP-1) (Munoz-Gamez et al., 2009). Inhibition of autophagy resulted in increased doxorubicin-induced cell death, suggesting that autophagy may function in conjunction with PARP-1 in the DNA repair mechanism to prevent cell death. Autophagy induction is associated with the activation of another DNA damage sensor, ataxia telangiectasia mutated (ATM). ATM is a serine/threonine kinase that serves as a checkpoint-specific damage sensor that is activated in response to DNA damage. Upon DNA damage, transcription factor FOXO3a detaches from DNA to interact with ATM in the nucleus to promote DNA repair (Tsai et al., 2008). DNA damage inducing agents, including ROS production and the chemotherapy agent BO-1051, activate ATM and subsequently induce autophagy as a response to DNA damage (Alexander et al., 2010; Chen et al., 2011).
Autophagy has also been shown to produce a cytoprotective adenosine triphosphate (ATP) surge as an adaptive response against DNA damage (Katayama et al., 2007). After exposure to DNA damaging agents etoposide or temozolomide in malignant glioma cell lines, the induction of autophagy was associated with a cytoprotective surge of ATP generated by oxidative phosphorylation, suggesting that autophagy may be enhancing catabolic metabolism and ATP levels, in order to maintain cell survival in the midst of DNA damage (Katayama et al., 2007).
The tumor-suppressor protein p53 regulates a variety of cell response pathways including cell cycle arrest, apoptosis, and metabolism in response to genotoxic stress. Recent findings have demonstrated a bidirectional role of p53 in autophagy regulation. Under basal, non-stressed conditions, cytoplasmic p53 inhibits autophagy through an mTOR/ AMPK-dependant pathway (Tasdemir et al., 2008b). In p53 deficient conditions (p53 knockout mutants or depletion of p53 protein levels), autophagy is restored and promotes cell survival (Tasdemir et al., 2008a). Inhibition of autophagic flux also induces p53-dependant cell death (Maclean et al., 2008; Zaidi et al., 2001). Conversely, p53 has also been shown to transcriptionally activate autophagy through activation of several autophagy inducers including DRAM1 and Sestrin2 (Budanov and Karin 2008; Crighton et al., 2006). One interpretation emerging from these studies is that cytoplasmic and nuclear p53 may have opposing effects on autophagy.
5.3: Interaction Between Autophagy and Other Stress-Compensatory Systems
Autophagy pathways connect with other cytoprotective pathways, including the antioxidant response pathway regulated by the transcription factor, Nrf2 (nuclear factor, erythroid derived 2, like 2). The scaffold protein, p62, also known as sequestosome-1, is responsible for the selective sequestration of polyubiquinated and other damaged cellular proteins and organelles into aggregates, targeting them to the lysosome for degradation via autophagy (Seibenhener et al., 2007). In autophagy-deficient cells p62 accumulates due to a block in its autophagic degradation, allowing it to interact with other signaling molecules, including Keap1 from the Nrf2/Keap-1 complex (Komatsu et al., 2010; Lau et al., 2010). Accumulation of p62 sequesters Keap1 in the cytoplasm, blocking the Keap1-Nrf2 interaction. This causes Nrf2 stabilization and translocation into the nucleus, activating transcription of Nrf2 target genes. This provides an additional adaptive response to cellular stress when autophagy is defective or unable to meet the demands of stressed cells.
NF-kB is a transcription factor that integrates a diverse array of stress signals including immune signals and autophagy. NF-kB is activated through the phosphorylation of the NF-kB inhibitor (IkB) by IkB kinase (IKK) (Beg et al., 1993). IKK is activated under diverse stress signals, including ROS, DNA damage, and activation of death receptors. NF-kB can directly activate autophagy through the induction of Beclin 1 expression by NF-kB family member p65/RelA (Copetti et al., 2009). IKK can also stimulate autophagy in an NF-kB-independent pathway through activation of the AMPK and JNK1 pathways (Criollo et al., 2010). The interaction between the NF-kB and autophagy pathways illustrates that multiple compensatory pathways can act in concert in response to cellular stress.
5.4 Protein quality control
Principally, there are two protein degradation systems in eukaryotic cells: the ubiquitin/proteasome system (UPS) and the lysosomal system. The normal half-life of cellular proteins ranges widely, from minutes (e.g., the tumor suppressor p53) to days (e.g., myosin) (Kamijo et al., 1998; Martin et al., 1977). Virtually all nucleated cells have constitutive autophagic activity at low basal levels to perform the homeostatic function of protein turnover. Early studies defined two categories of proteins based on their degradation kinetics. Short-lived, rapidly degraded proteins were primarily degraded by the proteasome, whereas long-lived, slowly degraded proteins were degraded by the lysosome (Mortimore and Poso 1987). In addition to the basal degradation of long-lived cellular proteins, autophagy has also been shown to be responsible for the constitutive degradation of aged or damaged proteins and organelles (Elmore et al., 2001; Fortun et al., 2003; Iwata et al., 2006).
A theme repeated in this review is that autophagy can degrade damaged cellular components either by a bulk (non-selective) process or alternatively through a selective process targeting specific proteins and organelles for degradation. In order for a misfolded protein to be degraded by macroautophagy the damaged protein must be tagged and shuttled to the phagophore for degradation. One such adaptor protein involved in the recognition and shuttling of misfolded or aggregated proteins is p62. When denatured or improperly folded proteins accumulate and exceed the cell’s degradation capacity, aggregates of these misfolded proteins can form (Klemes et al., 1981). Most aggregated proteins are decorated with ubiquitin, and p62 has both LC3-II-binding and ubiquitin-binding domains, allowing it to act as a scaffold between ubiquitinated proteins and LC3-II in the membrane of the forming autophagosome (Bjorkoy et al., 2005; Pankiv et al., 2007). Recruitment of polyubiquitinated proteins for autophagic degradation can also be mediated by histone deacetylase 6 (HDAC6). The role of HDAC6 in autophagic degradation of misfolded proteins is unclear, but there is evidence suggesting that HDAC6 activity on microtubules is involved in the fusion of the autophagosome with the lysosome (Ding and Yin 2008). In vitro HDAC6 genetic knockdown studies have revealed that HDAC6 serves as a recruitment protein, required for the autophagic degradation of aggregated proteins by recruiting actin-remodelling machinery to ubiquitinated protein aggregates, which facilitates the fusion of the autophagosome to the lysosome and subsequent clearance of the aggregated proteins (Lee et al., 2010).
In addition to macroautophagy, CMA has also been shown to selectively degrade individual damaged cellular proteins. Exposed hydrophobic regions on damaged proteins present a danger to the cell because the hydrophobic regions can bind to normal proteins and disrupt essential protein-protein interactions (Sherman and Goldberg 2001). Chaperone proteins involved in CMA can recognize specific misfolded proteins by binding to exposed hydrophobic regions (which contain the consensus amino acid motif KFERQ) on the surface of the proteins and transferring those proteins to the CMA degradation system.
5.5: Intracellular Pathogen Defense
Autophagic machinery used to engulf and degrade cellular organelles and proteins can also be used for the selective delivery of microorganisms to the lysosome for degradation in a process termed xenophagy (Amano et al., 2006). Xenophagy can target extracellular bacteria that invade the cell (e.g., Group A Streptococcus) as well as true intracellular bacterial pathogens (e.g., Mycobacterium tuberculosis and Shigella flexneri) (Gutierrez et al., 2004; Nakagawa et al., 2004; Ogawa et al., 2005). In addition, xenophagy has also been shown to target newly synthesized virions during their exit from the nucleus through the cytoplasm (Talloczy et al., 2002). Outside of its direct role in pathogen elimination, xenophagy may also play a cytoprotective function through enhancing immune recognition of infected cells by the production of antigenic bacterial peptides (Paludan et al., 2005). Interestingly, several bacteria and viruses have the ability to co-opt components of the autophagic machinery to establish their own defense mechanism and provide a replicative niche that avoids destruction (Dorn et al., 2001; Pizarro-Cerda et al., 1998).
5.6: Antigen Processing and Presentation
Presentation of antigenic peptides by MHC class I and II molecules is an essential component of adaptive immunity. Autophagy plays a vital role in the generation of peptides and presentation of those peptides by MHC class molecules to epitope-specific T cells (Dorfel et al., 2005; Li et al., 2008; Paludan et al., 2005; Zhou et al., 2005). MHC class I molecules are limited to the recognition of endogenous antigens from viruses, tumors, or self-proteins for presentation to CD8+ T cells (Reviewed in (Jensen 2007)). Autophagy has not been implicated in playing a role in classical MHC class I antigen presentation, but it has been implicated in regulating class I-mediated cross presentation of exogenous antigens. In class I-mediated cross presentation, antigen presenting cells such as dendritic cells internalize and process extracellular antigens and display the resulting peptides on cell surface MHC class I molecules (Shen and Rock 2006). Modulation (induction or inhibition) of autophagy in HEK 293T cells expressing a model antigen, OVA, altered cross presentation in-vitro. The induction of autophagy enhanced the cross presentation of OVA to epitope-specific CD8 + T cells. Conversely, inhibition significantly reduced cross presentation (Li et al., 2008). Autophagy has also been implicated in playing a role in MHC class II antigen processing and presentation. Induction of autophagy due to serum starvation in B-cells resulted in an increase in MHC class II associated peptides derived from intracellular proteins (Dengjel et al., 2005). CMA has also been identified as playing a role in the selective processing and MHC-loading of exogenous or endogenous antigens during antigen presentation. B-cells with reduced levels of CMA proteins (Lamp2a or Hsc70) exhibit decreased presentation of cytoplasmic epitopes on MHC class II molecules. Conversely, an increase in cytoplasmic autoantigen presentation was observed upon over-expression of Lamp2a or Hsc70 (Zhou et al., 2005). In addition, B-cells deficient in Lamp2 had impaired antigen presentation of exogenous antigens and peptides to CD4+ T cells (Crotzer et al., 2010). Interestingly, class II presentation of peptides from endogenous proteins was detected in Lamp2 deficient cells, suggesting that Lamp2 levels may also affect the type of peptides displayed by MHC class II molecules.
6. Autophagy: A view from the organism
6.1: Autophagy and embryonic development
Autophagy is essential to mammalian development, serving roles in adaptation to the stress and starvation that occurs during embryogenesis, and, working in concert with apoptosis, in embryonic cell elimination. In mammals, whole organism genetic knockouts of essential autophagy genes (Beclin 1, Ambra1, Atg7, Atg5) are lethal, either resulting in death at early embryonic stages due to severe developmental defects, or within a few days of birth, failing to survive the starvation period during the transition from placental to gastrointestinal nutrition (Fimia et al., 2007; Komatsu et al., 2005; Kuma et al., 2004; Yue et al., 2003). Autophagy is induced after fertilization in the mouse and is essential for the preimplantation development (Tsukamoto et al., 2008). Its importance extends to the late stages of mammalian embryonic and postnatal development, including neurogenesis, cardiogenesis, hematopoiesis, osteogenesis, and folliculogenesis (Choi et al., 2010; Fimia et al., 2007; Nakai et al., 2007; Pua et al., 2007; Settembre et al., 2008). This role is not peculiar to mammals. Autophagy plays a major role in cellular remodeling during dauer formation in C. elegans, Drosophila metamorphosis, and in yeast sporulation (Denton et al., 2009; Deutschbauer et al., 2002; Melendez et al., 2003).
6.2: Autophagy and constitutive homeostasis
Protein quality control has an important role in all cells, but particularly in non-proliferative cell types that can not dilute damaged proteins through cytoplasmic expansion. In neural cells, autophagy plays a constitutive role in the bulk removal of unnecessary, damaged, or aggregated proteins. Loss of Atg7 in mice leads to neurodegeneration (Komatsu et al., 2006). Mice lacking Atg7 in the central nervous system display behavioral defects, including abnormal limb-clasping reflexes, reduction in coordinated movements, and death within 28 weeks of birth. In neurons of those mice, polyubiquitinated proteins accumulated as inclusion bodies, which increased in size and number with age. Similar observations have been made in mice with neural cell-specific deletion of Atg5 (Hara et al., 2006). These results highlight the importance of constitutive autophagic function in the survival of neural cells and prevention of neurodegenerative disorders through maintaining protein quality control. The importance of constitutive autophagy is not confined to the nervous system. Liver-specific deletion of Atg7 in mice resulted in progressive hepatomegaly accompanied by accumulation of aberrant organelles and heavily ubiquinated proteins (Komatsu et al., 2005). These studies highlight the fact that autophagy is important for the maintenance of normal hepatocyte function and the removal of damaged cellular components that can be deleterious to the cell.
6.3: Autophagy and homeostasis under stress
Autophagy also serves as a mechanism that allows organisms to survive under adverse conditions. At birth the trans-placental nutrient supply is suddenly interrupted and neonates go through a period of severe starvation until nutrient supply can be restored through maternal milk. Neonates adapt to this neonatal starvation period by inducing autophagy. Levels of autophagy in mice remain low during embryogenesis; however autophagy is immediately induced in various tissues after birth. Neonate mice deficient in Atg5 appear almost normal at birth, but die within 1-day (Kuma et al., 2004). Atg5 deficient pups, starved for 10-hours post delivery, had decreased amino acid concentrations in plasma and tissue in comparison to wide-type pups, presumably due to a lack in normal autophagic activity that is required to replenish amino acid pool during periods of nutrient deprivation. Starved pups also displayed signs of energy depletion including activation of AMPK and altered electrocardiograms, highlighting the importance of a sufficient amino acids reservoir in maintaining energy homeostasis.
Autophagy is also a necessary adaptation to starvation in adults, where it is essential to normal cardiac function. After 12 hours of starvation in adult mice, electron-dense vesicles, which increased in number during starvation, appeared in cardiomyocytes (Takemura et al., 2009). Treatment with BafA1 did not affect cardiac function in normally fed mice, but significantly depressed cardiac function and caused significant left ventricular dilation in starved mice, suggesting that autophagy plays a critical role in the maintenance of cardiac function during starvation.
Komatsu et al. generated conditional knockouts of the autophagy gene, Atg7 in the livers of recombinant mice (Komatsu et al., 2005). Livers isolated from nutrient starved knockout mice failed to show characteristics of autophagy. Atg7 deficiency also led to other abnormalities such as accumulation of deformed mitochondria and ubiquitin-positive aggregates in the livers in comparison to starved wild-type mice, suggesting that autophagy deficient mice cannot protect liver function during starvation. Degradation of long-lived proteins was also inhibited in knockout hepatocytes, independent of nutrient deprivation.
6.4: Autophagy and cancer
6.4.1: Autophagy and cancer causation
Cancer was one of the first diseases genetically linked to autophagy defects (Liang et al., 1999). Beclin 1 is monoallelically deleted in 40–75% of cases of human breast, ovarian, and prostate cancer (Aita et al., 1999). Furthermore, hemizygosity of BECN1 (the gene encoding Beclin 1) or complete loss of the gene encoding the UVRAG binding protein Bif-1 (regulates autophagy by forming a complex with Beclin 1 and UVRAG to activate class III PI3K), resulted in increased tumor susceptibility in mice (Qu et al., 2003; Takahashi et al., 2007). In addition, the ectopic expression of BECN1 or UVRAG has been shown to repress the growth of human cancer cell xenografts (Liang et al., 1999; Takahashi et al., 2007). Ambra1, a component of the autophagy activation Beclin 1/ PI3K complex, and Atg4, have also been shown to control cellular growth and exert tumor suppressive effects (Fimia et al., 2007; Marino et al., 2007). Thus, in certain experimental models, autophagy appears to be a tumor-suppressive process.
To complicate matters, however, in other settings autophagy appears to promote tumorogenesis, perhaps by enhancing cell survival under adverse conditions. During tumor growth, periods of poor vascularization lead to limited nutrient availability and hypoxic regions within tumors (Hirst et al., 1991). Under these circumstances, autophagy induction provides a survival advantage to the transformed cells, compensating for the metabolic stress caused by limited nutrients or oxygen (Degenhardt et al., 2006). Thus, the particular point during carcinogenesis at which autophagy is impaired or induced may determine whether it provides a growth advantage or a disadvantage.
6.4.2: Autophagy and cancer treatment
Autophagy induction in tumor cells in response to adverse conditions is one potential mechanism driving tumor resistance to antineoplastic compounds. Liu et al. in 2010 investigated the mechanism of hypoxia-induced resistance to the retinoid, N-(4-Hydroxyphenyl)retinamide (4-HPR) in the human cervical cancer cell line HeLa (Liu et al., 2010). Under hypoxic conditions, 4-HPR induced autophagy through activation of HIF-1α, characterized by an increase in LC3 II protein levels and acidic vesicle formation. Co-treatment with autophagy inhibitors 3MA or chloroquine enhanced apoptosis and decreased cell viability in 4-HPR exposed cells, indicating that autophagy induction conferred hypoxia-induced resistance in HeLa cells. SKBR3/TzbR is a HER2-positive breast cancer cell line that exhibits acquired resistance to the Anti-HER2 monoclonal antibody trastuzumab (Tzb). Constitutive autophagic levels were significantly higher in the Tzb-conditioned cells resistant to Txb (TzbR) versus the parental Tzb-sensitive line (Vazquez-Martin et al., 2009). Knockdown of LC3 expression via siRNA reduced TzbR cell proliferation and increased sensitivity to Tzb treatment. Interestingly, a sub-population of Tzb-naïve parental cells accumulated LC3 punctate structures and decreased p62 expression after treatments with high-dose Tzb, likely promoting their own resistance. Translational research following this general theme has confirmed that genetic or pharmacological inhibition of autophagy can enhance cytotoxicity of cancer chemotherapeutic agents, making this an active area of research aimed at increasing the efficacy of chemotherapy agents (Amaravadi et al., 2007; Carew et al., 2007).
7. Is autophagy important in toxicology?
7.1: Xenobiotic stress can induce autophagy
Cellular damage (ER stress, oxidative stress, or DNA damage) induced by xenobiotics can result in the induction of autophagy as a cytoprotective mechanism to facilitate cell recovery and return to homeostasis. One significant group of environmental toxicants that have been shown to induce autophagy are metals including cadmium, chromium, and arsenic, which activate autophagy through a variety of mechanisms including the generation of ER-stress and oxidative stress. Human CD34++ hematopoietic progenitor cells exposed to 10 µM hexavalent chromium or cadmium showed substantial cell loss (Di Gioacchino et al., 2008). Surviving cells were positive for autophagosomes and autolysosomes, and had extensive organelle damage, suggesting that the induction of autophagy may have been compensatory to cadmium-induced damage. Autophagy induction is being studied as a potential biomarker of toxic metal exposure in blood cells (Di Gioacchino et al., 2008). Our research group has shown that sodium arsenite-induced toxicity in human lymphoblastoid cell lines (LCL) is strongly associated with the induction of autophagy. Arsenite-induced autophagy was also associated with increased protein levels of LC3-II, increased enzymatic activity of lysosomal protease cathepsin D, and coordinated induction of lysosomal gene expression corresponding with increased expression of the lysosome biogenesis master regulator gene, TFEB (Bolt et al., 2010a; Bolt et al., 2010b). In rat proximal convoluted tubule cells, cadmium exposure resulted in the induction of ER stress, which triggered cell proliferation and autophagy as an adaptive response against toxicity (Chargui et al., 2011). Ovarian cancer cell lines, exposed to increasing concentrations of arsenic trioxide displayed characteristics of autophagy in response to oxidative damage (Smith et al., 2010). In that model, arsenic trioxide induced autophagy conferred protection against cellular damage through a Beclin 1-independent pathway.
The importance of autophagy in toxicology is not confined to metal xenobiotics. The induction of autophagy in response to diverse anti-neoplastic compounds and DNA damaging agents was described in previous sections.
7.2: Autophagy can confer xenobiotic resistance
Autophagy induction in response to cellular stress induced by xenobiotic exposure can serve as a resistance mechanism to enable the cell to recover. Human lung epithelial fibroblasts (WI38) exposed to cadmium responded to ER stress through activation of the PERK arm of the UPR (Lim et al., 2010). A resistant WI38 cell line (RWI38) was generated by culturing WI38 cells in media with increasing concentrations of cadmium for six months, selecting for viable cells (Oh et al., 2009). RWI38 cells exposed to cadmium do not demonstrate ER stress or mitochondrial apoptosis, and are 4-fold resistance to cadmium toxicity in comparison the parent cell line. Cadmium exposure in RWI38 is associated with the induction of autophagy. This induction was demonstrated by an increase in Atg5 and LC3-II protein, and formation of a GFP-LC3 punctate fluorescence pattern.
Treatment of human renal epithelial cells with the immunosuppressive drug cyclosporine for 24 hours induced ER stress and the subsequent induction of autophagy. Inhibition of autophagy through siRNA gene silencing of the autophagy gene, Beclin 1, significantly increased tubular cell death, indicating that autophagy induction is a novel resistance mechanism against ER stress induced tubular damage (Pallet et al., 2008).
Rat hepatocytes can induce autophagy as a protective mechanism to minimize hepatotoxicity due to acetaminophen (APAP) overdose (Ni et al., 2011). Pharmacological inhibition of autophagy further exacerbated APAP-induced hepatotoxicity, implicating autophagy as an in vivo xenobiotic stress-compensatory mechanism. The induction of autophagy, likely activated through the generation of ROS and possible inhibition of mTOR, eliminated damaged mitochondria preventing further damage from occurring. This suggests that the induction of autophagy could be a novel therapeutic approach to preventing APAP-induced liver damage.
7.3: Xenobiotics can perturb / modulate autophagy
Xenobiotics can modulate the autophagy process, providing an opportunity to manipulate autophagy in order to achieve better therapeutic efficacy or conversely, to reduce toxicity and disease progression (Table 1). Treatment of cells with the mTOR inhibitor, rapamycin, is an established experimental method used to induce macroautophagy, however due to the lack of specificity the clinical implications of rapamycin treatment are limited (Ravikumar et al., 2004). Other mechanisms that regulate autophagy, independent of mTOR inhibition, have been reported using other xenobiotics. Calcium channel blockers, including verapamil and nimodipine, induce autophagy through the reduction of intracytosolic calcium levels (Williams et al., 2008). Lithium induces autophagy through the inhibition of inositol monophosphatase, which results in the reduction of myo-inositol-1,4,5-triphosphate (IP3) levels (Sarkar et al., 2005).
Table 1.
Autophagy Modulators
| Xenobiotic | Mechanism of Action |
Effect on Autophagy | References |
|---|---|---|---|
| 3-Methyladenine | PI3K Inhibitor |
Inhibit, block formation of autophagosome |
(Petiot et al., 2000) |
| Wortmannin | PI3K Inhibitor |
Inhibit, block formation of autophagosome |
(Petiot et al., 2000) |
| Vinblastine | Damages microtubules |
Inhibit, block fusion of autophagosome with lysosome |
(Kovacs et al., 1982) |
| Chloroquine | Lysosomotropic agent, weak base amine, alters lysosome pH |
Inhibit, Alter lysosome hydrolase activity and block fusion of autophagosome to lysosome |
(Boya et al., 2005) |
| Bafilomycin A1 | Vacuolar-type H+-ATPase Inhibitor, alters lysosome pH |
Inhibit, Alter lysosome hydrolase activity and block fusion of autophagosome to lysosome |
(Yamamoto et al., 1998) |
| Rapamycin | mTOR Inhibitor | Induce | (Ravikumar et al., 2004) |
| Verapamil and Nimodipine |
Calcium Channel Blocker |
Induce, Reduction of cytosolic Ca+2 |
(Williams et al., 2008) |
| Lithium | Inhibition of IMPase, Reduction in IP3 |
Induce | (Sarkar et al., 2005) |
Abbreviations: phosphoinositide-3-kinase (PI3K), mammalian target of rapamycin (mTOR), inositol monophosphatase (IMPase), myo-inositol-1,4,5-triphosphate (IP3)
Clomipramine, an FDA-approved tricyclic antidepressant, and its active metabolite, desmethylclomiramine (DCMI), modulate autophagic flux (Rossi et al., 2009). Treatment with DCMI caused a significant increase in LC3-II protein levels and GFP-LC3 puncta. The study revealed that the increase of autophagosome markers actually resulted from blocked degradation of autophagic cargo or inhibition of autophagic flux. These findings highlight an important caveat in the interpretation of an increase in autophagic structures and proteins as conclusive evidence of autophagy induction. Interestingly, DCMI-mediated inhibition of autophagic flux increased the cytotoxic effects of several chemotherapeutic agents, making it a promising candidate for use in conjunction with other chemotherapy agents to increase the efficacy of treatment. Our group determined that at cytotoxic concentrations (6 µM, 96 hrs), sodium arsenite treatment decreased autophagic flux in LCL. These findings suggest that arsenite can not only induce autophagy, but also block the breakdown of autophagic vesicles, resulting in the accumulation of autophagosomes inside of the cell (Bolt et al., 2010b). In the high-content screen of cytotoxic agents from the NCI panel, 26% of the chemicals that induced GFP-LC3 puncta failed to stimulate autophagic flux, suggesting that they actually block autophagy. Further investigation into these chemicals revealed that they all disrupted the microtubular network, which may disrupt the fusion of the autophagosome with the lysosome (Shen et al., 2011).
Several xenobiotics have a long history of experimental use in autophagy research because of their ability to modulate autophagy. 3-methyladenine and wortmannin are PI3K (class I and III) inhibitors that block autophagic sequestration of damaged cellular components (Petiot et al., 2000). Other inhibitory compounds block post-sequestration steps. Vinca alkaloids, including vinblastine and other microtubule damaging agents, block fusion of the autophagosome to the autolysosome (Kovacs et al., 1982). Alternatively, some compounds modulate autophagy by elevating lysosome pH. These include bafilomycin A1, which is a vacuolar-type H+-ATPase inhibitor, and chloroquine, a lysosomotropic weak base amine. Both alter lysosomal hydrolase activity and block fusion of the autophagosome to the lysosome (Boya et al., 2005; Yamamoto et al., 1998).
8: Concluding Remarks
Autophagy is a critical biological process that plays roles in constitutive and stress-induced cellular homeostasis. An increased research focus on autophagy is rapidly expanding our appreciation of the many biological processes impacted by this process. In this review we have highlighted facets of autophagy that underscore its importance in toxicology studies. Autophagy is positioned at the balance point between cellular compensation for xenobiotic stress, and stress-induced cell death. Xenobiotics can induce autophagy as well as impair autophagic induction. It is clear that defining the role of autophagy in xenobiotic-induced human toxicity will be difficult, given that the outcome of autophagy can depend on many things: the toxicant, the exposure duration, the cell type, the developmental stage, and the disease stage. But it is equally clear that failing to appreciate the role of autophagy in toxicology will limit our ability to fully explain how biological systems can be harmed by environmental toxicants.
References
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14(3):500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114(Pt 13):2491–2499. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- Ahlberg J, Glaumann H. Uptake--microautophagy--and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp Mol Pathol. 1985;42(1):78–88. doi: 10.1016/0014-4800(85)90020-6. [DOI] [PubMed] [Google Scholar]
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59(1):59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano A, Nakagawa I, Yoshimori T. Autophagy in innate immunity against intracellular bacteria. J Biochem. 2006;140(2):161–166. doi: 10.1093/jb/mvj162. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(2):326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol. 1962;12:198–202. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Thumm M. A genomic screen identifies AUT8 as a novel gene essential for autophagy in the yeast Saccharomyces cerevisiae. Gene. 2001;274(1–2):151–156. doi: 10.1016/s0378-1119(01)00614-x. [DOI] [PubMed] [Google Scholar]
- Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13(6):3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4(12):e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(6):1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt AM, Douglas RM, Klimecki WT. Arsenite exposure in human lymphoblastoid cell lines induces autophagy and coordinated induction of lysosomal genes. Toxicol Lett. 2010a;199(2):153–159. doi: 10.1016/j.toxlet.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt AM, Byrd RM, Klimecki WT. Autophagy is the predominant process induced by arsenite in human lymphoblastoid cell lines. Toxicol Appl Pharmacol. 2010b;244(3):366–373. doi: 10.1016/j.taap.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursch W, Karwan A, Mayer M, Dornetshuber J, Frohwein U, Schulte-Hermann R, Fazi B, Di Sano F, Piredda L, Piacentini M, Petrovski G, Fesus L, Gerner C. Cell death and autophagy: cytokines, drugs, and nutritional factors. Toxicology. 2008;254(3):147–157. doi: 10.1016/j.tox.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17(8):1595–1607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–322. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, Duranton C, Tauc M, Hofman P, Poujeol P, El May MV, Mograbi B. Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci. 2011;121(1):31–42. doi: 10.1093/toxsci/kfr031. [DOI] [PubMed] [Google Scholar]
- Chen LH, Loong CC, Su TL, Lee YJ, Chu PM, Tsai ML, Tsai PH, Tu PH, Chi CW, Lee HC, Chiou SH. Autophagy inhibition enhances apoptosis triggered by BO-1051, an N-mustard derivative, and involves the ATM signaling pathway. Biochem Pharmacol. 2011;81(5):594–605. doi: 10.1016/j.bcp.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Chiang HL, Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem. 1988;263(14):6797–6805. [PubMed] [Google Scholar]
- Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246(4928):382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Choi JY, Jo MW, Lee EY, Yoon BK, Choi DS. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil Steril. 2010;93(8):2532–2537. doi: 10.1016/j.fertnstert.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Clark SL., Jr Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol. 1957;3(3):349–362. doi: 10.1083/jcb.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contento AL, Xiong Y, Bassham DC. Visualization of autophagy in Arabidopsis using the fluorescent dye monodansylcadaverine and a GFP-AtATG8e fusion protein. Plant J. 2005;42(4):598–608. doi: 10.1111/j.1365-313X.2005.02396.x. [DOI] [PubMed] [Google Scholar]
- Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29(10):2594–2608. doi: 10.1128/MCB.01396-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, Tailler M, Delahaye N, Tesniere A, De Stefano D, Younes AB, Harper F, Pierron G, Lavandero S, Zitvogel L, Israel A, Baud V, Kroemer G. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29(3):619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotzer VL, Glosson N, Zhou D, Nishino I, Blum JS. LAMP-2-deficient human B cells exhibit altered MHC class II presentation of exogenous antigens. Immunology. 2010;131(3):318–330. doi: 10.1111/j.1365-2567.2010.03309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem. 1994;269(42):26374–26380. [PubMed] [Google Scholar]
- Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269(5 Pt 1):C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1(7):570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Hu W, Lim B, Dice JF. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell. 1998;9(8):1995–2010. doi: 10.1091/mbc.9.8.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83(3–4):301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1(2):66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102(22):7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19(20):1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99(24):15530–15535. doi: 10.1073/pnas.202604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gioacchino M, Petrarca C, Perrone A, Farina M, Sabbioni E, Hartung T, Martino S, Esposito DL, Lotti LV, Mariani-Costantini R. Autophagy as an ultrastructural marker of heavy metal toxicity in human cord blood hematopoietic stem cells. Sci Total Environ. 2008;392(1):50–58. doi: 10.1016/j.scitotenv.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3(4):295–299. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4(2):141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- Dorfel D, Appel S, Grunebach F, Weck MM, Muller MR, Heine A, Brossart P. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood. 2005;105(8):3199–3205. doi: 10.1182/blood-2004-09-3556. [DOI] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69(9):5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott A, Reiners JJ., Jr Suppression of autophagy enhances the cytotoxicity of the DNA-damaging aromatic amine p-anilinoaniline. Toxicol Appl Pharmacol. 2008;232(2):169–179. doi: 10.1016/j.taap.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15(12):2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol. 2008a;266:207–247. doi: 10.1016/S1937-6448(07)66005-5. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Fine structure of the autophagosome. Methods Mol Biol. 2008b;445:11–28. doi: 10.1007/978-1-59745-157-4_2. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy. 2005;1(3):141–145. doi: 10.4161/auto.1.3.2000. [DOI] [PubMed] [Google Scholar]
- Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23(33):10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J. 2003;375(Pt 1):75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11(7):526–534. [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270(5):2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst DG, Hirst VK, Joiner B, Prise V, Shaffi KM. Changes in tumour morphology with alterations in oxygen availability: further evidence for oxygen as a limiting substrate. Br J Cancer. 1991;64(1):54–58. doi: 10.1038/bjc.1991.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281(7):4035–4041. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- Jensen PE. Recent advances in antigen processing and presentation. Nat Immunol. 2007;8(10):1041–1048. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998;95(14):8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang KB, Zhu C, Yong SK, Gao Q, Wong MC. Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol Cancer. 2009;8:66. doi: 10.1186/1476-4598-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24(6):980–991. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14(3):548–558. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15(11):4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147(2):435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemes Y, Etlinger JD, Goldberg AL. Properties of abnormal proteins degraded rapidly in reticulocytes. Intracellular aggregation of the globin molecules prior to hydrolysis. J Biol Chem. 1981;256(16):8436–8444. [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3(3):181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5(4):539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S, Kaufman RJ, Weppler SA, Voncken JW, Lambin P, Koumenis C, Sonenberg N, Wouters BG. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25(5):1114–1125. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14(2):230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- Kovacs AL, Reith A, Seglen PO. Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp Cell Res. 1982;137(1):191–201. doi: 10.1016/0014-4827(82)90020-9. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10(5):602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J, Corcoran M, Bjorklund AC, Hultenby K, Lundin J, Heyman M, Soderhall S, Mazur J, Porwit A, Pandolfi PP, Zhivotovsky B, Panaretakis T, Grander D. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009;16(7):1018–1029. doi: 10.1038/cdd.2009.46. [DOI] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30(13):3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Kim NY, Suh YA, Lee C. Involvement of ROS in Curcumin-induced Autophagic Cell Death. Korean J Physiol Pharmacol. 2011;15(1):1–7. doi: 10.4196/kjpp.2011.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68(17):6889–6895. doi: 10.1158/0008-5472.CAN-08-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lim SC, Hahm KS, Lee SH, Oh SH. Autophagy involvement in cadmium resistance through induction of multidrug resistance-associated protein and counterbalance of endoplasmic reticulum stress WI38 lung epithelial fibroblast cells. Toxicology. 2010;276(1):18–26. doi: 10.1016/j.tox.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Liu XW, Su Y, Zhu H, Cao J, Ding WJ, Zhao YC, He QJ, Yang B. HIF-1alpha-dependent autophagy protects HeLa cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol Res. 2010;62(5):416–425. doi: 10.1016/j.phrs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118(1):79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36(12):2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282(25):18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- Martin AF, Rabinowitz M, Blough R, Prior G, Zak R. Measurements of half-life of rat cardiac myosin heavy chain with leucyl-tRNA used as precursor pool. J Biol Chem. 1977;252(10):3422–3429. [PubMed] [Google Scholar]
- Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. In situ detection of starvation-induced autophagy. J Histochem Cytochem. 2006;54(1):85–96. doi: 10.1369/jhc.5A6743.2005. [DOI] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103(15):5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301(5638):1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(Pt 9):1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999;18(14):3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–564. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, Menissier de Murcia J, Almendros A, Ruiz de Almodovar M, Oliver FJ. PARP-1 is involved in autophagy induced by DNA damage. Autophagy. 2009;5(1):61–74. doi: 10.4161/auto.5.1.7272. [DOI] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2011 doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307(5710):727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Oh SH, Lee SY, Choi CH, Lee SH, Lim SC. Cadmium adaptation is regulated by multidrug resistance-associated protein-mediated Akt pathway and metallothionein induction. Arch Pharm Res. 2009;32(6):883–891. doi: 10.1007/s12272-009-1610-6. [DOI] [PubMed] [Google Scholar]
- Oku M, Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 2010;277(16):3289–3294. doi: 10.1111/j.1742-4658.2010.07741.x. [DOI] [PubMed] [Google Scholar]
- Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem. 2005;280(36):31582–31586. doi: 10.1074/jbc.M506736200. [DOI] [PubMed] [Google Scholar]
- Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61(2):439–444. [PubMed] [Google Scholar]
- Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, Thervet E, Anglicheau D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4(6):783–791. doi: 10.4161/auto.6477. [DOI] [PubMed] [Google Scholar]
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307(5709):593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15(10):1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90(2):313–323. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goni I, Moreno E, Gorvel JP. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 1998;66(12):5711–5724. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5(3):393–396. doi: 10.4161/auto.5.3.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rieber M, Rieber MS. Sensitization to radiation-induced DNA damage accelerates loss of bcl-2 and increases apoptosis and autophagy. Cancer Biol Ther. 2008;7(10):1561–1566. doi: 10.4161/cbt.7.10.6540. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2(1):39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, Bampton ET, Glynn P, Bonanno G, Knight RA, Nicotera P, Melino G. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci. 2009;122(Pt 18):3330–3339. doi: 10.1242/jcs.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouschop KM, Ramaekers CH, Schaaf MB, Keulers TG, Savelkouls KG, Lambin P, Koritzinsky M, Wouters BG. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother Oncol. 2009;92(3):411–416. doi: 10.1016/j.radonc.2009.06.029. [DOI] [PubMed] [Google Scholar]
- Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275(35):27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15(1):105–112. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170(7):1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Geetha T, Wooten MW. Sequestosome 1/p62--more than just a scaffold. FEBS Lett. 2007;581(2):175–179. doi: 10.1016/j.febslet.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Arteaga-Solis E, McKee MD, de Pablo R, Al Awqati Q, Ballabio A, Karsenty G. Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes Dev. 2008;22(19):2645–2650. doi: 10.1101/gad.1711308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB Links Autophagy to Lysosomal Biogenesis. Science. 2011 doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101(52):18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Rock KL. Priming of T cells by exogenous antigen cross-presented on MHC class I molecules. Curr Opin Immunol. 2006;18(1):85–91. doi: 10.1016/j.coi.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Shen S, Kepp O, Michaud M, Martins I, Minoux H, Metivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene. 2011 doi: 10.1038/onc.2011.168. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29(1):15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- Smith DM, Patel S, Raffoul F, Haller E, Mills GB, Nanjundan M. Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells. Cell Death Differ. 2010;17(12):1867–1881. doi: 10.1038/cdd.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics. 2000;154(2):787–801. doi: 10.1093/genetics/154.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9(10):1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura G, Kanamori H, Goto K, Maruyama R, Tsujimoto A, Fujiwara H, Seishima M, Minatoguchi S. Autophagy maintains cardiac function in the starved adult. Autophagy. 2009;5(7):1034–1036. doi: 10.4161/auto.5.7.9297. [DOI] [PubMed] [Google Scholar]
- Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99(1):190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy. 2008a;4(6):810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008b;10(6):676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Chung YM, Takahashi Y, Xu Z, Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol. 2008;10(4):460–467. doi: 10.1038/ncb1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321(5885):117–120. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One. 2009;4(7):e6251. doi: 10.1371/journal.pone.0006251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23(1):33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17(12):5094–5104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281(40):30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Tuttle DL, Shi YJ, Ralph GS, Dunn WA., Jr Glucose-induced microautophagy in Pichia pastoris requires the alpha-subunit of phosphofructokinase. J Cell Sci. 1997;110(Pt 16):1935–1945. doi: 10.1242/jcs.110.16.1935. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi AU, McDonough JS, Klocke BJ, Latham CB, Korsmeyer SJ, Flavell RA, Schmidt RE, Roth KA. Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J Neuropathol Exp Neurol. 2001;60(10):937–945. doi: 10.1093/jnen/60.10.937. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4(3):378–380. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Xue T, Yang X, Zhu H, Ding X, Lou L, Lu W, Yang B, He Q. Autophagy plays an important role in sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol Appl Pharmacol. 2010;248(1):20–27. doi: 10.1016/j.taap.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, Brutkiewicz RR, Blum JS. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22(5):571–581. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]