

Abstract
Mutations in the GARS gene cause CMT2D and dSMA V—allelic disorders characterized by predominantly distal upper extremity weakness and atrophy, typically beginning during the second decade of life. Here, we report monozygotic twin girls with onset of weakness in infancy and a previously reported GARS mutation within the anticodon binding domain. The severity and remarkable similarity in phenotypes of these girls and the reported case suggest that mutations within the anticodon binding domain are more damaging to aminoacyl tRNA synthetase function than those within other domains of GARS.
Keywords: GARS, glycine aminoacyl tRNA synthetase, CMT2D, dSMA V, anticodon binding domain
Introduction
Mutations in the GARS gene (OMIM 600287), which encodes a class II aminoacyl tRNA synthetase responsible for catalyzing the esterification of glycine to glycine-specific tRNAs, have been shown to cause the allelic, autosomal dominant disorders Charcot-Marie-Tooth 2D (CMT2D, OMIM 601472) and distal spinal muscular atrophy type V (dSMA V, OMIM 600794) (Antonellis, et al., 2003). Both clinical phenotypes present with predominantly distal, upper extremity weakness and atrophy, and a slowly progressive course. The principle phenotypic difference between patients with CMT2D and dSMA V is that those with the former exhibit mild-to-moderate sensory deficits (vibration > light touch, pain, temperature), while patients with the latter show no sensory signs—at least until advanced stages of disease (Sivakumar, et al., 2005, Rohkamm, et al., 2007). Both phenotypes typically become symptomatic during the second decade of life. However, there have been reports of infantile/childhood and late adult onset, indicating a wider phenotypic spectrum. (Sivakumar, et al., 2005, Motley, et al., 2009).
To date, 11 disease-causing mutations have been reported in humans. While most lie within the WHEP-TRS or catalytic domains, James, et al., 2006 described two patients with mutations within the anticodon binding domain, one of whom was the first published case of infantile onset dSMA V. Here we present a second family with an anticodon binding domain mutation and infantile onset of weakness, similar to a patient reported by James et al. (2006).
Case Report
Monozygotic twin girls were born at 31 6/7 weeks gestation without evidence of neuromuscular disease. By 6 months of age, neither child had been observed moving toes or feet. At 16 months, both girls had delayed motor milestones and were losing motor skills (e.g. ability to bounce while supported and to army crawl). They did not have a pincer grasp, and used their heads to push buttons on toys. On examination, both girls were <1st percentile for both height and weight. They were cognitively normal but had marked diffuse hypotonia, symmetrical diffuse weakness, and absent or diminished deep tendon reflexes (DTRs). Serum CK was normal (101 U/L). SMN1 testing was normal.
A muscle biopsy in Twin A at 18 months of age revealed wide variation in fiber size (5 – 40 μm) (Fig. 1A) and striking type I fiber predominance (Fig. 1B, 1C) — features most consistent with a congenital myopathy. EMG/NCV studies on Twin B revealed decreased ulnar nerve amplitudes, p-waves, and fibrillations, consistent with a motor neuronopathy.
Figure 1.
Frozen sections of skeletal muscle biopsy from Twin A at 18 months of age stained with hematoxylin and eosin (A) as well as immunoperoxidase staining for slow (B) and fast (C) myosin heavy chain. A wide variation in fiber size (ranging from 5 – 40 μm in diameter) as well as a striking type I fiber predominance can be appreciated.
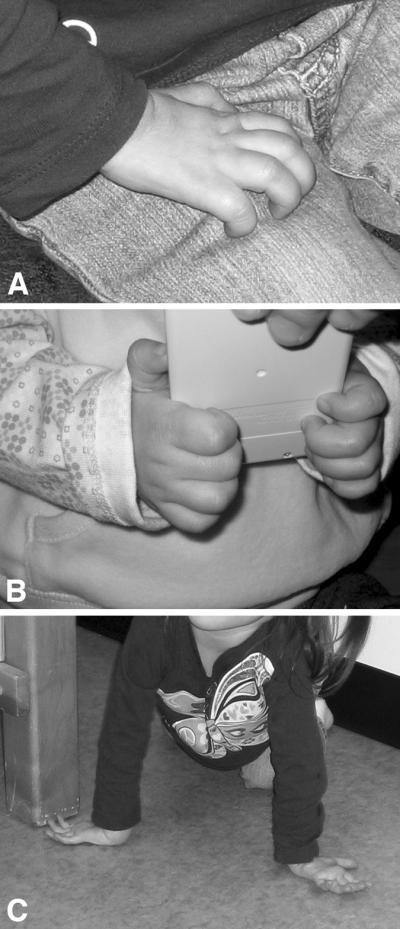
By 23 months, hand weakness had progressed (Fig. 2A, 2B) and they had intrinsic hand muscle wasting. They crawled by placing the dorsal surfaces of one or both hands against the floor (Fig. 2C). At age 3 years, 9 months, both girls lacked foot/toe movement and had limited use of their fingers. They walked only with walkers. Both have hyperlordosis and twin B has scoliosis. Vocal cord dysfunction was documented in both girls, and was clinically apparent (inspiratory stridor) in one.
Figure 2.

(A) The resting hand of Twin B shows extension at the metacarpal-phalangeal joints and flexion to approximately 90° at the proximal and distal interphalangeal joints. Intrinsic hand musculature wasting is also evident. (B) The palmar grip of one of the girls as she holds a small calculator. (C) Twin B shown crawling on the dorsal surfaces of both hands.
Genetic Analysis
PCR amplification and bi-directional DNA sequencing of the GARS gene revealed that Twins A and B have a missense G652A (c.1955G>C) mutation (Athena Diagnostics). Parental sequencing of GARS was normal. This G652 is highly conserved across species (to Drosophila melanogaster). While SIFT analysis predicts this genetic change is tolerated, PolyPhen-2 (HumVar) analysis predicts this variation is possibly damaging.
Discussion
We report a second family with a severe, infantile onset presentation of CMT2D/dSMA V due to a G652A mutation in the anticodon binding domain of GARS. The present cases are quite similar to the previously reported patient with this mutation (proband in Family 1 in James et al., 2006), whose phenotype consists of infantile onset, marked delay in motor milestones, hand weakness, distal wasting of legs with inability to walk independently, hyperlordosis, and scoliosis. Our patients have the additional finding of vocal cord dysfunction, which was not documented by James et al.
This mutation causes a complete loss of the enzyme's synthetase activity in a functional assay, perhaps explaining the early onset and severe phenotype (Talbot and Davies, 2007). One other mutation in the anticodon binding domain of GARS has been reported: c.2260C>T (S581L), affecting a mother and her son (Family 2 in James, et al., 2006). While the mother had a clinical presentation more typical of CMT2D/dSMA V (second decade onset), her son became symptomatic at 4 years of age. Interestingly, both had foot deformities. The few patients described suggest that mutations in the anticodon binding domain of GARS are likely to cause a severe, early onset disease phenotype.
Muscle biopsy in our family did not manifest the typical pathologic features of childhood neuropathy or motor neuron disease, instead showing scattered atrophic and hypertrophic fibers with type I fiber predominance (Fig. 1). These features suggest the possibility of a combined pathogenesis involving motor neurons and skeletal muscle.
Due to the twins' young age, an accurate sensory exam has not been possible. Therefore, they are not yet classified as CMT2D or dSMA V. This distinction however, might not be clinically important, as there is evidence of both inter- and intra-familial phenotypic variation in individuals with GARS mutations (Del Bo, et al., 2006), and some patients with late stage dSMA V eventually develop sensory signs (Sivakumar, et al., 2005 and Rohkamm, et al., 2007).
Table 1.
| Twins A and B | Proband of Family 1 in James, et al. (5) | Average CMT2D phenotype | Average dSMA V phenotype | |
|---|---|---|---|---|
| Onset | Infantile | 6 months | 2nd decade (4,9) | 2nd decade (3) |
| 1st Symptoms | No spontaneous movement of feet or toes. Gross developmental delay with some regression of skills. |
“floppy” feet | Hand weakness +/− cramping and pain (9) | UE in 6/9 LE in 1/9 UE+LE in 2/9 (3) |
| Inheritance | De novo | De novo | Autosomal Dominant or de novo | Autosomal Dominant or de novo |
| Mutation with location | GARS: G652A in Anticodon Binding Domain | GARS: G598A in Anticodon Binding Domain |
GARS: G240R, E71G, P244L, I280F, D500N, S581L. Predominantly in catalytic domain, centered around the dimer interface (3,7) |
GARS: L129P, E71G, H418R, G526, D500N. BSCL2: N88S (3) |
| Distal axonal neuropathy with UE>LE | UE symptoms = LE symptoms | No, LE symptoms > UE symptoms | Yes, but 20/20 developed bilateral foot and peroneal weakness and atrophy within an average of 3.3 yrs after hand atrophy (9) | 8/12 with UE+LE motor deficits 6/12 with UE+LE atrophy (3) |
| Course | Slowly Progressive | Slow-to-moderate progression | Slowly Progressive | Slowly Progressive |
| Limited use of fingers/hands | Yes: finger extension difficult; impaired grip | Yes: impaired finger grip at age 7 yrs | Reported intrinsic hand muscle wasting, but no clear report of decreased hand function | Reported intrinsic hand muscle wasting, but no clear report of decreased hand function |
| Pes Cavus | No, flat feet | No, flat feet | 16/20 (9) | 5/12 subjects (3); 4/38 (9) |
| LE Distal Wasting | Yes--at 2 yrs | Yes--at 3 yrs | 7/20 (9) | 1/12 subjects (3); 12/40 (9) |
| UE DTRs | 1–2+ | Absent | Absent (4) | Absent in 5/12 (3) |
| LE DTRs | Absent | Absent | Absent or decreased in 17/20 (4,9) | Absent or decreased in 1/12 (3); 25/40 (9) |
| Pyramidal Signs | No | No | None or rare (8,9) | 5/40 (9); “Some” (3); “Common” (8) |
| Sensory Signs | None to date (4 years) | None by age 7 years | Yes: vibration deficits common in most CMT2D patients; decreased touch, pain, and temperature sensation in some (9) | Rarely, but in advanced stages of disease--average of 30 yrs after symptom onset (8,9) |
| Hyperlordosis | Yes (severe in Twin B) | Yes, severe | None Reported | None Reported |
| Scoliosis | No in Twin A; Yes in Twin B | Yes | Reported in some: 4/60 (9) | 3/12 (3) |
| Independent Ambulation | Not to date, but walk with AFOs and reverse walker | No | Yes, but 5/60 developed need for assistance in advanced stages of disease (9) |
Acknowledgements
JME, SAM, and KDM receive support from a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (MDCRC) grant from the National Institutes of Health (Bethesda, MD) (NIH U54 NS053672). JME is a medical student research fellow at the MDCRC.
We graciously thank the parents of the reported girls for their cooperation and assistance in helping us obtain clinical photographs. We also wish to express our gratitude to Christina Trout, MSN, for organizing correspondence with the family as this manuscript was being prepared. Finally, thank you to Joel Carl for assistance in assembling the figures.
References
- 1.Antonellis A, Ellsworth R, Sambuughin N, Puls I, Abel A, Lee-Lin S, Jordanova A, Kremensky I, Christodoulou K, Middleton L, Sivakumar K, Ionasescu V, Funalot B, Vance J, Goldfarb L, Fischbeck K, Green E. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am.J.Hum.Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Del Bo R, Locatelli F, Corti S, Scarlato M, Ghezzi S, Prelle A, Fagiolari G, Moggio M, Carpo M, Bresolin N, Comi G. Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology. 2006;66:752–754. doi: 10.1212/01.wnl.0000201275.18875.ac. [DOI] [PubMed] [Google Scholar]
- 3.Dubourg O, Azzedine H, Ben Yaou R, Pouget J, Barois A, Meininger V, Bouteiller D, Ruberg M, Brice A, LeGuern E. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721–1726. doi: 10.1212/01.wnl.0000218304.02715.04. [DOI] [PubMed] [Google Scholar]
- 4.Ionasescu V, Searby C, Sheffield V, Roklina T, Nishimura D, Ionasescu R. Autosomal dominant Charcot-Marie-Tooth axonal neuropathy mapped on chromosome 7p (CMT2D) Human Molecular Genetics. 1996;5:1373–1375. doi: 10.1093/hmg/5.9.1373. [DOI] [PubMed] [Google Scholar]
- 5.James P, Cader M, Muntoni F, Childs A-M, Crow Y, Talbot K. Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology. 2006;67:1710–1712. doi: 10.1212/01.wnl.0000242619.52335.bc. [DOI] [PubMed] [Google Scholar]
- 6.Motley W, Talbot K, Fischbeck K. GARS axonopathy: not every neuron's cup of tRNA. Trends in Neurosciences. 2009;33:59–66. doi: 10.1016/j.tins.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nangle L, Zhang W, Xie W, Yang X-L, Schimmel P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. PNAS. 2007;104:11239–11244. doi: 10.1073/pnas.0705055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohkamm B, Reilly M, Lochmüller H, Schlotter-Weigel B, Barisic N, Schöls L, Nicholson G, Pareyson D, Laurà M, Janecke A, Miltenberger-Miltenyi G, John E, Fischer C, Grill F, Wakeling W, Davis M, Pieber T, Auer-Grumbach M. Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. Journal of the Neurological Sciences. 2007;263:100–106. doi: 10.1016/j.jns.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivakumar K, Kyriakides T, Puls I, Nicholson G, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs J, Zamba-Papanicolaou E, Ionasescu V, Dalakas M, Green E, Fischbeck K, Goldfarb L. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128:2304–2314. doi: 10.1093/brain/awh590. [DOI] [PubMed] [Google Scholar]
- 10.Talbot K, Davies K. Spinal muscular atrophies and hereditary motor neuropathies. In: Eisen A, Shaw P, editors. Handbook of Clinical Neurology, Vol 82 (3rd series). Motor Neuron Disorders and Related Diseases. Elsevier; 2007. pp. 141–153. [DOI] [PubMed] [Google Scholar]