

Abstract
Recently, missense mutations in titin-associated proteins have been linked to the pathogenesis of dilated cardiomyopathy (DCM). The objective of this study was to search for novel disease-associated mutations in the two human titin-binding proteins myopalladin and its amino-terminal-interacting partner cardiac ankyrin-repeat protein (CARP). In a cohort of 255 cases with familial and sporadic DCM, we analyzed the coding regions and all corresponding intron flanks located in the MYPN and CARP-encoding ANKRD1 gene. Two heterozygous missense mutations were detected in the MYPN gene (p.R955W and p.P961L), but neither of these mutations was found in 300 healthy controls. Both mutations were located in the α-actinin-binding region of myopalladin. Endomyocardial biopsies from the p.R955W carrier showed normal subcellular localization of myopalladin and α-actinin in cardiac myocytes, while their regular sarcomeric staining pattern was significantly disrupted in the p.P961L carrier, indicating that disturbed myofibrillogenesis and altered sarcomere assembly are the cause of the disease. In the ANKRD1 gene, we identified synonymous base exchanges (c.108T>C and c.-79C>T, respectively), but no non-synonymous mutations. In summary, we have identified novel missense mutations in the third immunoglobulin-like domain of myopalladin, which have either no or profound effects on the molecular composition of the sarcomere. According to our epidemiological data, the prevalence of ANKRD1 mutations seems to be lower than that of its binding partner myopalladin, indicating the clinical significance of myopalladin for the functional integrity of the sarcomeric apparatus and the protection against DCM.
Keywords: myopalladin, cardiac ankyrin-repeat protein, CARP, point mutation, single-nucleotide polymorphism, dilated cardiomyopathy
Introduction
Many efforts made towards the understanding of the pathophysiology of dilated cardiomyopathy (DCM) have been devoted to the identification of novel disease-causing mutations. Epidemiological studies addressing the prevalence of gene mutations in DCM patients have demonstrated that inherited forms account for at least one-third of all DCM cases.1, 2, 3, 4 Familial cases frequently show a monogenic origin, and transmission indicates autosomal dominant or X-linked inheritance pattern with incomplete and age-dependent penetrance.3 To date, positional cloning and candidate gene screening have led to the identification of numerous heterozygous mutations in more than 40 different genes, most of which encode proteins important for the structural integrity and function of cardiomyocytes, such as regulatory proteins of the sarcomere, the nuclear envelope or intracellular homeostasis.1, 2, 5 In particular, defects in proteins required for force transduction and force transmission of the sarcomeric apparatus seem to promote the development of DCM.
Titin and several titin-interacting proteins have been involved in the mechanical stretch response of myofibrils in striated muscle. The giant protein titin, also known as connectin, has a key role in maintaining the structural integrity and elasticity of the sarcomere architecture, permitting the muscle cell to stretch and then return to its resting length with no disruption in sarcomeric organization. It functions as a molecular scaffold for anchoring α-actinin in the Z-disk, and there is growing evidence that titin is linked to several signaling pathways. Titin and its 145-kDa interacting partner myopalladin located in the sarcomeric Z-band both bind to cardiac ankyrin-repeat protein (CARP), an early differentiation marker of cardiac myogenesis expressed during embryonic and fetal heart development and in end-stage heart failure in the adult heart.6, 7, 8, 9 CARP is localized in the cytoplasm in the sarcomeric I-region through its interaction with the N2A domain of titin and the N-terminal domain of myopalladin. In addition, CARP was detected in the nucleus, where it functions as a transcriptional regulator involved in gene expression.8 This nucleocytoplasmic distribution supports the hypothesis that CARP shuttles between the cytosolic and nuclear compartment to directly transmit titin filament-based strain signals into transcriptional responses.7, 10 Similar to CARP, myopalladin has also been detected in the nucleus; however, it is unclear whether it shuttles in a complex with CARP or via different translocation pathways.8, 11 Yeast two-hybrid screens have mapped the α-actinin-binding site to the carboxy-terminal region of myopalladin, which encompasses the third to the fifth immunoglobulin-like domains.11
Recently, mutations in the myopalladin-encoding MYPN and the CARP-encoding ANKRD1 gene (also termed ankyrin-repeat domain 1-encoded cardiac adriamycin responsive protein) have been reported in patients with DCM.3, 12, 13, 14, 15 As titin-binding proteins have been proposed to link mechanical load sensing in sarcomeres to myocardial gene expression, we extended the search for novel point mutations in a heterogeneous, but clinically well-characterized cohort of familial and sporadic DCM cases. Based on a mutational screening approach, we here present additional genetic variants identified in the two titin-associated proteins, which were thought to be most probably disease causing.
Patients and methods
Clinical evaluation
A total of 255 unrelated, consecutive patients with DCM were included in this study. All study participants had been referred to the Department of Cardiology at the University of Marburg for clinical assessment of heart failure symptoms. The diagnosis of DCM was based on accurate medical history, physical examination, blood sampling, chest X-ray, 12-lead electrocardiography, and transthoracic M-mode, two-dimensional and Doppler echocardiography. Holter-ECG and serum creatine kinase levels were obtained when possible. In each patient, heart catheterization was routinely performed for angiographical exclusion of coronary artery disease and, in the same procedure, endomyocardial biopsies were obtained for histological examination. Patients were considered eligible for the study, if, in the absence of secondary causes of heart failure, the echocardiographically measured left-ventricular ejection fraction (LVEF) was <45% and/or the left-ventricular end-diastolic diameter (LVEDD) was >117% of the expected value. Patients were classified as familial cases according to the guidelines of the Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy if at least two first-degree relatives in the same family were affected by heart failure.16 Cases were considered sporadic if no evidence of familial disease was observed or when no relatives could be clinically evaluated. Patients who fulfilled the diagnostic criteria for DCM were invited to participate in the study and written informed consent was acquired. Pharmacotherapy of heart failure included angiotensin-converting enzyme inhibitors or angiotensin-II type-1 receptor blockers, beta blockers, aldosterone receptor antagonists, and cardiac glycosides according to the guideline of the European Society of Cardiology.17, 18 An independent control sample (n=300) consisted of blood donors from the local Blood Transfusion Service who came from the same geographical area as the DCM study cohort. All control subjects were free of disease symptoms, as judged by detailed medical examination. From each study participant, blood samples were obtained in EDTA anticoagulant-containing tubes. Genomic DNA was isolated from blood cells and stored at −80 °C until further use. All study subjects gave their signed informed consent for genotyping and detailed prospective phenotyping. The study protocol conforms to the principles outlined in the Declaration of Helsinki and was approved by the German Heart Failure Network and the local Ethics Committee.
Mutation analysis
Genomic desoxyribonucleic acid was isolated from peripheral white blood cells of all subjects using a DNA isolation kit (QIAamp DNA Blood Mini kit from Qiagen, Hilden, Germany) according to the manufacturer's instruction. To amplify DNA fragments from each exon, including the flanking intron sequences, PCRs were performed in a total volume of 25 μl, including 10 × PCR buffer, 1.5 mℳ MgCl2, 0.15 mℳ of each dNTP, 0.5 mℳ of each specific primer pair, 100 ng of genomic DNA and 1 U of Taq DNA polymerase. The PCR conditions were 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 60–62 °C for 30 s, and extension at 72 °C for 45 s with a final elongation step at 72 °C for 7 min. Primers were designed on the basis of the GenBank sequence database provided by the National Center for Biotechnology Information (NM_032578 for MYPN and NM_014391 for ANKRD1, respectively) using the Primer3 software (SourceForge, Mountain View, CA, USA). The sequences of oligonucleotides used in this study are listed in Table 1.
Table 1. Primer pairs used in the study for amplifying coding sequences from the MYPN and ANKRD1 gene, respectively.
| Mypn2.1F: | 5′-(GxCy)n-ACATAGACTTGTTATTCCCTAGAAATG-3′ |
| Mypn2.1R: | 5′-CGCCCGCCCGCCCGCTCCTGGCAGAAGTTAGGCTC-3′ |
| Mypn2.2F: | 5′-GGCCGGCGGCCGCCGGCAGACTGGCCATCAATTACGAC-3′ |
| Mypn2.2R: | 5′-(GxCy)n-CAGGGATAGGGATGGGAACAG-3′ |
| Mypn2.3F: | 5′-GGCCGGCGGCCGCCGGCAGACTGGCCATCAATTACGAC-3′ |
| Mypn2.3R: | 5′-(GxCy)n-CAGGGATAGGGATGGGAACAG-3′ |
| Mypn3F: | 5′-GCCCGCCGGCCGCCGGCGCTCATTTAAGAGAATATCTGGAGCC-3′ |
| Mypn3R: | 5′-(GxCy)n-CAAGAGCGCATGGTAGAGGAG-3′ |
| Mypn4F: | 5′-(GxCy)n-ATGAATAAATCTTATGTCGTGTTTAGGAACC-3′ |
| Mypn4R: | 5′-GGCCGCGGAGCCACCCTTCTTAAGTTC-3′ |
| Mypn5F: | 5′-(GxCy)n-GTTCTTACACCTGTAAGCAGTGATGCC-3′ |
| Mypn5R: | 5′-CCCATGCAAGATGGTCATGGTC-3′ |
| Mypn6F: | 5′-(GxCy)n-GGTTTGTTATGTCTCACTATCCATC-3′ |
| Mypn6R: | 5′-GGCCGGCGGCCGCCGGAACTTTGGGTGCTGTGGTATC-3′ |
| Mypn7F: | 5′-GCGCGCATTCTTCTTATCATTATAGATG-3′ |
| Mypn7R: | 5′-(GxCy)n-AATTTAAAGTATTAACTTACTT -3′ |
| Mypn8F: | 5′-GCGTCTATAGGAAGCTTCTGATGAAC-3′ |
| Mypn8R: | 5′- (GxCy)n-ACTTCATTATGTATTCTTGTATA-3′ |
| Mypn10F: | 5′-CGCGCGCTCAGGTCCTGTGATTGTCCTT-3′ |
| Mypn10R: | 5′-(GxCy)n-AAGCGATTCATCCTACTTTTTACTTACA-3′ |
| Mypn11.1F: | 5′-(GxCy)n-CATTGTTTGAAAGGTGAGGC-3′ |
| Mypn11.1R: | 5′-GCGCTGGAGATTTGGTTTGCACAG-3′ |
| Mypn11.2F: | 5′-GCGCGCAGTGAGCAAAGAAAGCCTCTTAG-3′ |
| Mypn11.2R: | 5′-(GxCy)n-ATCGACTCTTAACTAGCATCT-3′ |
| Mypn12F: | 5′-ACCGCTGGTTTCTGGTTG-3′ |
| Mypn12R: | 5′-(GxCy)n-TTCAATAAACCTTCTAAATCC-3′ |
| Mypn14F: | 5′-(GxCy)n-CCTCACCCCAGACCCTAGTTC-3′ |
| Mypn14R: | 5′-CCGCATGACATCAACCGCTCCTTC-3′ |
| Mypn15F: | 5′-(GxCy)n-ATTTCACGGTGTTCTGGTCC -3′ |
| Mypn15R: | 5′-GGCCAGTACTTTGGTGCTCACG-3′ |
| Mypn16F: | 5′-(GxCy)n-ATATCTGAACATGCTTGTTGT-3′ |
| Mypn16R: | 5′-GCGCGCCTAAGCATGGGTGCTGGA-3′ |
| Mypn18F: | 5′-(GxCy)n-TTCAAATAAGAATGACCCTTCTCC-3′ |
| Mypn18R: | 5′-CCGCCGGCCGGCGGCCGCCGGCGCAACAAGCCCCACTGAG-3′ |
| Mypn19F: | 5′-GCGCGCGCGCGCGGTATATTCTCTCTTTTTCTCC-3′ |
| Mypn19R: | 5′-(GxCy)n-GTTCAACTAACAGCATTACAC-3′ |
| Mypn20F: | 5′-GCGCGCGCACCTCTGCAGCGTACATGC-3′ |
| Mypn20R: | 5′-(GxCy)n-GCTCTCTTACACAGCAGGTACC-3′ |
| (GxCy)n=5′-CCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCGCGGCCCCCCCCCG-3′ | |
| Ankrd1 1F: | 5′-CAACTCCAGGGATTCCTTCC-3′ |
| Ankrd1 1R: | 5′-CTGAAAGCAAAGGGCATTTC-3′ |
| Ankrd1 2F: | 5′-GAAGAATGACCCAGGACGAG-3′ |
| Ankrd1 2R: | 5′-ATTTCCATTCCCTACCCCAG-3′ |
| Ankrd1 3/4F: | 5′- TTTATCTCAGACCTTGCAGATTTAG-3′ |
| Ankrd1 3/4R: | 5′-CAGGTGGAAGGACTTTGGC-3′ |
| Ankrd1 5F: | 5′-GCTTCATAGCTCTTAAAAGGGTTTAC-3′ |
| Ankrd1 5R: | 5′-TTTTCCGGAGCTTCATATAGTC-3′ |
| Ankrd1 6/7F: | 5′-CAGGTTCTAATTCCCCAGACC-3′ |
| Ankrd1 6/7R: | 5′-TGATTCCAAATACAAAAGCAATG-3′ |
| Ankrd1 8F: | 5′-GACCCACTTCCTGGCCTC-3′ |
| Ankrd1 8R: | 5′-ATTGTGGGAGTCGTTTCACC-3′ |
| Ankrd1 9F: | 5′-CCTAATCCTTACCTCATGAAACAC-3′ |
| Ankrd1 9R: | 5′-GGCCATGCCTTCAAAATG-3′ |
Genotyping for the MYPN gene was carried out by denatured gradient gel electrophoresis using a 20–60% urea/formamide gradient in 8% acrylamide, 0.5 × Tris/acetic acid/EDTA buffer (300 V, 60 °C for 6 h). The gels were stained for separated DNA fragments with ethidium bromide. The amplified PCR products were randomly sequenced to validate the genotyping assay. The MYPN mutation in codon 955 was confirmed by incubating the amplified PCR product with 3 U of the restriction enzyme MspI (New England Biolabs, Ipswich, MA, USA) for 4 h, leaving the 404-base pair (bp) PCR product of the mutant intact, while cleaving the wild-type C allele. The mutation in codon 961, which introduced a new cleavage site for BfaI, yielded additional 290- and 114-bp digestion fragments only if the mutation was present. Enzyme digests obtained from restriction fragment length polymorphism analyses were visualized on 2% agarose gels.
Sequence variations in the exons of the ANKRD1 gene were detected by means of single-strand conformational polymorphism (SSCP) analysis and didesoxy fingerprinting (ddF). For SSCP analysis, the PCR-amplified DNA products were heated to 95 °C for 5 min and quenched on ice to produce almost complete denaturation. Strand separation was achieved by loading 3 μl of each sample onto an 8% polyacrylamide gel. The running conditions of gel electrophoresis were adjusted as needed to give optimal separations for individual sequences. Usually, samples were run at 4 °C with a 250-V constant voltage for ∼7 h. Abnormal SSCP patterns were analyzed on an automated sequencer based on didesoxy chain termination (Seqlab, Göttingen, Germany). DdF was performed using radiolabeled didesoxy-ATP terminator and thermostable polymerase. Approximately 100-ng purified PCR product from each exon was reamplified for 30 cycles using the same parameters as above. After the samples had been mixed with the manufacturer's stop solution (Applied Biosystems, Carlsbad, CA, USA), a 4-μl aliquot was denatured for 5 min at 95 °C and immediately cooled on ice before electrophoresis through a 6%, 37.5:1 non-denaturing polyacrylamide gel. Autoradiography was performed on vacuum-dried gels by overnight exposure on Biomax MR films (Kodak, Rochester, NY, USA).
Identified mutations were confirmed by sequencing on an independent PCR product generated from a second blood sample taken at a later time point. Sequencing data including electropherograms were visualized using the Chromas software (version 2.33, Technelysium Pty, South Brisbane, QLD, Australia). Evidence in favor of a causative role of a mutation was derived from amino-acid substitution prediction programs, which considered the phylogenetic conservation of the affected amino-acid residue, such as Polymorphism Phenotyping (PolyPhen-2) and Mutation Taster. A novel missense mutation was assumed to be possibly disease-causing if the two programs independently indicated a damaging effect.
Immunohistochemical analysis
Subcellular localization of myopalladin and α-actinin in cardiomyocytes was analyzed using indirect immunofluorescence microscopy on endomyocardial biopsy samples. Cryosections obtained from DCM patients with (n=2) or without MYPN mutations (n=4) were blocked for 45 min in 25% fetal calf serum in phosphate-buffered saline (FCS/PBS) and then stained for 2 h at room temperature with either a polyclonal anti-rabbit antibody against myopalladin (a friendly gift from Dr Labeit, Mannheim, Germany) or a monoclonal antibody against α-actinin (purchased from Sigma, Taufkirchen, Germany) diluted 1:1000 in FCS/PBS. In control experiments, the primary antibody was omitted. After three washes in PBS, the samples were incubated with Cy3-conjugated secondary antibodies (Dianova, Hamburg, Germany) for 45 min followed by nuclear staining with Hoechst dye. Then samples were rinsed three times in PBS and finally mounted in fluorescence mounting medium (Southern Biotech, Birmingham, AL, USA). Images were captured by fluorescence microscopy performed on a Leica DM5000B microscope (Wetzlar, Germany) equipped with appropriate fluorescence filters. Images were obtained with a CCD camera and further processed with the Leica QWin software.
Results
Baseline characteristics of the study cohort
Of the 255 study patients with DCM, 187 (73.3%) were male and 68 (26.7%) were female (Table 2). Occurrence was familial in 57 patients, but sporadic and non-familial in the other 198 cases. The mean age in the total study population was 50.2 years with a SD of 12.8 years. The mean LVEF at study inclusion was 29.6±8.4%. The echocardiographically measured, increased LVEDD at baseline in the total study cohort (67.7±8.4 mm) was also observed in the two groups of familial and non-familial DCM patients. At follow-up 12 months after study recruitment, the parameters of the left-ventricular function had generally improved, as judged from an increase in the mean LVEF (39.5±12.7%) and a reduced LVEDD (64.3±9.4 mm), which was probably the consequence of adequate medical therapy. Again, there were no statistical differences in the improvement of left-ventricular function during follow-up between the two groups of sporadic and familial cases (for details, see Table 2).
Table 2. Baseline characteristics of the total study cohort including the two subgroups of patients with familial and sporadic, non-familial DCM, respectively.
| Total study cohort (n=255) | Familial DCM cases (n=57) | Sporadic DCM cases (n=198) | |
|---|---|---|---|
| Age (years) | 50.2±12.8 | 51.3±12.4 | 49.9±12.9 |
| Sex (m, %) | 73.3 | 66.7 | 75.8 |
| LVEF (%) | 29.6±8.4 | 28.4±9.5 | 29,9±8,0 |
| LVEDD | 67.7±8.4 | 68.0±6.9 | 67.6±8.8 |
| LVEF FU (%) | 39.5±12.7 | 37.2±13.2 | 40.1±12.4 |
| LVEDD FU (mm) | 64.3±9.4 | 64.6±7.6 | 64.2±9.9 |
Abbreviations: DCM; dilated cardiomyopathy, FU; follow-up; LVEDD; left-ventricular end-diastolic diameter, LVEF; left-ventricular ejection fraction.
All variables except sex are given as means and SD's.
Mutational analysis of the MYPN gene
Screening of the human MYPN-coding sequence in the DCM patients led to the identification of numerous known SNPs (Table 3). We, additionally, detected two novel heterozygous missense mutations in the MYPN gene, both of which were localized in exon 13, resulting in a prevalence of 0.8% (2/255). Each variant was independently confirmed by means of direct DNA sequencing as well as restriction fragment length polymorphism analysis using MspI and BfaI, respectively (Figure 1). Neither of these missense mutations was found in the 300 healthy blood donors.
Table 3. Number of patients from the total study cohort with SNPs detected in exon sequences and their flanking intron regions in the human MYPN gene.
| Exon number | Nucleotide exchange | Amino-acid change | No. of patients | SNP |
|---|---|---|---|---|
| 3 | c.1078+47T>C | — | 9 | rs28429181 |
| 4 | c.1130+17G>A | — | 44 | rs2817760 |
| 5 | c.1134C>T | p.I378I | 6 | rs145701607 |
| 5 | c.1178T>C | p.V393A | 2 | rs11596653 |
| 5 | c.1210T>C | p.L404L | 1 | — |
| 10 | c.1884C>G | p.F628L | 5 | rs10823148 |
| 11 | c.2072G>A | p.S691N | 2 | rs10997975 |
| 11 | c.2120G>A | p.S707N | 1 | rs7916821 |
| 11 | c.2409C>G | p.S803R | 1 | rs3814182 |
| 12 | c.2565-21A>G | — | 72 | rs7097776 |
| 13 | c.2886C>T | p.V962V | 1 | rs10733838 |
| 14 | c.2926-30A>G | — | 1 | rs71584497 |
| 17 | c.3403C>A | p.P1135T | 163 | rs7079481 |
Abbreviation: SNPs, single-nucleotide polymorphisms.
Figure 1.

Two missense mutations in the MYPN gene detected in a population of patients with DCM. (a–c) Identification of the myopalladin mutation p.R955W in a 44-year-old male DCM patient. DNA sequencing demonstrated the presence of a heterozygous nucleotide substitution in exon 13, resulting in an amino-acid exchange in position 955 (a). The p.R955W mutation was confirmed by means of denatured gradient gel electrophoresis (DGGE, b) and restriction fragment length polymorphism analysis (RFLP) using MspI (c). (d–f) Genetic analyses demonstrating the presence of a p.P961L mutation in a 33-year-old male DCM patient. The C>T nucleotide exchange in the corresponding codon shown in an electropherogram (d) was confirmed using DGGE (e) and RFLP with BfaI (f). As controls, DNA samples from other DCM patients were included in the respective gels, with the index patient marked with an arrow.
A substitution of arginine by tryptophan at amino-acid position 955 (p.R955W) in the MYPN gene was found in a 44-year-old Caucasian male patient presenting with a reduced left-ventricular function (LVEF 24%, LVEDD 68 mm), whose mother had died of heart failure resulting from DCM (Figures 1a-c). The software programs PolyPhen-2 and Mutation Taster predicated this point mutation to be damaging and disease-causing with probability scores above 0.96.
The second mutation (c.2882C>T) was located at amino-acid position 961 (Figures 1d–f). The carrier of the p.P961L mutation was a 33-year-old Caucasian male patient, who showed a severely compromised left-ventricular systolic function with reduced LVEF (15%) and increased LVEDD (82 mm). As a maternal uncle of the index patient had died of heart failure, familial DCM was diagnosed. The proline to leucine exchange in this position was predicated by Mutation Taster to be disease-causing with a probability score of P>0.999, and also PolyPhen-2 estimated its phenotypic consequence as damaging (P=0.978).
Immunohistochemical analysis of endomyocardial biopsies showed normal staining patterns for myopalladin and α-actinin in cardiac myocytes from the carrier of the p.R955W mutation as well as from four further, randomly chosen DCM patients (Figure 2). In contrast, the ordered subcellular assembly of these two cytoskeletal proteins was significantly altered in myocardial tissue samples from the p.P961L carrier. In this patient, the periodic localization of myopalladin and α-actinin along sarcomeres, observed in cardiomyocytes from other DCM patients, was no longer detectable. The abnormal distribution of myopalladin and its interacting partner α-actinin in the p.P961L carrier resulted in an altered cytoskeletal organization in cardiomyocytes with a severely disrupted sarcomeric architecture.
Figure 2.

Cardiac myocytes from the carrier of the p.P961L mutation showed a disrupted sarcomeric distribution of myopalladin and α-actinin. Endomyocardial biopsies obtained from three DCM patients expressing either wild-type or mutant myopalladin (p.R955W and p.P961L) were immunohistochemically stained with either a polyclonal antibody directed against myopalladin or a monoclonal α-actinin antibody. Specific immunoreactivity was detected by incubating the samples with Cy3-labeled secondary antibodies, while nuclei were stained with Hoechst dye.
Detection of variations in the ANKRD1 gene
In the total study cohort, seven variations were detected in various intronic regions of the ANKRD1 gene. In two DCM patients, we identified a SNP, unreported so far, in exon 1 (c.-79C>T). Four different variations were found in intron 4, three of which were deletions of 16, 21, and 26 bps, respectively, and the fourth was a known base exchange in nucleotide position 2268 (rs56177217). SNP rs3839939 in intron 7 was detected in 35 patients and SNP rs3939 in exon 9 in 70 patients. In the entire coding sequence of the ANKRD1 gene, we identified only one rare synonymous SNP, which was a base exchange in exon 2 located in codon 36 (c.108T>C; rs145211719). The carrier of this heterozygous synonymous mutation was a 65-year-old male patient (Figure 3). However, no disease-related mutation in the ANKRD1 gene was detected in the total study population.
Figure 3.
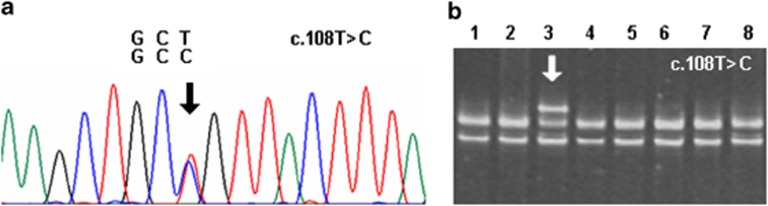
Nucleotide exchange in exon 2 of the CARP-encoding ANKRD1 gene as detected in a 65-year-old male DCM patient. (a) Electropherogram demonstrated the heterozygous nucleotide exchange c.108 T>C in codon 36. (b) SSCP analysis showed the presence of an additional, lower migrating band corresponding to the index patient (in lane 3 marked with an arrow), which was absent in other DCM patients.
Discussion
As a number of missense mutations in genes encoding for titin and titin-associated proteins have recently been reported to be responsible for DCM,3, 12, 13, 14, 15, 19, 20, 21, 22, 23, 24, 25 we searched in a well-characterized sample of DCM patients for unknown SNPs and/or mutations in the human MYPN and ANKRD1 genes. In order to extend our knowledge on disease-causing mutations in these two proteins expressed in heart tissue, we screened the coding sequences and corresponding intron flanks of the two genes and found two novel non-synonymous mutations in the MYPN gene and only one rare synonymous SNP in the coding region of the ANKRD1 gene. Interestingly, the identified two MYPN missense mutations were located in exon 13 and affected two highly conserved amino-acid residues in the third immunoglobulin-like domain. The argininyl residue 955 in human myopalladin is conserved in chimpanzee, rhesus monkey, mouse, chicken, and zebrafish, whereas glycyl is substituted for arginine in the cat and a lysyl residue is present in the equivalent position in Takifugu rubripes (Figure 4a). The homologous proline 961 in the human myopalladin is even more conserved as it can be found in all vertebrate variants sequenced so far, suggesting an important role of this residue in the structural stability of the immunoglobulin-like fold and possibly also in protein–protein interactions. While crystallographic or NMR data on the folding of the third immunoglobulin-like domain of myopalladin are not yet available,26 sequence similarity to the third immunoglobulin-like domain of palladin underlines the significance of a prolyl residue at this position for accurate domain folding (Figure 4b).
Figure 4.

(a) Amino-acid residues R955 and P961 in the coding sequence of myopalladin are conserved between vertebrate species. Aligned wild-type sequences of the third immunoglobulin-like domain of myopalladin from different species and the mutant variants in DCM patients detected in our study sample (marked in red when mutated and in blue when conserved) are shown. (b) Cartoon structure of the third immunoglobulin-like domain of palladin as created by PyMOL, including the side chain of the prolyl residue 1017, which is homologous to P961 in the myopalladin sequence. (c) Peptide sequence alignment of the five immunoglobulin-like domains of myopalladin including third immunoglobulin-like domain of palladin. Mutants identified in this study and their corresponding wild-type residues are marked with red and blue, respectively.
Palladin, a member of the recently discovered palladin/myotilin/myopalladin family of actin- and α-actinin-binding proteins, shares many features in common with myopalladin such as a periodic distribution along microfilaments.27, 28, 29, 30, 31 The largest palladin isoform, with a molecular weight of ∼200 kDa, has a similar domain arrangement as demonstrated for myopalladin with two amino-terminal and three carboxy-terminal immunoglobulin-like domains.31 Remarkably, the sequence motif 961PVTFTC966 in the third immunoglobulin-like domain of myopalladin is identical to the amino-acid sequence 1017–1021 in the corresponding palladin domain, for which structural data have recently been reported (PDB 2DM2). The available high-resolution NMR structure of the third immunoglobulin-like domain of palladin suggests that proline 1017 is required for the overall folding of a hallmark β-sheet by allowing the formation of backbone hydrogen bonds between neighboring anti-parallel β-strands.31 From this structural observation, we assume that substituting this amino-acid residue may critically disturb the characteristic ‘sandwich' structure of this domain, resulting in altered binding properties of the entire domain (Figure 4c). Two independent computer programs predicted the phenotypic consequences of an amino-acid exchange at this position as disease-causing with high probability scores. In line with these calculations, the carrier of the p.P961L mutation, a 33-year-old male patient, suffered from symptoms of heart failure, and his medical examination showed a severely reduced left-ventricular systolic function, which was interpreted as a characteristic sign of early-onset DCM.
As the functional significance of these genetic variations is currently unknown, we performed a histological analysis on left-ventricular endomyocardial biopsies. These immunofluorescence studies on the subcellular localization of myopalladin and α-actinin confirmed the critical role of proline 961 in sarcomere organization. The periodic distribution of the two proteins in cardiac myocytes was significantly disrupted in endomyocardial biopsy samples obtained from the carrier of the p.P961L mutation, while the sarcomeric architecture was preserved in the patient carrying the p.R955W mutation and four other DCM patients used as controls. This observation demonstrates that the third immunoglobulin-like domain of myopalladin is required for its incorporation into the sarcomeric structure and that proline 961 has a key role in this process.
In their recently published study, Purevjav et al15 reported on a p.F954L mutation in a 17-year-old Caucasian female patient with DCM, in whom the disease-causing amino-acid exchange was only one single codon upstream of our mutation. Furthermore, the authors described a p.R955Q mutation in normal controls (rs149887823). Compared with the p.P961L mutation, a nucleotide exchange in codon 955 appears to be less critical for the cardiac function of myopalladin, as the replacement of arginine by glutamine was identified in subjects with normal left-ventricular dimensions at the time of study enrollment. Nevertheless, assessment of the impact of the p.R955W mutation, and also of p.R955Q, predicted damaging effects on the phenotypic level suggestive of disease induction. The presence of an aromatic side chain instead of a positively charged arginine residue as in the wild-type protein most probably affects the gross topology of this domain.
Interestingly, the first three missense mutations in the MYPN gene, which had been described in DCM patients, were located in the fourth and fifth immunoglobulin-like domains.3 The p.R1088H mutation reported in a case of familial DCM is located only one residue, amino-terminally, in the homologous fourth immunoglobulin-like domain as aligned to the corresponding position in the third domain, in which we detected our point mutation p.P961L.31 Duboscq-Bidot et al3 have shown that the p.R1088H mutation resulted in a decreased localization of myopalladin to the Z-band area of the left-ventricular cardiac myofibrils, similar to that which we found in the heart tissue from the p.P961L mutation carrier. The other two myopalladin mutations identified by these authors (p.P1112L and p.V1195M) led to sarcomere disorganization and premature cell death in cultured rat neonate cardiomyocytes transfected with the corresponding mutant expression vectors.3 Recently, four other point mutations have been detected exclusively in DCM patients (p.I213V, p.Y339F, p.A611T, and p.A882T), and only one of these mutations (p.Y339F) was predicted as damaging.15 The p.Y20C variant reported by these authors was not restricted to DCM patients as it was also found in a patient with hypertrophic cardiomyopathy. Moreover, transgenic mice with cardiac-restricted overexpression of p.Y20C mutant showed disrupted intercalated disks and developed hypertrophic cardiomyopathy.15 It would provide strong evidence for the pathological role of the MYPN mutations identified in our study if knock-in mouse models with the corresponding substitution mutations of the homologous amino acids were to develop DCM.
In contrast to the MYPN gene, we found no mutations and only one synonymous SNP in the CARP-encoding ANKRD1 gene. The low prevalence of mutations in the coding sequence of the ANKRD1 gene reported in this study population was unexpected as a recently published genetic investigation found five missense mutations in 231 independent cases of familial DCM.12 However, our sample was clinically more heterogeneous as suggested from a higher percentage of non-familial cases compared with the previous study. Thus, further studies in DCM patients with greater sample sizes will have to be conducted in order to extend the genetic and epidemiological analysis of CARP mutations.
Although we are confident that all blood donors from our local Blood Transfusion Service used as healthy controls were free of disease as assessed by medical examination, we do not have valid information about their ethnic background. Thus, we cannot entirely exclude the possibility that our observation might have been affected by an artefact caused by ethnic inhomogeneity. However, from the genotyping of candidate genes, we have confirmed that the allelic distribution of known SNPs in our control sample closely resembles those reported in other European populations and substantially deviates from non-European samples. Furthermore, the low number of immigrants from non-European countries in the rural geographical area of Marburg suggests that there is, if any, only a negligible selection bias between cases and controls.
In summary, in this study we confirm the disease-associated role of the titin- and α-actinin-binding protein myopalladin in the pathogenesis of DCM and identify novel missense mutations located in conserved regions within the third immunoglobulin-like domain. Histological evidence of disrupted sarcomere organization in heart tissue and structural considerations underline the importance of this domain and, in particular, key residues within this domain, for protein–protein interactions in cardiomyocytes. The frequency of CARP mutations encoded by the ANKRD1 gene was lower than in its binding partner myopalladin, pointing to the clinical significance of myopalladin for the functional integrity of the sarcomeric apparatus and the protection against DCM.
Acknowledgments
We gratefully thank Dr Labeit, Klinikum Mannheim, Germany, for valuable reagents. The research on this subject was funded by a grant from the Von Behring-Röntgen-Stiftung to TM and by the Federal Ministry of Education and Research 01GI 0205 – TP9a to SP and BM.
The authors declare no conflict of interest.
References
- Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364:1643–1656. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- Chang AN, Potter JD. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail Rev. 2005;10:225–235. doi: 10.1007/s10741-005-5252-6. [DOI] [PubMed] [Google Scholar]
- Duboscq-Bidot L, Xu P, Charron P, et al. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res. 2008;77:118–125. doi: 10.1093/cvr/cvm015. [DOI] [PubMed] [Google Scholar]
- Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012;33:296–304. doi: 10.1093/eurheartj/ehr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmüller S, Erdmann J, Binner P, et al. Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail. 2011;13:1185–1192. doi: 10.1093/eurjhf/hfr074. [DOI] [PubMed] [Google Scholar]
- Jeyaseelan R, Poizat C, Baker RK, et al. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem. 1997;272:22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development. 1997;124:793–804. doi: 10.1242/dev.124.4.793. [DOI] [PubMed] [Google Scholar]
- Miller MK, Bang ML, Witt CC, et al. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Torrado M, Iglesias R, Nespereira B, Centeno A, López E, Mikhailov AT. Intron retention generates ANKRD1 splice variants that are co-regulated with the main transcript in normal and failing myocardium. Gene. 2009;440:28–41. doi: 10.1016/j.gene.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Mikhailov AT, Torrado M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int J Dev Biol. 2008;52:811–821. doi: 10.1387/ijdb.082655am. [DOI] [PubMed] [Google Scholar]
- Bang ML, Mudry RE, McElhinny AS, et al. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153:413–427. doi: 10.1083/jcb.153.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboscq-Bidot L, Charron P, Ruppert V, et al. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur Heart J. 2009;30:2128–2136. doi: 10.1093/eurheartj/ehp225. [DOI] [PubMed] [Google Scholar]
- Purevjav E, Varela J, Morgado M, et al. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J Am Coll Cardiol. 2010;56:1493–1502. doi: 10.1016/j.jacc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purevjav E, Arimura T, Augustin S, et al. Cardiomyopathies due to mutations in the myopalladin gene: Genotype-phenotype correlation. Progress Pediatric Cardiol. 2011;31:139–140. [Google Scholar]
- Purevjav E, Arimura T, Augustin S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21:2039–2053. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Cleland J, Dargie H, et al. Task force for the diagnosis and treatment of chronic heart failure of the European Society of Cardiology. Guidelines for the diagnosis and treatment of chronic heart failure: executive summary (update 2005): The Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology. Eur Heart J. 2005;26:1115–1140. doi: 10.1093/eurheartj/ehi204. [DOI] [PubMed] [Google Scholar]
- Zugck C, Franke J, Gelbrich G, et al. Implementation of pharmacotherapy guidelines in heart failure: experience from the German Competence Network Heart Failure. Clin Res Cardiol. 2012;101:263–272. doi: 10.1007/s00392-011-0388-y. [DOI] [PubMed] [Google Scholar]
- Arimura T, Nakamura T, Hiroi S, et al. Characterization of the human nebulette gene: a polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum Genet. 2000;107:440–451. doi: 10.1007/s004390000389. [DOI] [PubMed] [Google Scholar]
- Itoh-Satoh M, Hayashi T, Nishi H, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- Knöll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- Mohapatra B, Jimenez S, Lin JH, et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab. 2003;80:207–215. doi: 10.1016/s1096-7192(03)00142-2. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192–2201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi T, Inagaki N, et al. Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. J Muscle Res Cell Motil. 2005;26:367–374. doi: 10.1007/s10974-005-9018-5. [DOI] [PubMed] [Google Scholar]
- Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Yang H, Xue X, Huang Q, Bartlam M, Chen S. Expression, crystallization and preliminary X-ray studies of the immunoglobulin-like domain 3 of human palladin. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:556–558. doi: 10.1107/S1744309106016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey CA, Rachlin A, Moza M, Arneman D, Carpen O. The palladin/myotilin/myopalladin family of actin-associated scaffolds. Int Rev Cytol. 2005;246:31–58. doi: 10.1016/S0074-7696(05)46002-7. [DOI] [PubMed] [Google Scholar]
- Rachlin AS, Otey CA. Identification of palladin isoforms and characterization of an isoform-specific interaction between Lasp-1 and palladin. J Cell Sci. 2006;119:995–1004. doi: 10.1242/jcs.02825. [DOI] [PubMed] [Google Scholar]
- Dixon RD, Arneman DK, Rachlin AS, et al. Palladin is an actin cross-linking protein that uses immunoglobulin-like domains to bind filamentous actin. J Biol Chem. 2008;283:6222–6231. doi: 10.1074/jbc.M707694200. [DOI] [PubMed] [Google Scholar]
- Goicoechea SM, Arneman D, Otey CA. The role of palladin in actin organization and cell motility. Eur J Cell Biol. 2008;87:517–525. doi: 10.1016/j.ejcb.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey CA, Dixon R, Stack C, Goicoechea SM. Cytoplasmic Ig-domain proteins: cytoskeletal regulators with a role in human disease. Cell Motil Cytoskeleton. 2009;66:618–634. doi: 10.1002/cm.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]