

Abstract
Fibrosis is a major cause of morbidity and mortality worldwide. Currently, therapeutic options for tissue fibrosis are severely limited, and organ transplantation is the only effective treatment for end-stage fibrotic disease. However, demand for donor organs greatly outstrips supply, and so effective anti-fibrotic treatments are urgently required. In recent years the integrin family of cell adhesion receptors have gained prominence as key regulators of chronic inflammation and fibrosis. Fibrosis models in multiple organs have demonstrated that integrins have profound effects on the fibrotic process. There is now abundant in vivo data demonstrating critical regulatory roles for integrins expressed on different cell types during tissue fibrogenesis. In this review we will examine the ways in which integrins regulate these processes and discuss how the manipulation of integrins using function blocking antibodies and small molecule inhibitors may have clinical utility in the treatment of patients with a broad range of fibrotic diseases.
Keywords: Integrins, fibrosis, TGFβ
Introduction
Fibrosis represents a massive health care burden worldwide. Chronic tissue injury with fibrogenesis results in disruption of tissue architecture, organ dysfunction and eventually organ failure. Our therapeutic repertoire for the treatment of tissue fibrosis is severely limited and organ transplantation is currently the only effective treatment in end-stage fibrotic disease. However, organ transplantation has several disadvantages including limited donor organ availability, high cost, co-morbidities in potential recipients and on a global scale, organ transplantation can only be offered to a small percentage of the patients suffering from the complications of fibrosis. Therefore there is an urgent imperative to develop effective anti-fibrotic therapies.
A universal feature of tissue fibrogenesis is the complex interplay between the inflammatory, epithelial, myofibroblast and extracellular matrix components of the wound healing response1,2,3. Furthermore, the pericellular extracellular matrix is a highly dynamic environment known to exert profound influences on cell behaviour. Many of the key cell-cell and cell-matrix interactions which regulate fibrosis are mediated by members of the integrin family of cell adhesion molecules, of which there are 24 known members in humans (noncovalent α/β heterodimers composed from 18 different α subunits and 8 β subunits). Integrins represent a major mode of communication between the extracellular matrix, inflammatory cells, fibroblasts and parenchymal cells and hence are intimately involved in the processes that govern the initiation, maintenance and resolution of tissue fibrosis. Integrins are transmembrane proteins and are major receptors for cell adhesion to extracellular matrix proteins and cell-cell adhesion4. These molecules can therefore mediate the translation of spatially fixed extracellular signals into a wide variety of changes in cell behavior including cell adhesion, migration, proliferation, differentiation and apoptosis4,5. In addition to their direct effects on cellular proliferation and survival, integrins can also potentiate signals from soluble growth and survival factors. For example, nearly all of the pro-fibrogenic cytokine transforming growth factor beta 1 (TGFβ1) is secreted and bound to the extracellular matrix in a latent form, and therefore conversion to an active form is an important step in the regulation of TGFβ1 activity. In recent years it has become clear that a subset of the integrin family (αv integrins) play a key role in the activation of latent TGFβ1. Specifically, the integrins αvβ3, αvβ5, αvβ6 and αvβ8 have been shown to bind the RGD sequence in the latency associated peptide (LAP) of TGF-β1 and -β3, and have the potential to activate latent TGF-β6,7,8,9,10. In this review we will highlight recent data demonstrating the profound effects of integrins in modulating the fibrotic process via activation of TGFβ, and how pharmacologic manipulation of specific integrins may lead to the development of new antifibrotic treatments.
Lung fibrosis
αv integrin-mediated activation of latent TGFβ
Secreted transforming growth factor beta 1 (TGFβ1) is a major pro-fibrogenic cytokine and a key regulator of fibrosis in multiple organs11,12,13. Therefore, the molecular pathways that regulate TGFβ1 activity and signaling are attractive targets for novel anti-fibrotic therapies. There are three mammalian isoforms of TGFβ, and all are synthesized as precursor proteins that are processed by proteolytic cleavage in the endoplasmic reticulum and assembled as a non-covalent complex of a disulfide linked homodimer of the mature cytokine (a short C-terminal fragment) and a disulfide linked homodimer of a larger amino terminal fragment called the latency associated peptide (LAP), forming the “small latent complex”. In this form the associated LAP homodimer prevents the mature C-terminal fragment from binding to its receptors and inducing TGFβ’s known effects. This “small latent complex” is further modified in the endoplasmic reticulum by disulfide linkage to another family of gene proteins called latent TGFβ binding proteins, which, upon secretion, are themselves chemically cross-linked to the extracellular matrix, to store and tether TGFβ in a latent form in the extracellular space. Much of the regulation of TGFβ biology thus occurs at the level of extracellular activation of this stored latent complex14,15.
Because the active form of TGFβ is non-covalently linked to the latency associated peptide and easily dissociates upon changes in temperature or pH15, in vitro examination of TGFβ activation has been difficult. Therefore, the in vivo mechanisms of matrix-bound latent TGFβ conversion into an active cytokine is a subject of intense research. Two of the three mammalian TGFβ isoforms (TGFβ1 and 3) can be activated by members of the integrin family that interact with a linear arginine-glycine-aspartic acid (RGD) motif present in the latency associated peptide6,7,16. Inhibition and blockade of two of these integrins (αvβ6 and αvβ8) phenocopies all of the developmental effects of loss of TGFβ1 and 317, suggesting that these two integrins are required for most or all important roles of these TGFβ isoforms during development. However, the mechanisms of TGFβ activation that contribute to tissue pathology in adults are less well understood.
In the lung, the αvβ6 integrin is minimally expressed in alveolar epithelial cells at baseline but is rapidly induced in this cell type following lung injury18. Evidence supporting an important role for the αvβ6 integrin in TGFβ1 activation came from observation of the phenotype of β6 integrin subunit knockout mice. These mice develop exaggerated inflammatory responses in the lungs and skin, reminiscent of, but less severe than the exaggerated inflammation seen in mice homozygous for a null mutation of TGFβ119. Furthermore, following treatment with bleomycin (a widely used inducer of pulmonary fibrosis), β6 null mice develop exaggerated inflammation but are dramatically protected from subsequent pulmonary fibrosis6. β6 inhibition (both by genetic knockout and blockade by anti-αvβ6 antibodies) was also protective in radiation-induced pulmonary fibrosis20. The αvβ6 integrin can bind directly to the LAP of TGFβ1 and TGFβ316 and cells expressing αvβ6 generate TGFβ1 activity in vitro that can be completely inhibited by β6 blocking antibodies. In addition, microarray analysis of the lungs of wild type or β6 null mice following intratracheal instillation of bleomycin identified a large group of TGFβ-inducible genes that were induced at substantially lower levels in β6 knockout mice21. Taken together, these data demonstrate that αvβ6 integrin expression on lung epithelial cells is a major regulator of TGFβ1 activation during lung fibrosis.
Activation of TGFβ1 was inhibited by blockade of actin polymerization6 and by Rho kinase inhibition22, suggesting a role for force generation by the actin cytoskeleton. Indeed, the recently solved crystal structure of the small latent complex of TGFβ1 demonstrated that mechanical force generated by integrins is a common mechanism for activating latent TGFβ123. Shi and colleagues found that crystals of dimeric porcine proTGF-β1 revealed a ring-shaped complex, a novel fold for the prodomain (LAP) of TGFβ1, and demonstrated that the prodomain shields the growth factor from recognition by receptors and alters its conformation. Furthermore, complex formation between αvβ6 integrin and the prodomain of TGFβ1 was insufficient for TGFβ1 release, and force-dependent activation of TGFβ1 required unfastening of a “straitjacket” that encircles each growth factor monomer.
Myofibroblasts are a further cell type intrinsically involved in the fibrotic process, as they are the major source of extracellular matrix proteins during organ scarring. These contractile cells express several αv integrins and force generated by the actomyosin cytoskeleton can be transmitted to the extracellular matrix by αv integrins. Elegant in vitro studies of myofibroblasts have shown that these cells can utilize alternative αv integrins to activate TGFβ1, and demonstrates that myofibroblasts can liberate and activate TGFβ1 from pre-existing and self-generated deposits in the extracellular matrix by transmitting their high contractile force to the large latent complex through αvβ5 integrin and as yet unidentified β1 and 3 integrins10.
The integrin αvβ8 is also capable of binding to and activating TGFβ17. This was an unexpected finding, as αvβ6-mediated activation was found to depend critically on sequences within the β6 cytoplasmic domain6, however the β8 cytoplasmic domain and the β6 cytoplasmic domain are completely divergent. In addition, even deletion of the β8 cytoplasmic domain did not diminish αvβ8-mediated TGFβ1 activation, suggesting that these integrins (which both bind to the same RGD sequence in the TGFβ1 and TGFβ3 latency associated peptides) might activate the TGFβ1 latent complex by differing mechanisms. Further work demonstrated this to be the case. In contrast to αvβ6 mediated activation of TGFβ1, which depends on direct cell-cell contact, αvβ8-mediated activation releases active TGFβ1 into the culture medium of αvβ8 expressing cells. In addition, whereas αvβ6-mediated activation is completely resistant to inhibition by a variety of protease inhibitors, metalloprotease inhibitors abolish αvβ8-mediated TGFβ1 activation, and transfection studies in cells demonstrated a role for the protease MT1-MMP (MMP14) in this process. Therefore αvβ8 appears to activate TGFβ1 by presenting latent complexes to cell-surface metaloproteases which degrade the latency associated peptide and release free TGFβ1 into the extracellular milieu. An important role for αvβ8-mediated TGFβ1 activation in vivo is supported by studies of β8 knockout mice. Some of these mice die in mid-gestation from a defect in vascular development reminiscent of that seen in some TGFβ1 null mice24. Mice that survive to birth die soon after from brain haemorrhage that could be explained by loss of developmental vascular effects of TGFβ1. Furthermore, many of these mice have a cleft palate, a prominent feature in TGFβ-3 knockout mice25.
These data strongly suggest that the αvβ8 integrin is an important regulator of TGFβ1 and TGFβ3 activation in vivo, but does manipulation of this integrin have any modulatory effect on the fibrotic process? Previous studies have shown that αvβ8 expression is increased in the airway fibroblasts of COPD (chronic obstructive pulmonary disease) patients and expression correlated with the extent of airway wall fibrosis. Furthermore, αvβ8-mediated activation of TGFβ1 by COPD fibroblasts increased pro-fibrogenic differentiation26. Recently studies conducted by the same group have examined the role of fibroblast αvβ8 in murine airway fibrosis27. Kitamura et al. demonstrated that conditional deletion of lung fibroblast αvβ8 inhibited airway fibrosis in both IL-1β and ovalbumin-induced murine models of airway fibrosis. Furthermore, deletion of αvβ8 reduced TGFβ1 activation by cultured mouse lung fibroblasts. Extending their studies to human lung fibroblasts, the authors also found that IL-1β enhanced αvβ8-dependent TGFβ activation, collagen expression and pro-inflammatory gene expression in COPD compared with normal lung fibroblasts.
α3β1-mediated regulation of lung fibrosis
In recent years the origin of myofibroblasts in pulmonary fibrosis has been intensely studied, with potential sources including resident fibroblasts, circulating progenitors and epithelial-mesenchymal transition (EMT)28,29,30. Because the extracellular matrix is a key regulator of alveolar epithelial cell responses to TGFβ1 (and this cytokine is a potent inducer of EMT in vitro), Kim and colleagues investigated the role of the prominent epithelial integrin α3β1 (a laminin receptor known to co-localise with E-cadherin and β-catenin at adherens junctions31) in a mouse model of pulmonary fibrosis using mice with conditional epithelial cell-specific deletion of α3 integrin expression32. Despite a normal response to acute bleomycin-induced lung injury, these mice demonstrated a reduction in lung myofibroblasts and type I collagen and did not progress to fibrosis. To investigate whether this phenotype was secondary to a reduction in EMT, the authors examined β-catenin signalling as β-catenin has been implicated in EMT. They found that in primary alveolar epithelial cells α3 integrin was required for β-catenin phosphorylation at tyrosine residue 654 (Y654), formation of a pY654-β-catenin/p-SMAD2 complex, and initiation of EMT both in vitro and in vivo during fibrosis following bleomycin-induced lung injury. Furthermore, analysis of human lung tissue from idiopathic pulmonary fibrosis (IPF) patients demonstrated pY654-β-catenin-pSMAD2 complexes and accumulation of pY654-β-catenin in myofibroblasts. This suggests that alveolar epithelial integrin-dependent crosstalk between β-catenin and Smad signaling is important during the evolution of lung fibrosis, and that EMT plays a role in the development of lung fibrosis. However, it should also be noted that a number of recent cell fate mapping studies in multiple organs including the lung, have shown that EMT does not directly contribute to the pool of collagen-producing myofibroblasts during fibrogenesis in vivo33,34,35. The molecular mechanisms of integrin-mediated regulation of lung fibrosis are summarized in Figure 1.
Figure 1. Mechanisms of integrin-mediated regulation of lung fibrosis.
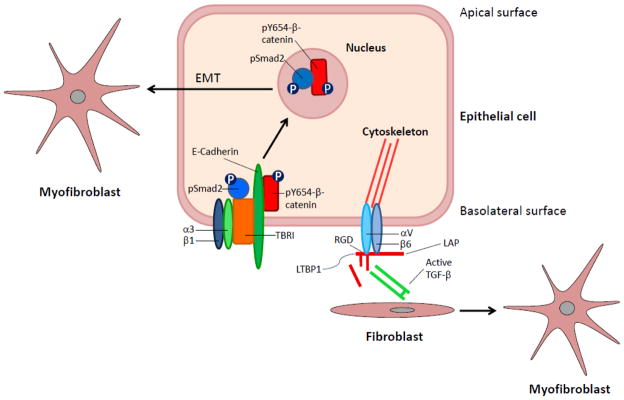
α3β1-mediated promotion of myofibroblast formation: In uninjured alveolar epithelial cells α3β1 co-localises with TGFβ receptor I (TBRI), E-cadherin and β-catenin. In the presence of TGFβ1, α3 integrin is required for β-catenin phosphorylation at tyrosine residue 654 (Y654), which is necessary for formation of a pY654-β-catenin/p-SMAD2 complex. This pY654-β-catenin/p-SMAD2 complex then translocates to the nucleus and induces EMT (epithelial-mesenchymal transition). αvβ6-mediated activation of latent TGFβ: αvβ6 binds to the RGD sequence in the LAP of TGFβ1 and 3. This complex is tethered by a disulfide linkage to LTBP1, which is essential for TGFβ activation. Binding alone is insufficient to activate latent complexes. Activation requires extracellular signals that lead to epithelial cell contraction and induction of a conformational change in the latent complex. This conformational change presents the active site on the mature TGFβ dimer to TGFβ receptors on adjacent cells, such as fibroblasts.
Liver fibrosis
Integrin αvβ6 mRNA expression is increased in patients with fibrotic liver disease secondary to a variety of aetiologies (primary biliary cirrhosis, alcohol-induced, hepatitis B and C) and expression increases with fibrosis stage in hepatitis C36. Furthermore αvβ6 expression is virtually absent in normal liver but is significantly upregulated in rodent models of liver fibrosis36,37. Using the bile duct ligation model of acute biliary fibrosis Wang et al.37 demonstrated that bile duct obstruction induces a marked increase in cholangiocyte αvβ6 expression. Furthermore, biliary fibrosis is reduced by 50% in β6 integrin null mice compared to wild type controls, and administration of a blocking antibody to αvβ6 significantly decreased acute fibrosis after bile duct ligation. A recent study has also examined the effect of a small molecule inhibitor of αvβ6 (EMD527040) during biliary fibrosis38. Biliary fibrosis was studied in rats after bile duct ligation and in Mdr2(abcb4)−/− mice. Differing doses of EMD527040 were given to rats from week 2 to 6 after BDL and to Mdr2(abcb4)−/− mice from weeks 4 to 8. EMD527040 reduced bile duct proliferation and peribiliary collagen deposition by 40–50%, decreased pro-fibrotic gene expression and up-regulated fibrolytic genes.
Hepatic stellate cells (Ito cells, liver specific pericytes) are the major source of extracellular matrix proteins during hepatic fibrogenesis39,40, and therefore represent an important target in the development of anti-fibrotic therapies for liver fibrosis. Zhou et al. examined the possibility that stellate cell fate is influenced by the extracellular matrix through the intermediary of αvβ3 integrin41. αvβ3 was expressed by rat and human culture-activated liver myofibroblasts, and blockade of this integrin inhibited stellate cell proliferation and increased apoptosis of cultured stellate cells. A recent study using cilengitide (an antagonist mainly selective for αvβ3 and αvβ5, with less potency towards αvβ6) demonstrated a 30% increase in hepatic collagen in two models of liver fibrosis (bile duct ligation and thioacetamide (TAA)-induced)42.
Kidney fibrosis
αvβ6 integrin expression is low in the normal kidney, but marked induction of this integrin occurs in a wide range of renal diseases associated with chronic inflammation and fibrosis. Human biopsy samples from membranous glomerulonephritis, diabetes mellitus, IgA nephropathy, Goodpasture’s syndrome, Alport syndrome and lupus all demonstrated prominent αvβ6 staining in the epithelial lining of dilated and damaged tubules43. To assess the potential regulatory role of αvβ6 in renal fibrosis, Hahm et al. investigated the effects of function-blocking αvβ6 antibodies and genetic ablation of the β6 subunit using a mouse model of Alport syndrome (Col4A3−/− mice). αvβ6-blocking antibody treatment attenuated accumulation of activated fibroblasts and deposition of interstitial collagen matrix, and similar inhibition of renal fibrosis was observed in β6-deficient Alport mice. Renal fibrosis is also decreased in β6 null mice following unilateral ureteric obstruction44, further demonstrating that αvβ6 plays a central regulatory role in the pathogenesis of kidney fibrosis.
Skin fibrosis
In recent years there have been a number of studies focusing on the role of the αv integrins in skin fibrosis and wound healing. Systemic sclerosis or scleroderma is an acquired disease typically leading to fibrosis of the skin and internal organs3. αvβ3 and αvβ5 expression are upregulated on human scleroderma fibroblasts and both of these integrins are involved in activation of latent TGFβ1 in primary cultures of these cells. Furthermore, treatment of scleroderma fibroblasts with anti-αvβ3 and αvβ5 antibodies reduced type I procollagen expression8,9,45,46,47. A role for αvβ6 in skin wound healing has also been examined. αvβ6 expression is strongly upregulated in the epidermis of human chronic wounds. Furthermore, transgenic mice harboring the human β6 integrin gene under the control of the cytokeratin 14 promoter (to target constitutive expression of the αvβ6 integrin in epidermal basal cells) develop spontaneous chronic skin wounds surrounded by progressive fibrosis48. In addition, aged β6 null mice demonstrate a significant delay in wound healing when compared to age-matched controls49.
Skin scleroderma can be modeled in mice by repetitive subcutaneous injection of bleomycin. To investigate the role of β1 integrin in cutaneous sclerosis Liu et al. generated mice with fibroblast specific deletion of the β1 integrin (using mice expressing a tamoxifen-inducible Cre recombinase driven by the mouse collagen type 1, alpha 2 promoter)50. Bleomycin treatment induced marked cutaneous thickening and fibrosis in control mice, however fibroblast specific deletion of β1 integrins resulted in resistance to bleomycin-induced skin fibrosis.
Table 1 summarizes the murine fibrosis models which have demonstrated a role for integrins in the regulation of fibrosis in vivo. Clearly these studies cannot be translated directly to human disease, but they do offer very useful insights into the molecular mechanisms driving fibrosis, allowing potential therapeutic targets to be identified.
Table 1.
Analysis of integrin function in transgenic mouse models of fibrosis
| Integrin | Organ | Model of fibrosis | Manipulation | Summary | Reference |
|---|---|---|---|---|---|
| αvβ6 | Lung | Bleomycin | Global knockout of αvβ6 | Absence of β6 markedly attenuates bleomycin-induced lung fibrosis via a reduction in activation of latent TGFβ1 | Munger et al. (1999)6 |
| αvβ6 | Lung | Radiation | Global knockout of αvβ6 | Absence of β6 protects against radiation-induced lung fibrosis | Puthawala et al.(2008) (2008)20 |
| αvβ6 | Kidney | Mouse model of Alport syndrome (Col4A3−/− mice) | Global knockout of αvβ6 | Absence of β6 attenuates renal fibrosis in β6-deficient Alport mice | Hahm et al. (2007)43 |
| αvβ6 | Kidney | Unilateral ureteric obstruction (UUO) | Global knockout of αvβ6 | Absence of β6 attenuates UUO-induced renal fibrosis | Ma et al. (2003)44 |
| αvβ6 | Liver | Bile duct ligation | Global knockout of αvβ6 | Absence of β6 attenuates acute biliary fibrosis | Wang et al. (2007)37 |
| αvβ8 | Lung | IL-1β and allergen induced lung injury | Conditional knockout of αvβ8 in fibroblasts | Reduced airway remodelling and dampening of adaptive immunity | Kitamura et et al. (2011)27 |
| α3β1 | Lung | Bleomycin | Conditional knockout of α3β1 in epithelial cells | Decreased lung fibrosis via a reduction in β-catenin/Smad signalling and EMT | Kim et al. (2009)32 |
| β1 | Skin | Subcutaneous injection of bleomycin | Conditional knockout of β1 in fibroblasts | Deletion of β1 integrin protects against bleomycin-induced skin fibrosis | Liu et al. (2009)50 |
Therapeutic targeting of integrins
Although TGFβ1 is a promising target for the treatment of fibrotic diseases, all of the currently available methods for inhibiting TGFβ target all three mammalian isoforms. TGFβ inhibitors therefore have the potential for important unintended side effects. One concern relates to the potential for carcinogenesis as TGFβ1 has an anti-proliferative effect on most epithelial cell types. This is particularly relevant with regard to advanced liver fibrosis in humans, as most hepatocellular carcinomas originate from underlying cirrhotic liver tissue. Secondly, owing to the critical role of TGFβ1 in immunosuppression (TGFβ1 null mice die at an early age from massive multi-organ inflammation51,52, generalized blockade of TGFβ activity may also lead to excessive autoimmunity and inflammation which could be highly detrimental in a patient with advanced fibrosis and limited organ reserve. Therefore inhibition of TGFβ1 signaling at specific sites, via inhibition of specific integrins, may yield the desired anti-fibrotic effects without the unwanted side-effects of pan-TGFβ blockade.
Specific blocking antibodies to αvβ6 have shown therapeutic promise in a wide range of pre-clinical models of fibrosis including lung fibrosis20,53, renal fibrosis43,44 and peri-biliary fibrosis37,54. Furthermore, in the lung low doses of αvβ6 blocking antibodies can prevent bleomycin-induced or radiation-induced pulmonary fibrosis in mice, without causing inflammation20,53. A monoclonal antibody targeting αvβ6 (clone 6.3G9) has been humanized as STX-100, and is currently being evaluated in phase 2 clinical trials for the treatment of patients with idiopathic pulmonary fibrosis. As noted above, pre-clinical data also suggest that targeting α3β1, αvβ3, αvβ5, αvβ8 or the β1 integrin on fibroblasts that regulate cutaneous fibrosis could hold promise for treatment of fibrotic diseases, however much less is currently known about the risk/benefit ratios of any of these interventions.
Conclusions
In recent years it has become apparent that integrins have profound effects on fibrosis in multiple organs. There is now abundant in vivo data demonstrating critical regulatory roles for integrins expressed on different cell types during the fibrotic process. The component parts of tissue fibrogenesis are exquisitely complex, and these studies highlight the important cross-talk between epithelia, tissue myofibroblasts and the cells of the immune system during the evolution and resolution of fibrosis. Strategies to manipulate integrins, such as antibody blockade and small molecule inhibitors, will hopefully yield effective anti-fibrotic therapies.
Highlights.
Tissue fibrosis is a major healthcare burden worldwide.
Integrin-mediated activation of latent TGFβ is a major mechanism driving fibrosis.
Pharmacologic manipulation of integrins may lead to new antifibrotic treatments.
Acknowledgments
The authors acknowledge the support of the Wellcome Trust (to NH), grants HL53949, HL102292 from the NHLBI and AI077439 from the NIAID (to DS).
Footnotes
Conflict of interest statement: Dean Sheppard is a co-owner of patents covering targeting the αvβ6 integrin for treatment of pulmonary fibrosis
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond) 2007;112:265–280. doi: 10.1042/CS20060242. [DOI] [PubMed] [Google Scholar]
- 3.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 6.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 7.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol. 2005;175:7708–7718. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 9.Asano Y, Ihn H, Yamane K, Jinnin M, Tamaki K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am J Pathol. 2006;168:499–510. doi: 10.2353/ajpath.2006.041306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 12.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 14.Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, Rifkin DB. TGF-beta latency: biological significance and mechanisms of activation. Stem Cells. 1997;15:190–197. doi: 10.1002/stem.150190. [DOI] [PubMed] [Google Scholar]
- 15.Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51:1376–1382. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 16.Annes JP, Rifkin DB, Munger JS. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65–68. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 17.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122:227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J Histochem Cytochem. 1993;41:1521–1527. doi: 10.1177/41.10.8245410. [DOI] [PubMed] [Google Scholar]
- 19.Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV, Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, Devitt ML, Horan GS, Weinreb PH, Lukashev ME, Violette SM, Grant KS, Colarossi C, Formenti SC, Munger JS. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82–90. doi: 10.1164/rccm.200706-806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A. 2000;97:1778–1783. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, Laurent GJ, Davis GE, Chambers RC, Matthay MA, Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–1614. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, Barzcak A, Xiao Y, Erle DJ, Nishimura SL. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007;117:3551–3562. doi: 10.1172/JCI32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, Wolters P, Hill A, Marks JD, Lou J, Pittet JF, Gauldie J, Baron JL, Nishimura SL. Mouse, human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J Clin Invest. 2011;121:2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chattopadhyay N, Wang Z, Ashman LK, Brady-Kalnay SM, Kreidberg JA. alpha3beta1 integrin-CD151, a component of the cadherin-catenin complex, regulates PTPmu expression and cell-cell adhesion. J Cell Biol. 2003;163:1351–1362. doi: 10.1083/jcb.200306067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:1475–1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, Stanger BZ, Wells RG. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53:1685–1695. doi: 10.1002/hep.24206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, Kolb A, Friess H, Schuppan D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48:453–464. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 37.Wang B, Dolinski BM, Kikuchi N, Leone DR, Peters MG, Weinreb PH, Violette SM, Bissell DM. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified rat liver fat-storing cells in culture divide and contain collagen. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- 40.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279:23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 42.Patsenker E, Popov Y, Stickel F, Schneider V, Ledermann M, Sagesser H, Niedobitek G, Goodman SL, Schuppan D. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, Sheppard D, Gardner HA, Violette SM. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, Sheppard D, Fogo AB. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Increased expression levels of integrin alphavbeta5 on scleroderma fibroblasts. Am J Pathol. 2004;164:1275–1292. doi: 10.1016/s0002-9440(10)63215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Involvement of αvβ5 integrin-mediated activation of latent transforming growth factor β1 in autocrine transforming growth factor β signalling in systemic sclerosis fibroblasts. Arthritis & Rheumatism. 2005;52:2897–2905. doi: 10.1002/art.21246. [DOI] [PubMed] [Google Scholar]
- 47.Asano Y, Ihn H, Jinnin M, Mimura Y, Tamaki K. Involvement of alphavbeta5 integrin in the establishment of autocrine TGF-beta signaling in dermal fibroblasts derived from localized scleroderma. J Invest Dermatol. 2006;126:1761–1769. doi: 10.1038/sj.jid.5700331. [DOI] [PubMed] [Google Scholar]
- 48.Häkkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, Larjava H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol. 2004;164:229–242. doi: 10.1016/s0002-9440(10)63113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.AlDahlawi S, Eslami A, Häkkinen L, Larjava HS. The alphavbeta6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen. 2006;14:289–297. doi: 10.1111/j.1743-6109.2006.00123.x. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, Kapoor M, Denton CP, Abraham DJ, Leask A. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum. 2009;60:2817–2821. doi: 10.1002/art.24801. [DOI] [PubMed] [Google Scholar]
- 51.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, Goyal J, Feghali-Bostwick CA, Matteson EL, O’Hara C, Lafyatis R, Davis GS, Huang X, Sheppard D, Violette SM. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 54.Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP. The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol. 2010;177:2837–2849. doi: 10.2353/ajpath.2010.100425. [DOI] [PMC free article] [PubMed] [Google Scholar]