

Glucose production by the liver is essential to providing substrate to the brain during fasting. The inability of insulin to suppress hepatic glucose output is a major etiological factor in the hyperglycemia of type 2 diabetes mellitus (T2DM) and other diseases of insulin resistance1, 2. For fifty years, one of the few classes of therapeutics effective in reducing glucose production has been the biguanides, which include phenformin and metformin, the latter the most frequently prescribed drug for T2DM3. Nonetheless, the mechanism of action of biguanides remains imperfectly understood. The suggestion a decade ago that metformin reduces glucose synthesis via activation of the enzyme AMP-activated protein kinase (AMPK) has recently been seriously challenged in genetic loss of function experiments4. Here we provide a novel mechanism by which metformin antagonizes the action of glucagon, thus reducing fasting glucose levels. Metformin leads to the accumulation of AMP and related nucleotides, which inhibit adenylate cyclase, reduce levels of cyclic AMP and protein kinases A (PKA) activity, abrogate phosphorylation of critical protein targets of PKA, and block glucagon-dependent glucose output from hepatocytes. These data support a mechanism of action for metformin involving antagonism of glucagon and suggest an approach to the development of antidiabetic drugs.
Biguanides exert their major effect through inhibition of liver glucose production, though enhanced glucose disposal has been noted in some studies5, 6. Despite the wide acceptance of metformin as a first line therapeutic for diabetes, its mechanism of action remains unclear. Metformin inhibits mitochondrial respiratory Complex 1, thus reducing hepatic energy charge; ten years ago it was suggested that metformin functioned through activation of the kinase AMPK7–10. This has been challenged recently in experiments utilizing livers and primary hepatocytes lacking either AMPK or its upstream activating enzyme LKB14. Since both glycogenolysis and gluconeogenesis are controlled during the fasting state in part by the hormone glucagon, whose abnormal secretion in T2DM is a major factor in the pathophysiology of hyperglycemia, we explored the idea that metformin produces its effects by inhibiting glucagon signaling pathways11–13.
Binding of glucagon to its receptor on the hepatocyte plasma membrane leads to activation of adenylyl cyclase (AC), production of the second messenger cyclic AMP (cAMP), and stimulation of protein kinase A (PKA), which phosphorylates protein targets that work in concert to increase hepatic glucose output12. We tested in primary mouse hepatocytes a panel of compounds that activate AMPK by decreasing or mimicking reduced energy charge for inhibition of glucagon-dependent increases in cAMP. Treatment with all agents increased AMPK phosphorylation and antagonized the glucagon-dependent elevation in cAMP (supplemental figure 1a–b). Phenformin exhibited a dose dependent inhibition of glucagon-induced cAMP accumulation with a half maximal inhibitory concentration of ~150 μM, which correlated well with intracellular AMP (figure 1a–b). To more closely mimic the chronic treatment of diabetic patients with biguanides, we tested 24-hour exposure of hepatocytes to phenformin or metformin for reduction of glucagon-increased cAMP levels. Following long-term treatment, phenformin was effective at significantly lower concentrations, with concentrations of 10 μM or greater causing significant reductions in glucagon-stimulated increases in cAMP levels (figure 1c). Metformin also inhibited cAMP accumulation at concentrations of 125 μM or greater, levels slightly higher than in serum of diabetic patients after taking a 1 mg dose of metformin but probably similar to those accumulated in splanchnic tissues (figure 1d)14, 15. In rats, administration of a therapeutic dose of 50 mg/kg leads to levels in liver greater than 250 μM14, 15. Like phenformin, therapeutic concentrations of metformin elicited significant increases in AMP levels in primary hepatocytes (supplemental figure 2a–c). Glucagon did not alter adenine nucleotide levels or activate AMPK in primary hepatocytes and was without effect on the changes produced by metformin (supplemental figure 2a–d).
Figure 1. Biguanides inhibit cAMP production.

A. Primary hepatocytes were incubated with the indicated phenformin concentrations for 2 hours, 5 nM glucagon for 15 minutes, lysed, and assayed for total cellular cAMP and protein. N=4 for each point. B. Primary hepatocytes incubated with the indicated concentration of phenformin for 2 hours were extracted with perchloric acid and cellular nucleotides quantified by HPLC. N=4 for each point. C–D. Primary hepatocytes were incubated with the indicated concentration of phenformin (C) or metformin (D) for 24 hours, treated with 5 nM glucagon, lysed, and assayed for total cellular cAMP. N=4 for each point. E. Primary hepatocytes were incubated with the indicated concentrations of phenformin for 2 hours, treated with 5 nM glucagon, lysed, and PKA kinase activity determined. N=6 for 0 and 1000 μM phenformin groups, N=4 for 100 and 300 μM phenformin groups. F. Primary hepatocytes were incubated with phenformin for 2 hours, then glucagon, and protein was analyzed by western blot with the phospho-PKA substrate motif antibody, total and phospho-PFKFB1 antibodies, and total and phospho IP3R antibodies. Error bars represent standard error.
To determine the effects of phenformin on the kinetics of activation of PKA, we employed the AKAR3 FRET reporter16. Primary hepatocytes were infected with adenovirus encoding AKAR3 and 18 hours later confocal images were acquired over time (supplemental figure 3a). Glucagon increased the FRET ratio (FRET/CFP) of AKAR3 in the cytoplasm of hepatocytes within 1 minute and reached a maximum at 2 minutes, thereafter exhibiting only minimal decay for 15 minutes. Phenformin both delayed the rise in PKA activity and accelerated its decay (supplemental figure 3b). The concentration dependence of inhibition of PKA activity measured biochemically was similar to blockade of cAMP accumulation in primary hepatocytes (Figure 1e). AICAR, an adenosine mimetic that can be phosphorylated to form the AMP mimetic ZMP, also antagonized the glucagon-dependent increase of PKA activity in isolated hepatocytes (supplemental figure 4a).
Treatment of primary hepatocytes with phenformin inhibited the glucagon-dependent phosphorylation of cellular proteins at PKA substrate motifs (figure 1f). Phenformin also antagonized phosphorylation of the PKA substrates PFKFB1 and the IP3R, as revealed by Western blots using phospho-specific antibodies against these proteins (figure 1f and supplemental figure 5a). If reductions in levels of endogenous cAMP are important to the actions of biguanides, we would expect the latter to block PKA target phosphorylation in response to glucagon but not a membrane permeable analog of cAMP, SP-8br-cAMPS-AM. Glucagon induced phosphorylation of the PKA target proteins PFKFB1, CREB, and IP3R in primary hepatocytes; phenformin activated AMPK and inhibited phosphorylation of these PKA target proteins (figure 2a and supplemental figure 6a). In contrast, SP-8br-cAMPS-AM concentrations that elicited comparable phosphorylation of the PKA substrates were not antagonized by phenformin (figure 2b and supplemental figure 6b). These results demonstrate that phenformin inhibits the glucagon- but not SP-8br-cAMPS-AM, dependent phosphorylation of PKA substrates, most consistent with the biguanide working at a signaling step upstream from the activation of PKA. Both glucagon and SP-8Br-cAMPS-AM increased glucose output from primary hepatocytes; however, treatment of hepatocytes with therapeutic concentrations of metformin prevented glucagon- but not SP-8Br-cAMPS-AM-stimulated increases in glucose output (figure 2b). Of note, higher concentrations of metformin reduced basal as well as glucagon- and SP-8Br-cAMPS-AM- dependent glucose output. If the major actions of biguanide were mediated by decreases in cAMP, one would expect the drug to be ineffective in cells in which PKA has already been inhibited. In hepatocytes overexpressing a dominant negative PKA regulatory subunit that is unable to bind cAMP, PKA activity was necessary for glucagon stimulation of glucose output, and biguanides did not reduce the rate of glucose output any further (supplemental figure 6c).
Figure 2. Biguanides inhibit glucagon signaling.
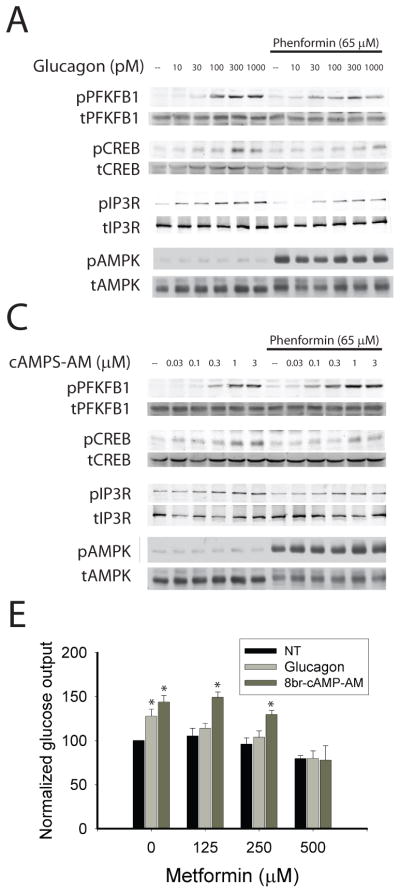
A–B. Primary hepatocytes were cultured for 18 hours in the presence or absence of 65 μM phenformin and for 15 minutes with the indicated concentrations of glucagon (A) or the cell permeable PKA agonist SP-8Br-cAMPS-AM (B). Western blot analysis of total and phosphorylated PFKFB1, CREB, IP3R, and AMPK. E. Cells were treated with the indicated concentration of metformin and either 1 nM glucagon or 3 μM SP-8Br-cAMPS-AM for 14 hours and then glucose output measured for 5 hours. Data represent the means of three experiments, N=6 for each experiment. Error bars represent standard error.
As the biguanides and other drugs used above activated AMPK in parallel to the reduction in cAMP, we asked whether the kinase was required for the effects of biguanides. Mice homozygous for the floxed alleles of both catalytic α1 and α2 subunits of the AMPK complex were infected with adeno-associated virus expressing Cre recombinase and western blots confirmed deletion of AMPK α protein and loss of phenformin-dependent phosphorylation of the AMPK substrate Acetyl-CoA Carboxylase (ACC) (figure 3a). In hepatocytes lacking any detectable AMPK activity, phenformin blocked glucagon-dependent cAMP accumulation in a manner indistinguishable from that in control cells (figure 3b). AICAR also reduced cAMP in the absence of AMPK (supplemental figure 4b). These data show that the effects of biguanides on cAMP metabolism are independent of AMPK.
Figure 3. Mechanism of biguanide effect on cAMP production.

A. AMPK α1lox/lox/α2lox/lox mice were infected with AAV-TBG-GFP or AAV-TBG-Cre virus and 14 days later primary hepatocytes were isolated. Cells were treated with the indicated concentrations of phenformin for 2 hours followed by 5 nM glucagon for 15 minutes. A. Total cellular protein was analyzed by western blot for total and phosphorylated T172 AMPK and total and phospho-S79 ACC. B. Hepatocytes were lysed and total cellular cAMP levels were quantified by ELISA. N=4 for all points. C. Primary hepatocytes were incubated with the indicated concentrations of phenformin for 2 hours and 50 μM RO-20-1724 (PDE4i) for the final 30 minutes. Cells were then treated with 5 nM glucagon for 15 minutes, lysed, and total cellular cAMP was assayed. N=4 for all points. D. The membrane fraction of primary hepatocytes was isolated by differential centrifugation and used in assays for Adenylyl Cyclase activity in the presence of the indicated AMP and ATP concentrations, 100 nM glucagon, and 100 μM GTP. N=6 for all points. Error bars represent standard error.
To determine whether reduction of the glucagon-dependent increase in cAMP was caused by activation of cAMP phosphodiesterase (PDE), we employed pharmacological inhibitors of PDEs. If inhibition of cAMP degradation did not also suppress the effect of phenformin on cAMP, it would imply that the site of action was unlikely to be PDE. Exposure of glucagon-treated hepatocytes to IBMX (a non-specific PDE inhibitor) or Ro-20-1724 (a specific PDE4 class inhibitor) but not cilostamide (a specific PDE3 class inhibitor) increased cAMP levels, indicating that PDE4 is the major cAMP-degrading enzyme in these cells (supplemental figure 7a). Even in the presence of IBMX or Ro-20-1724, phenformin inhibited glucagon-stimulated accumulation of cAMP (supplemental figure 7a). Moreover, Ro-20-1724 did not affect the dose responsiveness of hepatocytes to phenformin (figure 3c). To further resolve phenformin’s site of action, we measured its effects on cAMP accumulation in response to forskolin, which binds to and stimulates adenylyl cyclase independent of activated Gα protein. Both phenformin and AICAR blocked cAMP accumulation by both glucagon and forskolin to the same extent, indicating that the effects of these drugs were unlikely to be due to modulation of glucagon receptor or G protein signaling (supplemental figure 4c and 7b).
Since phenformin blocked glucagon-induced cAMP production at concentrations that correlated well with its effect on AMP levels, we sought to determine whether AMP could directly inhibit adenylyl cyclase, as has been reported previously17–20. Forskolin and glucagon stimulated adenylyl cyclase activity about four fold in membranes isolated from primary mouse hepatocytes (supplemental figure 8a). AMP inhibited glucagon-stimulated adenylyl cyclase activity when ATP was present at 160μM, which represents typical conditions for the assay, as well as the more physiological concentration of 1.28 mM ATP (figure 3d). In order to determine whether AMP accumulated in the livers of metformin-treated mice to levels sufficient to inhibit adenylyl cyclase, we measured hepatic AMP in fasted wild type mice. Consistent with a recent report, we found the concentration of AMP to be 2.3 mM and to rise to 2.9 mM following treatment with metformin in mice fed normal chow diet (Supplemental table 1)21, 22. Since these values seemed unphysiologically high, we also measured nucleotides in primary hepatocytes (supplemental table 2). In untreated cells, the AMP concentration was 215 μM, which correlated well with a value of 300 μM reported by Masson and Quistorff in perfused rat liver as determined by 31P NMR23. AMP in hepatocytes rose to over 1 mM in a dose-dependent manner upon treatment with phenformin (supplemental table 2).
To test whether the effects of biguanides on the accumulation of cAMP in primary hepatocytes were representative of actions in vivo, we examined the effects of metformin following injection of glucagon into mice. Glucagon rapidly increased blood glucose, an effect that was blunted by pre-administration of metformin (figure 4a). Gavage of mice with metformin increased both AMPK and ACC phosphorylation, indicating a decrease in the cellular energy charge of the liver (supplemental figure 9a). Most importantly, metformin abrogated the subsequent glucagon-dependent increase in hepatic cAMP, PKA activity, and phosphorylation of PKA substrates, demonstrating in vivo blockade of this pathway by biguanides (figure 4b–c). In order to assess the effect of metformin on the action of endogenous glucagon, fasted mice were given an oral gavage of water or 250 mg/kg metformin and 1 hour later blood glucose determined and hepatic tissue collected. This dose of metformin caused a significant drop in the blood glucose levels and an elevation in hepatic AMP (supplemental figure 9b, supplemental table 1), which correlated well with a concomitant reduction in hepatic cAMP content (figure 4d–f). We next tested the effects of metformin in diabetic mice fed a high fat diet for 10 weeks. These mice had elevated fasting glucose levels and hepatic AMP compared to chow fed animals (supplemental table 1 and supplemental figure 10a–b). Fasted HFD mice were given an oral gavage of water or 250 mg/kg metformin and 1 hour later blood glucose determined and hepatic tissue collected. Metformin significantly lowered blood glucose levels and elevated the hepatic AMP (supplemental figure 10c and supplemental table 1). Importantly, treatment of these mice with metformin led to a reduction in phosphorylation of two key PKA target proteins, PFKFB1 and the IP3R, and increased AMPK phosphorylation (figure 4g–I and supplemental figure 10d). The lack of change in Akt phosphorylation indicated that metformin was not affecting insulin responsiveness acutely (supplemental figure 10d–e).
Figure 4. Biguanides antagonize glucagon signaling in vivo.

A. Mice were gavaged with 500 mg/kg metformin and 1 hour later injected intraperitoneally with 200 μg/kg glucagon, and glucose levels were measured at the indicated times N=6 for water/pbs and metformin/glucagon, N=7 for water/glucagon and metformin/pbs. B–C. Fed mice were fasted for 1 hour and gavaged with water or 500 mg/kg body weight of metformin. One hour later mice were injected intraperitoneally with 2 mg/kg glucagon, and liver tissue was collected five minutes later. Liver was analyzed for B. total hepatic cAMP by ELISA (N=3 for each group) and C. total hepatic PKA activity (N=7, 8, 6, 7 for water/pbs, water/glucagon, metformin/pbs, metformin/glucagon, respectively). D–F. 18 hour fasted mice were gavaged with water or 250 mg/kg metformin, one hour later liver tissue was collected, and hepatic metabolites were extracted with perchloric acid and total hepatic (D.) AMP and (E.) cAMP levels were assayed. N=12 and 9 for water and metformin groups. G–I. Mice fed a high fat diet for 10 weeks were fasted overnight, gavaged either water or 250 mg/kg metformin and after 1 hour and liver tissue was collected for Western blot analysis of the phosphorylation status of (G.) AMPK, (H.) PFKFB1, and (I.) IP3R. N=3 for each group. Error bars represent standard error.
Understanding the mechanism by which the drug metformin reduces hepatic glucose output is a problem of considerable importance. Using a variety of biochemical and cell biological methods, we have confirmed earlier reports of an effect of biguanides on cAMP24–27. Furthermore, we have shown that it is through an elevation in intracellular AMP that metformin substantially abrogates the activation of adenylyl cyclase by glucagon. This results in a reduction in the phosphorylation of key substrates important for maintaining hepatic glucose output. For nearly 40 years it has been recognized that molecules containing an adenine moiety bind to the ‘P-site’ of adenylyl cyclase and inhibit its activity17. Though the endogenous P site ligand has been suggested to be AMP, the physiological or pharmacological significance of this regulatory event has not previously been recognized17–20, 28. We now provide support for the idea that therapeutic levels of metformin induce a mild energetic stress in hepatocytes, resulting in an increase in AMP concentration to levels capable of directly inhibiting adenylyl cyclase. These studies suggest that the P site of adenylate cyclase might represent a novel target for the development of therapeutics for the treatment of insulin resistance and T2DM.
Experimental Methods
A detailed experimental section is available online. Primary hepatocytes were isolated by collagenase perfusion as described previously29. Adenine nucleotides were extracted from cells and liver with perchloric acid and measured by ion pair RP-HPLC. cAMP in primary hepatocytes and frozen liver tissue was measured by ELISA (GE Healthcare) using the manufacturer’s lysis buffer. PKA activity was assayed in cell lysates as PKI sensitive Kemptide phosphorylation. PKA-FRET activity probes were used to examine intracellular PKA activity on a spinning disc confocal microscope16. Adenylyl Cyclase assays were performed using Adenosine-5′-triphosphate [α-32P] (American Radiolabeled Chemicals) and quantifying cAMP as previously described30. Glucose output studies in primary hepatocytes from fasted mice were carried out in Krebs buffer containing gluconeogenic substrates (20 mM lactate, 2 mM pyruvate, 10 mM glutamine) and were quantified using hexokinase based glucose assays (Sigma). For in vivo experiments metformin was gavaged at the indicated dosage and glucagon was injected intraperitoneally at the indicated dosages. Tissues were collected rapidly from anesthetized mice and frozen in precooled metal clamps. All results are expressed as the mean ± SEM. All two group comparisons were deemed statistically significant by unpaired 2 tailed student’s t-test if p<0.05.
Extended experimental methods section
Reagents and materials
Reconstituted GlucaGen (Novo Nordisk) was used for all glucagon studies. Phosphorylase Kinase B antibody was purchased from Acris Antibodies Inc. Total PFKFB1 antibody was purchased from Santa Cruz Biotechnology. The rabbit anti-PFKFB1 phospho-S33 antibody was produced by Cell Signaling Technology, Inc. All other antibodies were purchased from Cell Signaling Technology, Inc. The SP-8Br-cAMPS-AM was purchased from AXXORA, LLC (San Diego, CA). All additional reagents were purchased from Sigma Aldrich. Adeno-associated virus expressing the PKA-DN Rab mutant of the PKA RIα subunit from a liver specific TBG promoter was produced and purified in the University of Pennsylvania Viral Vector Core.
Mice
Mice were housed in a facility on a 12-h light-dark cycle with free access to food and water. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Blood glucose values were measured with OneTouch Ultra glucose analyzer. Mice were sacrificed either by rapid cervical dislocation, or for studies examining nucleotide levels, by anesthetization with 2,2,2-Tribromoethanol. Liver samples were rapidly removed and frozen in precooled metal tongs and frozen in liquid nitrogen. Time from liver ischemia to frozen tissue was approximately 30 or 5 seconds, for cervical dislocation or anesthetic based methods, respectively.
Primary hepatocytes
Primary hepatocytes were isolated from mice using a modified two-step perfusion method using Liver Perfusion Media and Liver Digest Buffer (Invitrogen). Cells were plated in collagen I coated 6- or 12-well plated (at 2 or 1 million cells per well, respectively) in M199 media + 10% FBS + Pen/Strep + 1 nM Insulin/100 nM T3/100 nM Dexamethasone. After 3 hours of attachment, the media was replaced with fresh media and the cells were incubated in culture overnight. Experiments were performed 18–24 hours after isolation of the cells. For 24 hour treatments of hepatocytes, compounds were added after the 3 hour attachment period.
Quantification of adenine nucleotides
Adenine nucleotides were extracted from primary hepatocytes as described previously29. For quantification of liver nucleotides, samples were harvested from anesthetized mice and immediately frozen. Approximately 50 mg pieces of liver were weighed and nucleotides were extracted with 0.5 M Perchloric acid. The insoluble materials were pelleted by centrifugation and the soluble fraction was neutralized with 0.25 volumes 2M KOH, 1M PO4 pH 7.8. Neutralized samples were stored at −80° C. Perchloric acid soluble materials were separated by isocratic elution in an ion-pairing reverse phase HPLC system (buffer used was 200 mM KH2PO4 pH 6.25, 5 mM Tetrabutylammoniumphosphate (TBAP), 3 % acetonitrile). AMP, ADP, and ATP peak areas were calculated and converted to molar amounts through comparison to standard curves generated from quantification of known quantities of pure nucleotides separated under identical conditions. Values of nmols/mg protein were converted to concentrations using the cell volume of primary hepatocytes22.
cAMP quantification
cAMP was assayed using a cAMP ELISA kit (GE Healthcare) according to the manufacturer’s alternative lysis protocol. For primary hepatocytes, cells in 12 well plates were lysed with 250 μL of lysis buffer 1B and cAMP quantified. For liver samples. cAMP was measured by ELISA, and normalized to total liver weight.
In vitro PKA assays
Total soluble lysates from either primary hepatocytes or liver were obtained using lysis buffer (1% Triton-X100, 150 mM NaCl, 20 mM PO4 pH 7.4). PKA activity was assayed using ATP-γ-P32 using a PKA Kinase Assay Kit (Millipore) as Kemptide phosphorylation that was sensitive to PKI inhibition.
Microscopy
Primary hepatocytes were plated on coverslip bottom tissue culture plates coated with collagen I and infected with adenovirus expressing the PKA FRET reporter AKAR316. 18 hours after infection the media was changed to imaging media (M199 media lacking sodium bicarbonate and phenol red supplemented with 20 mM HEPES pH 7.4) and cells were studied using a Zeiss confocal microscope. FRET (442 nm excitation/560 nm emission), CFP (442 nm excitation/480 nm emission), and YFP (523 nm excitation/560 nm emission) were acquired at 20 second intervals. 5 nM Glucagon was added as 500 uL of 50 nM glucagon in prewarmed imaging buffer to 2 mL of media. The FRET ratio ((FRET-background)/(CFP-background)) was taken from a random segment of the cytoplasm and nucleus, with one region of interest per cell, and over 40 regions of interest contributing to each condition. Ratios were normalized to the first 4 minutes of acquisition time.
Glucose output assays
Primary hepatocytes were isolated from 12 hour fasted mice and plated in M199 media with 10%FBS for 4 hours. After attachment the media was replaced with serum-free M199 media with 100 nM Dexamethasone and 100 nM T3, as well as the indicated drugs. After 14 hours the cells were washed with glucose output media (118 mM NaCl,4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.2 mM CaCl2, 20 mM NaCO3, 25 mM HEPES pH 7.4, and 0.025% BSA), and glucose output was measured in fresh glucose output media supplemented with gluconeogenic substrates (20 mM lactate, 2 mM pyruvate, 10 mM glutamine) for 5 hours. Glucose output was normalized to total protein and expressed as the percent of untreated basal glucose output.
Adenylyl Cyclase assays
Adenylyl cyclase activity was measured as previously described 30. Membrane preparations were obtained from primary hepatocytes in 15 cm plates. Cells were washed with PBS and lysed with hypotonic buffer (10 mM Tris pH 7.4, 5 mM EDTA) and 10 strokes of a dounce homogenizer. Unlysed cells and nuclei were pelleted and removed by centrifugation at 3,000 × g. The supernatant was centrifuged at 12,000 × g to pellet membranes, which were resuspended in lysis buffer and frozen at −80° C. Adenylyl cyclase assays were performed under various ATP concentrations with tracer quantities of Adenosine-5′-triphosphate [α-32P] (American Radiolabeled Chemicals) with the following final assay conditions: 15 mM HEPES pH 7.4, 200 mM NaCl, 1 mM EGTA, 10 mM MgCl2, 1 mM IBMX, 10 mM phosphocreatine, 60 U/mL Creatine Kinase (Sigma Aldrich). Assays were performed at 30° C for 10 minutes and stopped with the addition of an equal volume of stop solution (2% SDS, 1 mM ATP, 1 mM cAMP, ~2000 cpm cAMP [2,8 3H] (American Radiolabeled Chemicals)). 800 mL of water was added and this solution was purified by chromatography using Dowex 50WX8-200 (Sigma Aldrich) and Neutral alumina (Sigma Aldrich) columns. cAMP produced was measured as cAMP [32P] recovered corrected for cAMP [2,8 3H]. Specific activity of ATP (cpm/total ATP) was determined before and after incubation with purified membranes and AC assay buffer in the presence and absence of AMP. No significant reduction in specific activity or total ATP was observed (supplemental figure 8b–c).
Statistical analysis
All results are expressed as the mean ± SEM. All two group comparisons were deemed statistically significant by unpaired 2 tailed student’s t-test if p<0.05. All in vitro studies are either compilation of three independent experiments or representative of 3 independent experiments. Number of mice used in each in vivo condition is indicated in the figure legends
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1 DK56886 and PO1 DK49210 (M.J.B.) and F32 DK079572 (R.A.M), the Association pour l’Etude des Diabètes et des Maladies Métaboliques (ALFEDIAM) (to M. Foretz), the Programme National de Recherche sur le Diabète (PNRD) (to M. Foretz and B. Viollet), and the Institut Benjamin Delessert (to M. Foretz). Microscopy was performed in the University of Pennsylvania Cell and Developmental Biology Microscopy Core Facility. The Transgenic/Knockout, Mouse Phenotyping, Viral Vector, and Biomarker Cores of the University of Pennsylvania Diabetes and Endocrinology Research Center (NIH grant P30 DK19525) were instrumental in this work.
Footnotes
Author Contributions.
R.A.M. and Q.C. performed experiments; R.A.M. and M.J.B. designed experiments and wrote the manuscript. J.X. generated the phospho-S33 PFKFB1 antibody. M.F. and B.V. generated the AMPK α1 and α2 floxed alleles. J.X., M.F., and B.V. critically read the manuscript.
References
- 1.DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature of type 2 (non-insulin-dependent) and type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1982;23:313–9. doi: 10.1007/BF00253736. [DOI] [PubMed] [Google Scholar]
- 2.Postic C, Dentin R, Girard J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 2004;30:398–408. doi: 10.1016/s1262-3636(07)70133-7. [DOI] [PubMed] [Google Scholar]
- 3.Nathan DM, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203. doi: 10.2337/dc08-9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Inhibition of hepatic gluconeogenesis by metformin in the absence of LKB1-AMPK pathway: Role of hepatic energy state. Journal of Clinical Investigation. 2010;120 doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inzucchi SE, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338:867–72. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 6.Goodarzi MO, Bryer-Ash M. Metformin revisited: re-evaluation of its properties and role in the pharmacopoeia of modern antidiabetic agents. Diabetes Obes Metab. 2005;7:654–65. doi: 10.1111/j.1463-1326.2004.00448.x. [DOI] [PubMed] [Google Scholar]
- 7.El-Mir MY, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 8.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Alessio D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab. 2010;13(Suppl 1):126–32. doi: 10.1111/j.1463-1326.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 12.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284:E671–8. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 13.Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker GT, et al. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12:235–46. doi: 10.1111/j.1365-2125.1981.tb01206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24:49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- 16.Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–21. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- 17.Fain JN, Pointer RH, Ward WF. Effects of adenosine nucleosides on adenylate cyclase, phosphodiesterase, cyclic adenosine monophosphate accumulation, and lipolysis in fat cells. J Biol Chem. 1972;247:6866–72. [PubMed] [Google Scholar]
- 18.Blume AJ, Foster CJ. Mouse neuroblastoma adenylate cyclase. Adenosine and adenosine analogues as potent effectors of adenylate cyclase activity. J Biol Chem. 1975;250:5003–8. [PubMed] [Google Scholar]
- 19.Londos C, Preston MS. Regulation by glucagon and divalent cations of inhibition of hepatic adenylate cyclase by adenosine. J Biol Chem. 1977;252:5951–6. [PubMed] [Google Scholar]
- 20.Johnson RA, Yeung SM, Stubner D, Bushfield M, Shoshani I. Cation and structural requirements for P site-mediated inhibition of adenylate cyclase. Mol Pharmacol. 1989;35:681–8. [PubMed] [Google Scholar]
- 21.Berglund ED, et al. Hepatic energy state is regulated by glucagon receptor signaling in mice. J Clin Invest. 2009;119:2412–22. doi: 10.1172/JCI38650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoll B, Gerok W, Lang F, Haussinger D. Liver cell volume and protein synthesis. Biochem J. 1992;287(Pt 1):217–22. doi: 10.1042/bj2870217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masson S, Quistorff B. The 31P NMR visibility of ATP in perfused rat liver remains about 90%, unaffected by changes of metabolic state. Biochemistry. 1992;31:7488–93. doi: 10.1021/bi00148a008. [DOI] [PubMed] [Google Scholar]
- 24.Gawler DJ, Wilson A, Houslay MD. Metformin treatment of lean and obese Zucker rats modulates the ability of glucagon and insulin to regulate hepatocyte adenylate cyclase activity. J Endocrinol. 1989;122:207–12. doi: 10.1677/joe.0.1220207. [DOI] [PubMed] [Google Scholar]
- 25.Torres TP, et al. Impact of a glycogen phosphorylase inhibitor and metformin on basal and glucagon-stimulated hepatic glucose flux in conscious dogs. J Pharmacol Exp Ther. 2011;337:610–20. doi: 10.1124/jpet.110.177899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu B, Pugazhenthi S, Khandelwal RL. Effects of metformin on glucose and glucagon regulatedgluconeogenesis in cultured normal and diabetic hepatocytes. Biochem Pharmacol. 1994;48:949–54. doi: 10.1016/0006-2952(94)90365-4. [DOI] [PubMed] [Google Scholar]
- 27.Zhang T, et al. Mechanisms of metformin inhibiting lipolytic response to isoproterenol in primary rat adipocytes. J Mol Endocrinol. 2009;42:57–66. doi: 10.1677/JME-08-0130. [DOI] [PubMed] [Google Scholar]
- 28.Fain JN, Malbon CC. Regulation of adenylate cyclase by adenosine. Mol Cell Biochem. 1979;25:143–69. doi: 10.1007/BF00235364. [DOI] [PubMed] [Google Scholar]
- 29.Miller RA, et al. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121:2518–28. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Post SR, Ostrom RS, Insel PA. Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol Biol. 2000;126:363–74. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.