

Abstract
α/β-hydrolase domain-containing 6 (ABHD6) represents a potentially attractive therapeutic target for indirectly potentiating 2-arachidonoylglycerol signaling, however, the enzyme is currently largely uncharacterized. Here we describe a five element, ligand-based pharmacophore model along with a refined homology model of ABHD6. Following a virtual screen of a modest database, both the pharmacophore and homology models were found to be highly predictive, preferentially identifying ABHD6 inhibitors over druglike noninhibitors. The models yield insight into the features required for optimal ligand binding to ABHD6 and the atomic structure of the binding site. In combination the two models should be very helpful not only in high throughput virtual screening, but also in lead optimization, and will facilitate the development of novel, selective ABHD6 inhibitors as potential drugs.
Introduction
The endocannbinoid 2-arachidonoylglycerol (2-AG; Fig. 1) is a potent agonist at both cannabinoid receptors (CB1 and CB2). Approximately 85% of brain 2-AG hydrolase activity in vivo can be attributed to monoacylglycerol lipase (MGL) (1–4). Two largely uncharacterized enzymes, α/β-hydrolase domain-containing 6 (ABHD6) and α/β-hydrolase domain-containing 12 (ABHD12), are responsible for the majority of the remaining 15% (1). MGL, ABHD6, and ABHD12 display different subcellular distributions, suggesting that they may be responsible for regulating distinct 2-AG pools in the nervous system (1). While ABHD6 accounts for only a small percentage (~ 4%) of total brain 2-AG hydrolysis, in neurons its efficacy is similar to that of MGL (5). Neuronal ABHD6 is located post-synaptically at the site of 2-AG production, where it acts as a rate-limiting control point for 2-AG accumulation and efficacy, its acute inhibition leads to activity-dependent accumulation of 2-AG (5). Enhanced tissue 2-AG levels are considered therapeutic against pain, inflammation, and neurodegenerative/neuroinflammatory disorders including Alzheimer’s and Parkinson’s diseases (6–10). Additionally, ABHD6 is differentially expressed in some cancer cell lines and has been linked to tumorigenesis (11, 12).
Figure 1.

2-arachidonoylglycerol (2-AG), the native substrate for ABHD6; WWL70, a potent and selective ABHD6 inhibitor; and 5 a non-selective ABHD6 inhibitor.
Of the enzymes involved in 2-AG degradation, MGL is the most well-characterized. The high-yield bacterial expression and purification of human MGL (hMGL) has been reported (13) along with the proteomic characterization of hMGL’s active site (14). Both apo and inhibitor-bound X-ray crystal structures are available (15, 16), and the enzyme’s structure has been studied with nuclear magnetic resonance techniques (17). MGL inhibitors of varying selectivities have been reported (18–21).
MGL and ABHD6 are both lipases with an α/β hydrolase fold, in which a core of β sheets is flanked by α helices with a highly conserved active-site GXSXG motif. Both enzymes contain a Ser-His-Asp catalytic triad (postulated as S148, D278, and H306 in ABHD6 (22)). In marked contrast to MGL, ABHD6 is otherwise largely uncharacterized, although a potent and selective ABHD6 inhibitor has been reported, WWL70 (23) (Fig. 1).
Selective ABHD6 inhibition is emerging as a potentially attractive therapeutic goal as obstacles arise with inhibition of MGL and ABHD12. It has been shown that long-term MGL inhibition, resulting in 2-AG overload, desensitizes CB1 transmission countering any effect of increased 2-AG level on CB1 mediated signaling (24, 25). As ABHD6 is responsible for far less net 2-AG hydrolysis than MGL, but displays equivalent efficacy to MGL in neurons, it is possible that selective ABHD6 inhibition may not be associated with this drawback. Additionally, the essential role that ABHD12 has been shown to play in both the central and peripheral nervous systems and the eye, dampen enthusiasm for pursuing ABHD12 as therapeutic target because of the potential risk of long-term adverse effects (26). Hence, ABHD6 may be a potentially more attractive therapeutic target for indirectly potentiating CB1 mediated 2-AG signaling over MGL and ABHD12.
Here we describe a five element, ligand-based pharmacophore model along with a refined homology model of ABHD6. We detail the structural requirements for ABHD6 inhibition and examine the enzyme’s active site. Following a virtual screen of a modest database, both the pharmacophore and homology models were found to be highly predictive. The ability to preferentially identify known ABHD6 inhibitors over druglike noninhibitors verifies the models. Knowledge of the features required for optimal ligand binding to ABHD6 along with an understanding of the atomic structure of the binding site will facilitate the development of novel, selective ABHD6 inhibitors as potential drugs.
Methods
Creation of the Ligand Database
40 carbamate compounds with known ABHD6 activity were chosen from the literature (20, 23, 27). 36 compounds were active (0.05 µM < IC50 < 7 µM) and four ligands were inactive (IC50 > 50 µM) (see Supporting Information). The average Tanimoto similarity coefficient for the actives is 0.26 (molecules with a coefficient below 0.4 are not considered to be similar). All inhibitors are thought to act by covalent modification of the catalytic Ser148. Molecular mechanics methods are unable to account for the intrinsic reactivity of an inhibitor and it may be quite possible for actives and inactives to satisfy the correct pharmacophore query or docking grid. However, these high-throughput approaches are highly useful for identifying ligands that complement the active site, which can help achieve selectivity and improve potency. Ideally we would prefer to have active compounds from more than one class; unfortunately the only compounds with ABHD6 IC50 data currently available are carbamates.
The 40 carbamate compounds (average molecular weight of 443) were seeded into a database of 1,000 decoy ligands with an average molecular weight of 400, taken from the Glide enrichment studies (28, 29). Multiple conformations of each compound for use with pharmacophore models were generated with the conformational import methodology from MOE (30). Additionally, all compounds were prepared for docking with Glide by using the LipPrep protocol (31) and the OPLS_2005 force field.
Ligand-based pharmacophore model
The 36 ABHD6 inhibitors and 4 known non-inhibitors were used to construct a series of pharmacophore models with the pharmacophore elucidator in MOE (30). The approach exhaustively searches for all pharmacophore queries that induce good overlay of the active molecules and separates actives from inactives. Each query was required to be satisfied by at least 90% of the known actives, with a maximum of five pharmacophore elements. The initial distances between features were set to integer multiples of 0.6 Å. The large numbers of resultant pharmacophore queries were clustered to a RMSD of 1.25 Å. The radii of the aromatic and hydrophobic elements was set to 1.4 Å, and to 1.0 Å for all other elements. This produced 19 pharmacophore models, with either four or five elements. The overlap score, rating the active molecules atomic overlap when satisfying the query, ranged from 28.06 – 25.57. Visualization revealed that those queries with a score below 27.40 did not have an appropriate overlap, these pharmacophore models were discarded, and nine models remained. Further selection focused on the three models with five elements. The models were very similar, so the five-element model with the highest overlap score (27.70) was selected for virtual screening.
Homology modeling
The sequence for human ABHD6 was taken from the SWISS–PROT protein sequence database (primary accession number Q9BV23). Initial analysis of the ABHD6 sequence indicates that first ~9 residues are extracellular, the next ~30 residues are involved in a transmembrane helix, and the remaining ~290 residues are intracellular. Homology modeling considered a truncated version of the protein consisting of the cytosolic amino acids (residues 40–337). The model of truncated ABHD6 was constructed using the native crystal structure of BphD from Burkholderia xenovorans LB400 (PDB ID: 2OG1) (32) as a template in Prime (33). An initial BLAST alignment between the two sequences was adjusted by taking secondary structure into account using SSpro and PSIPRED (34). This alignment was further refined manually to account for tertiary structure (22% identity, 42% homology, 5% gaps; Fig. 2).
Figure 2.
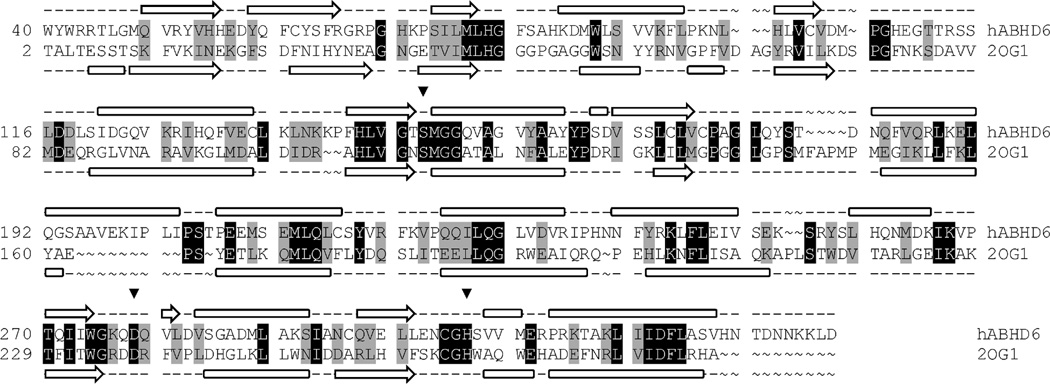
Modeling alignment for the sequences of human ABHD6 (Q9BV23) and BphD from Burkholderia xenovorans LB400 (PDB ID: 2OG1). Aligned query and template residues that are identical are highlighted in black; conserved residues (according to the BLOSUM62 scoring matrix), in gray. The predicted secondary structure for ABHD6 and the experimentally determined secondary structure for BphD are shown above and below the sequence, respectively. β sheets are denoted by an arrow, and α helices by a block. The residues of the catalytic triad are marked with an inverted triangle (▼).
The expect (E) value is an indicator of the accuracy of a homology model (35). The E-value describes the random background noise that exists between a sequence and template. While the alignment of hABHD6 with 2OG1 has only 22% identity, the E-value is 2e-17 which means the chance of an alignment as good as or better than this occurring by chance is tiny (2e-17). With such a low E-value for the alignment of hABDH6, it is likely that the overall fold will be correctly predicted. Additionally, analysis of the initial homology model shows that residues in the active site (defined as residues within 6 Å of Ser148 OG) share higher sequence homology (54% identity, 0% gaps).
When using templates with low sequence identity, loops are the regions which are most likely to be modeled inaccurately, and the extent of the inaccuracy increases with the length of the loop. After construction of the initial model loops which did not feature mainly homologous residues and or contained no gaps or insertions were refined using an ab initio loop prediction algorithm. The loop refinement step deletes the loop and reconstructs it from a backbone dihedral library; the loop is then exhaustively sampled to identify the lowest energy conformation. This method has been shown to achieve average RMSDs to the native structures of 0.43 Å for 5 residue loops and 0.84 Å for 8 residue loops (36).
As seen in Fig. 2 most loops are small and contain few gaps or insertions so should be modeled relatively accurately. Upon examination of the initial homology model there are, however, three loop segments of over 10 residues in length (excluding the C-terminus tail): M105-L119, C172-N182, and S194-S205. Loop S194-S205 is located in the lid region and is far from the catalytic site. Loop M105-L119 is also distant from S148. C172-N182 is in close proximity to the catalytic site, however the section which is nearest (C172-L176) is highly conserved with three of the five residues being identical to the template thus is likely to be modeled accurately. Following loop refinement, the protein underwent a truncated-Newton energy minimization, using the OPLS_2000 all-atom force field and a Generalized Born continuum solvation model.
Refinement
Homology models which are able to separate actives from decoys in prescreening enrichment studies on a small subset of ligands are more likely to be useful for virtual screening (37). An induced fit protocol (38) was used to generate a variety of ABHD6-inhibitor complexes. The seven most potent ABHD6 inhibitors (IC50 < 0.2 µM) were docked into the binding site of the ABHD6 homology model with Glide at the standard precision (SP) level (39). The van der Waals radii of the both the ligand and enzyme were scaled by 0.5. Up to 20 poses were carried forward for refinement with Prime. Residues within 5 Å of any ligand pose were refined; this consisted of a side-chain conformational search and optimization, followed by full minimization of the residues and the ligand. Complexes within 30.0 kcal/mol of the minimum energy structure were taken forward for redocking. The related ligand was redocked into each low-energy, induced-fit structure with default SP Glide settings (van der Waal radii scaling of 1.0 for ABHD6 and 0.8 for the ligand). This resulted in 83 ABHD6-inihibitor complexes. Models where the carbonyl of the ligand was further than 4 Å from the catalytic Ser148 hydroxyl were discarded, 19 complexes remained.
Each of the 19 complexes were prepared as docking receptors in Glide (39). The seven most potent ABHD6 inhibitors were prepared for docking using the LipPrep protocol (31) and the OPLS_2005 force field and docked to each grid at the SP level. Those models not yielding docking poses for all seven inhibitors were discarded, nine models remained.
In the BphD template structure, similar to other meta-cleveage product hydrolases, the catalytic His and Ser are not hydrogen bonded. However, it is more likely that for ABHD6, as with many other serine hydrolases, that a hydrogen bonding network exists between the catalytic triad to activate the serine hydroxyl for nucleophilic attack at the carbonyl carbon of the scissile bond. To reproduce this hydrogen bonding network, each of the nine models underwent further minimization, the three distance constraints were applied Ser148 Oγ – His306 Nε2 = 3.0 ± 0.4 Å, His306 Nε2 – Asp278 Oδ2 = 3.0 ± 0.4 Å, and Ser148 Oγ – ligand carbonyl carbon = 3.0 ± 1.0 Å.
Docking-based Virtual Screening
The full database of 1,040 compounds was docked to each of the nine modified ABHD6 models using the SP procedure in Glide (39). The successfully docked compounds were sorted based upon the Glide scores. This ranked list was then used to plot accumulation curves and generate enrichment metrics. The area under the accumulation curve ranged from 0.86 to 0.93 (a value greater than 0.90 is considered excellent whereas a value below 0.50 would be no better than random). After visualization of each of the structures the final ABHD6 model was selected.
Covalent Docking
The seven most potent ABHD6 inhibitors were covalently docked to the catalytic Ser148 of ABHD6 with a covalent docking module in Prime (33). This protocol forms the specified covalent bond and exhaustively samples the rotatable bonds of the ligand, producing a large number of potential poses. After clustering, the enzyme-ligand complexes are minimized and ranked by Prime energy.
Results and Discussion
Ligand-based pharmacophore model
The optimal pharmacophore model consisted of five elements; two hydrophobic, one aromatic and two hydrogen bond acceptor projections (which annotate the projected location of potential hydrogen bond donor atoms from a hydrogen bond acceptor based on implicit lone pair directions) (Fig. 3a). All 36 known ABHD6 inhibitors satisfied the query. Each of the ABDH6 inhibitors features a phenyl or biphenyl head-group attached to the carbamate oxygen. This head-group often includes electron-withdrawing substituents, most commonly a p -nitro group. The head-group is represented by an aromatic pharmacophore element. The carbonyl of the carbamate gives rise to the two hydrogen bond acceptor projections. The moiety of the tailgroup directly attached to the carbamate nitrogen, often a piperazine or piperidine ring, produces a hydrophobic pharmacophore element. The end of the tail group yields the second hydrophobic element.
Figure 3.

(a) Final ligand-based pharmacophore model with the potent, ABHD6 selective inhibitor WWL70 overlaid (grey carbons). (b) More stringent ligand-based pharmacophore model with the potent inhibitors 33e (cyan carbons) and JZL195 (magenta carbons) overlaid. Orange spheres require aromatic interactions, green spheres are hydrophobic, and cyan spheres are hydrogen-bond acceptor projections. Figures generated with the PyMOL Molecular Graphics System, Version 1.4.1 Schrödinger, LLC.
Virtual screening of 1,040 compounds including 36 known ABHD6 inhibitors against the final pharmacophore revealed that the model is highly predictive. The enrichment curve shows 92% of actives being recovered in the top 4% of the database, and all actives being recovered in the first 10.1 % of the database screened. The model was highly selective; the ROC curve shows a true positive rate of 0.97 with a false positive rate of only 0.04 (Fig. 4).
Figure 4.

Receiver operating characteristic (ROC) curve for the final ligand-based pharmacophore model is shown in red and for the more stringent model in blue. The curve for a random hit recovery is given in a solid black line.
After visualization of the overlap of known ABHD6 inhibitors with the pharmacophore query, it was clear that the majority of moieties satisfying the second hydrophobic element were also aromatic in character. To try to elucidate an even more selective model, this element was changed to require an aromatic ring (Fig. 3b). The enrichment curve is similar to the original model, with 92% of actives being recovered in the top 4% of the database. 97% of the actives were recovered in the first 5.6% of the database screened however one known inhibitor (5, (23)) does not satisfy the model. The more stringent model was more specific than the original with a true positive rate of 0.97 and with a false positive rate of just 0.02 (Fig. 4).
Homology model
To gain further insight into the requirements for ligand binding, a homology model of ABHD6 was created. The core domain (approximately residues 40–179 and 255–337) has the canonical α/β hydrolase fold and the lid (residues 180–254) is comprised of four α-helices. The ligand binding site is located between the two domains and contains the catalytic triad, Ser148, Asp278, His306 (Fig. 5). The binding site is largely hydrophobic with a smaller polar region (including Ser81, Ser148, Ser307, and His306) at the end. The nature of the ABHD6 binding pocket complements the composition of 2-AG with the head group occupying the hydrophilic region, while the aliphatic tail of the signaling lipid lies in the hydrophobic region. The presumptive oxyanion hole is formed by the backbone of Phe80 and Met149.
Figure 5.

Predicted tertiary structure of ABHD6 with a close up of the catalytic triad. Figure generated with the PyMOL Molecular Graphics System, Version 1.4.1 Schrödinger, LLC.
It is likely that many of the known ABHD6 inhibitors act by reversible or irreversible covalent modification of the catalytic Ser148. To gain insight into the features required for potent ABHD6 inhibition, the most potent inhibitors were covalently docked into ABHD6. The proposed mechanism for the covalent attachment was a SN2 reaction resulting in carbamylation of ABHD6 at catalytic Ser148 and elimination of the phenyl or biphenyl head-group from the ligand. In each docked pose, the ligand is found in the hydrophobic binding channel, with the carbonyl oxygen forming hydrogen bonds with the oxyanion hole (formed by the backbone of Phe80 and Met149) (Fig. 6a). The side-chain of Phe80 is involved in π stacking, in the T-shaped conformation, with an aromatic moiety in the ligand’s tail. Examination of the binding site indicates that the side-chain of Phe184 could form a similar π stacking stabilizing interaction with the head group of the ligand, prior to elimination of this group.
Figure 6.
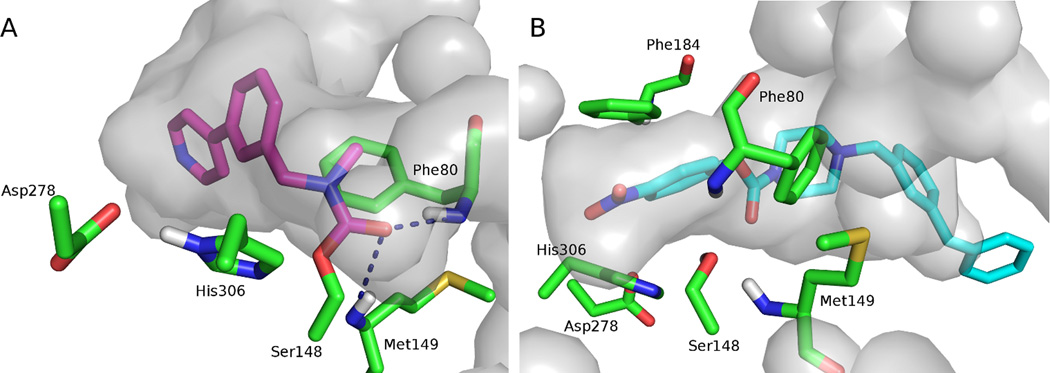
The ABHD6 binding pocket with the (a) predicted covalent binding mode of WWL70 (magenta carbons) and (b) non-covalent docking pose of JZL195 (cyan carbons). The pocket is shown in gray, and residues that may be important in the active site are shown in stick representation (green carbons). Figure generated with the PyMOL Molecular Graphics System, Version 1.4.1 Schrödinger, LLC.
Comparison of the ligand based pharmacophore model with the homology model of ABHD6 shows that the representations are congruous. The oxyanion hole is duplicated by the two hydrogen bond acceptor projections in the pharmacophore model. The side-chain of Phe184 corresponds to the aromatic pharmacophore element arising from the head-group of the ligands. The hydrophobic channel is echoed by the hydrophobic elements resulting from the tail-group, and the successful substitution to an aromatic element in the more stringent model is supported by the possibility of π stacking with the side-chain of Phe80.
While covalent docking may describe the ABHD6-ligand complex more accurately than non-covalent docking, it is time consuming and not suitable for high through-put screening. Hence, virtual screening was conducted with non-covalent docking, this approach can identify ligands that complement the active site, which can help achieve selectivity and improve potency (Fig. 6b).
Docking of the decoy database seeded with known ABHD6 inhibitors against the final homology model confirmed that the model is highly predictive. The enrichment curve shows 92% of actives being recovered in the top 12% of the database. The model is also selective; the ROC curve shows a true positive rate of 0.89 with a false positive rate of 0.06 (Fig. 7).
Figure 7.

Receiver operating characteristic (ROC) curve for the ABHD6 homology model is shown in blue. The curve for a random hit recovery is given in a solid black line.
Conclusions
A five element ligand-based pharmacophore model of ABHD6 has been validated and found to be highly predictive; identifying known ABHD6 inhibitors over drug like decoy ligands. Requirements for enzyme inhibition were further explored by the creation of a homology model which was also found to be highly predictive. The models are complementary and indicate that ABHD6 inhibitors should possess a hydrophilic tail, probably including an aromatic component, which may interact with Phe80. The head-group should be aromatic in nature to form favorable interactions with Phe184, with a polar tip, to complement the binding channel. The oxyanion hole, formed by the backbone of Phe80 and Met149, is available to stabilize formation of the tetrahedral intermediate. The models give insight into the requirements for ABHD6 inhibition and increases understanding of the enzyme’s substrate-interaction site. This will accelerate the identification of potent ABHD6 inhibitors which will be valuable as pharmacological probes and help define this enzyme’s role in the endocannabinoid signaling network.
Supplementary Material
Acknowledgement
This work has been supported by the National Institutes of Health, National Institute on Drug Abuse (grant DA027965). We thank Dr. David Janero for useful discussions.
Footnotes
Supporting Information Available: ABHD6 inhibitor data set used in this work.
References
- 1.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghafouri N, Tiger G, Razdan RK, Mahadevan A, Pertwee RG, Martin BR, et al. Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol. 2004;143:774–784. doi: 10.1038/sj.bjp.0705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 5.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–997. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol. 2007;152:787–794. doi: 10.1038/sj.bjp.0707425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hohmann AG. Inhibitors of monoacylglycerol lipase as novel analgesics. Br J Pharmacol. 2007;150:673–675. doi: 10.1038/sj.bjp.0707153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 9.Saario SM, Laitinen JT. Therapeutic potential of endocannabinoid-hydrolysing enzyme inhibitors. Basic Clin Pharmacol Toxicol. 2007;101:287–293. doi: 10.1111/j.1742-7843.2007.00130.x. [DOI] [PubMed] [Google Scholar]
- 10.Bisogno T, Di Marzo V. The role of the endocannabinoid system in Alzheimer's disease: Facts and hypotheses. Curr Pharm Design. 2008;14:2299–2305. doi: 10.2174/138161208785740027. [DOI] [PubMed] [Google Scholar]
- 11.Max D, Hesse M, Volkmer I, Staege MS. High expression of the evolutionarily conserved alpha/beta hydrolase domain containing 6 (ABHD6) in Ewing tumors. Cancer Sci. 2009;100:2383–2389. doi: 10.1111/j.1349-7006.2009.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Fei XW, Xu JX, Ji CN. An unannotated alpha/beta hydrolase superfamily member, ABHD6 differentially expressed among cancer cell lines. Mol Biol Rep. 2009;36:691–696. doi: 10.1007/s11033-008-9230-7. [DOI] [PubMed] [Google Scholar]
- 13.Zvonok N, Williams J, Johnston M, Panarinathan L, Janero DR, Li J, et al. Full mass spectrometric characterization of human monoacylglycerol lipase generated by large-scale expression and single-step purification. J Proteome Res. 2008;7:2158–2164. doi: 10.1021/pr700839z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zvonok N, Williams J, Johnston M, Panarinathan L, Karageorgos I, Janero DR, et al. Covalent inhibitors of human monoacylglycerol lipase: Ligand-assisted proteomic characterization of the catalytic site. Chem Biol. 2008;15:854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labar G, Bauvois C, Borel F, Ferrer J, Wouters J, Lambert DM. Crystal Structure of the Human Monoacylglycerol Lipase, a Key Factor in Endocannabinoid Signaling. Chembiochem. 2010;11:218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 16.Bertrand T, Augé F, Houtmann J, Rak A, Vallée F, Mikol V, et al. Structural Basis for Human Monoglyceride Lipase Inhibition. J Mol Biol. 2010;396:663–673. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 17.Karageorgos I, Tyukhtenko S, Zvonok N, Janero DR, Sallum C, Makriyannis A. Identification by nuclear magnetic resonance spectroscopy of an active-site hydrogen-bond network in human monoacylglycerol lipase (hMGL): implications for hMGL dynamics, pharmacological inhibition, and catalytic mechanism. Molecular Biosystems. 2010;6:1381–1388. doi: 10.1039/c004515b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandevoorde S. Overview of the chemical families of fatty acid amide hydrolase and monoacylglycerol lipase inhibitors. Curr Top Med Chem. 2008;8:247–267. doi: 10.2174/156802608783498005. [DOI] [PubMed] [Google Scholar]
- 19.Kapanda CN, Mucci GG, Labar G, Poupaert JH, Lambert DM. Bis(dialkylaminethiocarbonyl)disulfides as Potent and Selective Monoglyceride Lipase Inhibitors. J Med Chem. 2009;52:7310–7314. doi: 10.1021/jm901323s. [DOI] [PubMed] [Google Scholar]
- 20.Long JZ, Jin X, Adibekian A, Li WW, Cravatt BF. Characterization of Tunable Piperidine and Piperazine Carbamates as Inhibitors of Endocannabinoid Hydrolases. J Med Chem. 2010;53:1830–1842. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navia-Paldanius D, Savinainen JR, Laitinen JT. Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12) J Lipid Res. 2012;53:2413–2424. doi: 10.1194/jlr.M030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li WW, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 24.Lichtman AH, Blankman JL, Cravatt BF. Endocannabinoid Overload. Mol Pharmacol. 2010;78:993–995. doi: 10.1124/mol.110.069427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu PM, et al. Monoacylglycerol Lipase Activity Is a Critical Modulator of the Tone and Integrity of the Endocannabinoid System. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- 26.Fiskerstrand T, Brahim DHB, Johanssond S, M'Zahem A, Haukanes BI, Drouot N, et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am J Hum Genet. 2010;87:410–417. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachovchin DA, Ji TY, Li WW, Simon GM, Blankman JL, Adibekian A, et al. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 29.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 30.Molecular Operating Environment. Montreal, Canada: Chemical Computing Group Inc; 2010. Molecular Operating Environment. [Google Scholar]
- 31.LigPrep. New York, NY: Schrödinger, LLC; 2005. LigPrep. [Google Scholar]
- 32.Horsman GP, Ke J, Dai S, Seah SYK, Bolin JT, Eltis LD. Kinetic and Structural Insight into the Mechanism of BphD, a C—C Bond Hydrolase from the Biphenyl Degradation Pathway. Biochemistry. 2006;45:11071–11086. doi: 10.1021/bi0611098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prime. New York, NY: Schrödinger, LLC; 2012. Prime. [Google Scholar]
- 34.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 37.Xu MA, Lill MA. Utilizing Experimental Data for Reducing Ensemble Size in Flexible-Protein Docking. J Chem Inf Model. 2012;52:187–198. doi: 10.1021/ci200428t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des. 2006;67:83–84. doi: 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 39.Glide. New York, NY: Schrödinger, LLC; 2011. Glide. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.