

The ability to clone and express the variable (VH & VL) and constant (CH & CL) domains of monoclonal antibodies has made the construction of genetically engineered antibodies possible.[1-3] Antibody fragments, such as Fabs, have been fused to self-assembling protein domains such as leucine zipper regions of the transcription factors or to the CL and CH1 domains of IgG.[4, 5] Recombinant antibodies, referred to as single chain antibodies (scFv's), have been constructed by fusing the VH and VL domains of an antibody variable region (Fv) via a peptide linker. This advance resulted in the development of a variety of approaches for the preparation of the corresponding fusion relatives, diabodies (bivalent) and tandabs (tetravalent) consisting of a single polypeptide.[6-10] Although the current methods of preparing recombinant antibodies have a variety of advantages, in general control over their assembly and disassembly is not possible without resorting to conditions necessary for protein unfolding. Since the molecular weight and dimensions of engineered antibodies is a significant determinant of their in vivo avidity, biodistribution and pharmacokinetics, a method allowing these parameters to be temporally altered by chemically controlled assembly and disassembly would in principle enhance the therapeutic and diagnostc utility of recombinant antibodies[11, 12].
Recently, we have developed a protocol for the preparation of self-assembling protein macrocyclic oligomers, or nanorings, of dihydrofolate reductase fusion proteins (DHFR2) by chemical induction with bis-methotrexate (Bis-MTX). Self-assembling nanorings ranging in diameter from 7 to 30 nm could easily be prepared from two to eight DHFR2 monomers, respectively.[13] The nanoring size was found to be governed by the specific length and composition of the peptide inserted between the DHFRs. For example, when the linker peptide between the DHFR2 was a 13 amino acid flexible linker, dimer formation was observed almost exclusively after incubation with Bis-MTX. In comparison, octamers were observed when the peptide between the DHFR2 was a single glycine. Inspection of the x-ray crystal structure of the Bis-MTX dimerized DHFR suggested that proteins, such as scFv's, could be appended to DHFR2 fusion proteins without loss of their ability to undergo chemically induced macrocyclic oligomerization.[14] Thus, we hypothesized that scFv-DHFR2 fusion proteins could be induced by Bis-MTX to self-assemble into bivalent, tetravalent and octavalent antibody nanorings. To test our hypothesis, a DHFR2 scFv fusion protein that binds to the T-cell antigen, CD3 epsilon, of the human T-cell receptor was prepared. The binding affinities and cell surface behavior of the monovalent and bivalent scFv fusion protein were compared with those for the parental monoclonal antibody. (Figure 1a) In addition, we assessed the ability of trimethoprim, a non-toxic competitive inhibitor of DHFR and FDA approved antiobiotic, to carry out disassembly of the bivalent antibody nanoring. (Figure 1a)
Figure 1. Design and evolution of bivalent single-chain antibody.
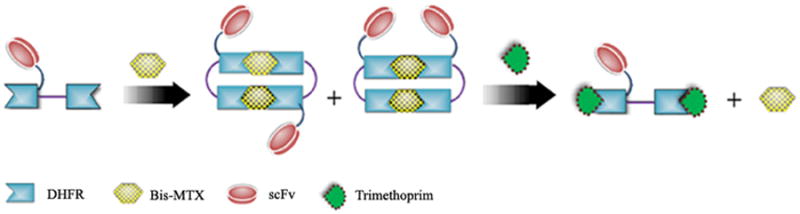

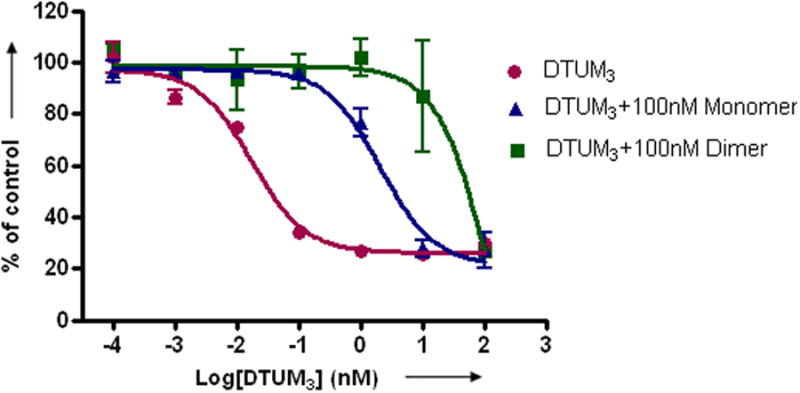
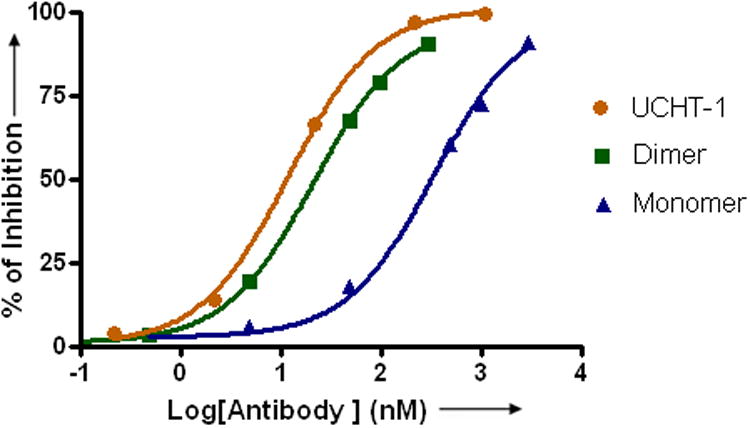
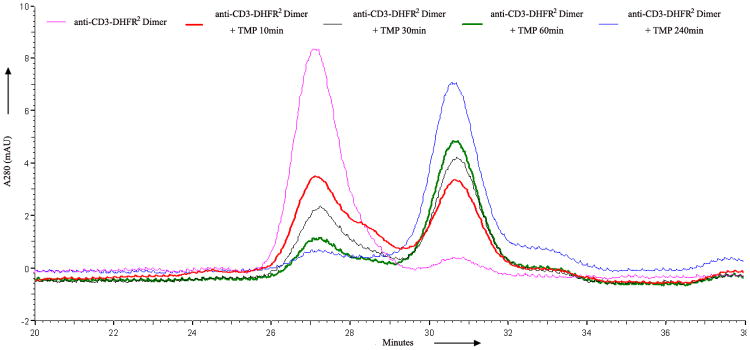
a) Scheme for the assembly and disassembly of bivalent single-chain antibody. b) Assembly of anti-CD3-DHFR2 dimer was characterized by size exclusion chromatography. Red curve showed anti-CD3-DHFR2 monomer, while green curve represented the induced dimerization reaction of anti-CD3-DHFR2 with Bis-MTX. c) Binding affinities of anti-CD3-DHFR2 monomer and dimer to HPB-MLT T leukemia cells were compared by in vitro cytotoxicity blocking assay. Plot displayed cytotoxicity of immunotoxin DTUM3 alone (purple dots), cytotoxicity of DTUM3 blocked by 100nM anti-CD3-DHFR2 monomer (blue triangles), and cytotoxicity of DTUM3 blocked by 100nM anti-CD3-DHFR2 dimer (green squares). d) Flow cytometric competitive binding assay was used to determine disassociation constant of anti-CD3-DHFR2 monomer (blue triangles), dimer (green squares) and UCHT-1 (orange dots). e) Disassembly of anti-CD3-DHFR2 dimer by trimethoprim at different time points was characterized with size exclusion chromatography. Light purple curve showed anti-CD3-DHFR2 dimer, while red, black, green, and blue curves displayed the disassembled anti-CD3-DHFR2 after incubating with trimethoprim for 10, 30, 60, and 240 mins.
To prepare the DHFR-DHFR-anti-CD3 scFv fusion protein, we first prepared the DHFR2 expression plasmid p13DD13CD3.1 by ligating the short double digested expression plasmid and anti-CD3 scFv encoding PCR product excised from Mo3.pGEMT.e by PCR followed by ligation. (See Supplementary Fig. 1 online) BL21 cells were transformed with p13DD13CD3.1 and substantial amounts of insoluble (inclusion bodies), but not soluble protein was observed. (See Supplementary Fig. 2 online) The sodium N-lauroyl-sarcosine (SLS) air oxidation method previously developed by Vallera et al.[15] for the preparation of bacterially expressed scFv's was employed; however, DHFR activity was not detectable, indicating that the fusion protein was incorrectly folded and unlikely to bind Bis-MTX.[16] Recognizing that each DHFR contained two cysteines and that the oxidative conditions necessary for scFv disulfide bond formation may result in inappropriate DHFR internal disulfide formation. we prepared the quadruple DHFR2 cysteine mutant (C85A C152S C257A C324S), since it had been previously shown that neither the DHFR mutant, C85A nor C152S resulted in loss of DHFR activity,[17] Upon solubilization and refolding by the sodium N-lauroyl-sarcosine (SLS) air oxidation method, the quadruple mutant was found to be soluble with the same specific DHFR activity as wild type DHFR (Data not shown), and easily purified by methotrexate affinity chromatography.[13]
The purified antibody fusion protein, anti-CD3-DHFR2 was incubated for 1 hr with one equivalent of Bis-MTX and analyzed by size exclusion chromatography (SEC). (Figure 1b) Dimeric anti-CD3-DHFR2 (138KD, 69%) was observed to elute from the Superdex G200 column at a retention time of 27 min, while monomeric anti-CD3-DHFR2 (68KD) eluted with a retention time of 30.5. A small amount of trimer (24.4 min, 3%) and monomer (28%) was observed. As observed previously for the oligomerization of DHFR2 fusion proteins containing a 13 amino acid linker, the larger species disappeared with time.[13] In addition, similar to DHFR2 containing a 13 amino acid linker, a small amount of remaining monomeric anti-CD3-DHFR2 (25%) was observable, even when incubated with as high as 5 equivalents of Bis-MTX.[13] (Data not shown) Given the slightly increased retention time (31 min vs. 30.5 min) relative to monomer not incubated with Bis-MTX and the observation that the amount of remaining monomer is considerably lower at high monomer concentrations, it is highly probable that this species results from intramolecular macrocyclization of anti-CD3-DHFR2. Relative to DHFR2, peak broadening of the anti-CD3-DHFR2 dimer was observed and is expected given the possibility of trans and cis isomer formation with respect to the position of the appended scFv's. The future development of strategies for DHFR heterodimerization should allow the preparation of exclusively either the cis or trans dimer isomer to be obtained. The stability of the self-assembled antibodies was assessed by incubating dimerized anti-CD3-DHFR2 at room temperature for 1 week followed by re-analysis by SEC. No appreciable change in the species distribution or amount of dimeric self-assembled antibody was observed. (See Supplementary Fig. 3 online)
To determine the binding affinity of the of anti-CD3-DHFR2 to CD3, we first compared the ability of the anti-CD3-DHFR2 monomer and dimer to protect CD3+ human T-leukemia HPB-MLT cells from the simultaneous treatment with the immunotoxin, diphtheria-anti-CD3 immunotoxin[18] (DTUM3, IC50 = 0.0337 ± 0.0116 nM). (Figure 1c) The anti-CD3 scFv was shown to be highly active by increasing the IC50 by greater than 100- fold (IC50 = 3.48 ± 1.33 nM). Protection from the immunotoxin was further enhanced by an additional 16- fold (IC50 = 55.7 ± 13.6 nM) when the cells were incubated with dimeric anti-CD3-DHFR2, thus affording and overall protection of approximately 1600- fold. Consistent with a fully functioning antibody mimic, the parental antibody, UCHT-1 (IC50 = 56.6 ± 2.9 nM), was found to provide the same degree of protection as dimerized anti-CD3-DHFR2. (See Supplementary Fig. 4 online)
To quantitatively determine the binding affinity of the chemically self-assembled diabody, the dissociation constants (Kd) for the monomic and dimeric anti-CD3-DHFR2 and UCHT-1 were evaluated with a flow cytometric competitive binding assay. (Figure 1d) Compared to the parental monoclonal antibody, UCHT1 (Kd = 1.8 ± 0.2 nM), dimeric anti-CD3-DHFR2 displayed a comparable Kd value of 3.5 ± 0.2 nM. Clearly, the bivalent antibodies have superior affinities over the monovalent scFv fusion protein, since the Kd value for monomeric anti-CD3-DHFR2 (Kd = 51 ± 9.8 nM) was found to be approximately 16- fold lower than the value for dimeric anti-CD3-DHFR2. Taken together, the results from the cellular protection and cell binding studies demonstrate that the self-assembled diabody is nearly identical in affinity to the parental monoclonal antibody.
Unlike other methods of preparing recombinant antibodies, one of the distinct advantages of a chemically induced self-assembly approach dependent on a bivalent ligand is the potential to temporally initiate disassembly by addition of a competitive monomeric ligand. Since trimethoprim is an FDA approved antibiotic that, like Bis-MTX, binds tightly to the DHFR active site, we investigated its ability to initiate DHFR oligomer disassembly by incubating dimeric anti-CD3-DHFR2 with a 10- fold excess of trimethoprim, followed by determination of the amount of dimer and monomer present at various time points. As can be seen in Figure 1e, disassembly was rapidly initiated with 40% conversion of the dimeric protein to monomer occurring in 10 min and approximately 100% conversion within 1 hr. The half-life for disassembly (t1/2) at 10 uM trimethoprim was found to be 18 min corresponding to a rate of 0.0385 min-1, (See Supplementary Fig. 5 online) Therefore, despite the lower affinity of trimethoprim[19] (Kd = 4.6 nM) relative to Bis-MTX[14] (Kd = 21 pM), rapid disassembly of the self-assembled antibodies can be achieved. The conversion of the cyclic oligomer to less stable intermediate linear species undoubtedly contributes to the observed rapid disassembly.
Confocal laser scanning microscopy was used to characterize the interaction of fluorescently labeled monomeric and dimeric anti-CD3-DHFR2 and UCHT-1 with living cells. (Figure 2) When incubated at 37°C, monomeric and dimeric anti-CD3-DHFR2 were found to first localize on the surface of CD3+ HPB-MLT cells, followed by intracellular uptake. Co-localization studies with a lysosomal specific red fluoresent dye, LysoTracker Red DND-99, identified these compartments as late endosomes. (Figure 3) Consistent with a mechanism relying on receptor and therefore energy dependent endocytosis, uptake of the recombinant antibodies was blocked by incubation at 4°C. (See Supplementary Fig. 6 online) No observable difference in the intracellular location of the recombinant antibodies and UCHT-1 was detectable. Consequently, both the monomeric and dimeric anti-CD3-DHFR2 and the parental monoclonal appear to interact with cells in a similar manner.
Figure 2.
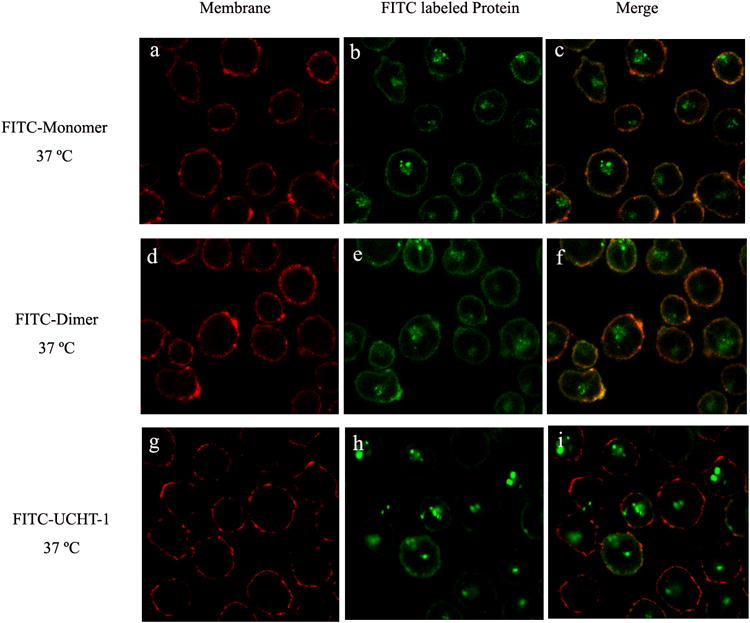
Confocal images of intracellular distribution of FITC-labeled anti-CD3-DHFR2 monomer, dimer or UCHT-1 at 37°C. HPB-MLT cells were incubated with FITC-labeled anti-CD3-DHFR2 monomer (a-c), FITC-labeled anti-CD3-DHFR2 dimer (d-f), or FITC-labeled UCHT-1 (g-i) for 2 hours at 37°C. Cell membranes were stained with concanavalin A-Alexa Fluor 594 (Red). Superposition of red channel and green channel were shown in images c, f, and i.
Figure 3.
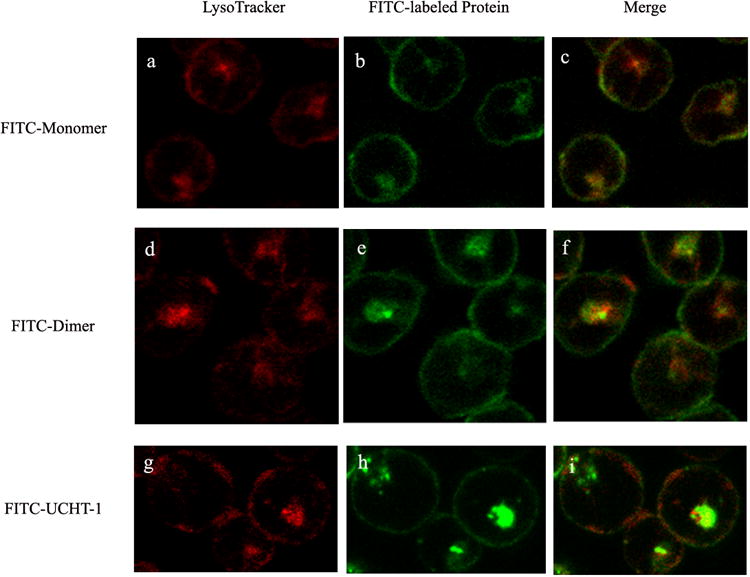
Confocal images of HPB-MLT T leukemia cells incubated with FITC-labeled anti-CD3-DHFR2 monomer (b) dimer (e) or UCHT-1 (h) for 30 mins at 37°C. Images a, d and g showed lysosomal compartments of the cells. Superposition of red channel and green channel were shown in images c, f, and i.
In conclusion, we have developed a modular method for the reversible preparation of stable recombinant antibodies by chemically induced self-assembly. Unlike previous recombinant antibody methods, our approach allows the oligomeric state of the antibody to be temporally controlled by a FDA approved drug. This is particularly important for not only controlling antibody avidity, but also molecular weight, a key determinant of antibody in vivo pharmacokinetics. Since we have previously demonstrated DHFR2 fusion proteins can be self-assembled into protein nanorings comprised of from two to eight subunits, we envision that our approach will allow preparation of antibody nanorings incorporating from two to eight scFv's. Our unique approach will then allow their disassembly into subunits with a common molecular weight of approximately 60 kDa, for rapid elimination.[2, 20] In addition, trifunctional Bis-MTX analogs are being prepared that are capable of delivering, fluorophores, radionuclides and drugs to scFv targeted tissues. The results of these studies will be reported in due course.
Supplementary Material
Footnotes
We thank National Institutes of Health and the Leukemia Research Foundation for financial support.
Contributor Information
Qing Li, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA).
David Hapka, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455 (USA).
DR. Hua Chen, Department of Therapeutic Radiology, University of Minnesota, Minneapolis, MN 55455 (USA)
DR. Daniel A. Vallera, Department of Therapeutic Radiology, University of Minnesota, Minneapolis, MN 55455 (USA)
DR. Carston R. Wagner, Email: wagne003@tc.umn.edu.
References
- 1.Pluckthun A, Pack P. Immunotechnology. 1997;3:83. doi: 10.1016/s1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 2.Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. J Immunol Methods. 2001;248:47. doi: 10.1016/s0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 3.Maynard J, Georgiou G. Annu Rev Biomed Eng. 2000;2:339. doi: 10.1146/annurev.bioeng.2.1.339. [DOI] [PubMed] [Google Scholar]
- 4.Kostelny SA, Cole MS, Tso JY. J Immunol. 1992;148:1547. [PubMed] [Google Scholar]
- 5.Muller KM, Arndt KM, Strittmatter W, Pluckthun A. FEBS Lett. 1998;422:259. doi: 10.1016/s0014-5793(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 6.Kipriyanov SM, Le Gall F. Mol Biotech. 2004;26:39. doi: 10.1385/MB:26:1:39. [DOI] [PubMed] [Google Scholar]
- 7.Holliger P, Prospero T, Winter G. Proc Nat Acad Sci U S A. 1993;90:6444. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alt M, Muller R, Kontermann RE. FEBS Lett. 1999;454:90. doi: 10.1016/s0014-5793(99)00782-6. [DOI] [PubMed] [Google Scholar]
- 9.Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, Von der Lieth CW, Matys ER, Little M. J Mol Biol. 1999;293:41. doi: 10.1006/jmbi.1999.3156. [DOI] [PubMed] [Google Scholar]
- 10.Schultz J, Lin YK, Sanderson J, Zuo YT, Stone D, Mallett R, Wilbert S, Axworthy D. Cancer Res. 2000;60:6663. [PubMed] [Google Scholar]
- 11.Bjorn Cochlovius MB, Welschof Martin. Mod Drug Discovery. 2003;33 [Google Scholar]
- 12.Jain M, Kamal N, Batra SK. Trends Biotechnol. 2007;25:307. doi: 10.1016/j.tibtech.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Carlson JCT, Jena SS, Flenniken M, Chou TF, Siegel RA, Wagner CR. J Am Chem Soc. 2006;128:7630. doi: 10.1021/ja060631e. [DOI] [PubMed] [Google Scholar]
- 14.Carlson JCT, Kanter A, Thuduppathy GR, Cody V, Pineda PE, McIvor RS, Wagner CR. J Am Chem Soc. 2003;125:1501. doi: 10.1021/ja026264y. [DOI] [PubMed] [Google Scholar]
- 15.Vallera DA, Brechbiel MW, Burns LJ, Panoskaltsis-Mortari A, Dusenbery KE, Clohisy DR, Vitetta ES. Clin Cancer Res. 2005;11:7920. doi: 10.1158/1078-0432.CCR-05-0725. [DOI] [PubMed] [Google Scholar]
- 16.Pineda P, Kanter A, McIvor RS, Benkovic SJ, Rosowsky A, Wagner CR. J Med Chem. 2003;46:2816. doi: 10.1021/jm034057i. [DOI] [PubMed] [Google Scholar]
- 17.Rajagopalan PTR, Zhang ZQ, McCourt L, Dwyer M, Benkovic SJ, Hammes GG. Proc Natl Acad Sci USA. 2002;99:13481. doi: 10.1073/pnas.172501499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallera DA, Todhunter D, Kuroki DW, Shu YQ, Sicheneder A, Panoskaltsis-Mortari A, Vallera VD, Chen H. Leukemia Res. 2005;29:331. doi: 10.1016/j.leukres.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Williams JW, Duggleby RG, Cutler R, Morrison JF. Biochem Pharmacol. 1980;29:589. doi: 10.1016/0006-2952(80)90381-0. [DOI] [PubMed] [Google Scholar]
- 20.Olafsen T, Kenanova VE, Sundaresan G, Anderson AL, Crow D, Yazaki PJ, Li L, Press MF, Williams LE, Wong JYC, Raubitschek AA, Shively JE, Wu AM. Cancer Res. 2005;65:5907. doi: 10.1158/0008-5472.CAN-04-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.