

Summary
In region‐specific forms of experimental autoimmune encephalomyelitis (EAE), lesion initiation is regulated by T‐cell‐produced interferon‐γ (IFN‐γ) resulting in spinal cord disease in the presence of IFN‐γ and cerebellar disease in the absence of IFN‐γ. Although this role for IFN‐γ in regional disease initiation is well defined, little is known about the consequences of previous tissue inflammation on subsequent regional disease, information vital to the development of therapeutics in established disease states. This study addressed the hypothesis that previous establishment of regional EAE would determine subsequent tissue localization of new T‐cell invasion and associated symptoms regardless of the presence or absence of IFN‐γ production. Serial transfer of optimal or suboptimal doses of encephalitogenic IFN‐γ‐sufficient or ‐deficient T‐cell lines was used to examine the development of new clinical responses associated with the spinal cord and cerebellum at various times after EAE initiation. Previous inflammation within either cerebellum or spinal cord allowed subsequent T‐cell driven inflammation within that tissue regardless of IFN‐γ presence. Further, T‐cell IFN‐γ production after initial lesion formation exacerbated disease within the cerebellum, suggesting that IFN‐γ plays different roles at different stages of cerebellar disease. For the spinal cord, IFN‐γ‐deficient cells (that are ordinarily cerebellum disease initiators) were capable of driving new spinal‐cord‐associated clinical symptoms more than 60 days after the initial acute EAE resolution. These data suggest that previous inflammation modulates the molecular requirements for new neuroinflammation development.
Keywords: autoimmunity, CD4 T cells, experimental autoimmune encephalomyelitis, inflammation
Introduction
Many excellent studies have examined the mechanisms that regulate entry of T cells into the central nervous system (CNS) during the initiation of neuroinflammatory disease but the mechanisms involved in secondary recruitment of T cells to already diseased CNS tissue remain largely unstudied. This is a concern, because it is clear that inflammation could have profound effects on the phenotype and function of CNS tissue cells and on T‐cell trafficking.1–3 Several pieces of evidence suggest that inflammatory conditioning could play a pivotal role in allowing the progression of various forms of autoimmunity, including multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), a model of MS.1–3 This possibility is particularly applicable to EAE because current models suggest that EAE is a two‐stage disease that makes use of a small initial CNS inflammation to drive the EAE pathology.4–6 Under these models, reactivation of small numbers of CNS‐specific T cells results in inflammatory conditioning of the CNS tissue allowing a secondary large‐scale immune cell infiltration. The requirement of multiple stages in disease initiation suggests that exacerbation of established disease or re‐induction of previously resolved disease may use previous inflammatory tissue conditioning and so not require molecules that may be vital to disease initiation. An understanding of the mechanisms involved in continuing or secondary inflammation would seem particularly important to illuminate clinically useful targets, as neuroinflammation is typically observed in clinical settings after disease initiation.
The clinical symptoms associated with autoimmune neuroinflammatory diseases such as MS and EAE are determined by the anatomic location of lesions within the CNS.7–11 In MS and EAE, myelin‐specific inflammation of the CNS results in discrete lesions within the context of a much larger field of myelin‐containing tissue. The formation of these lesions within specific CNS regions in turn determines the symptomatic effects of neuroinflammation. In this way, the mechanisms responsible for lesion localization play a vital role in determining the clinical outcomes of neuroinflammatory disease, with specific patterns of lesion formation predictive of both clinical manifestations and disease severity. Unfortunately, despite the clear importance of lesion localization in the MS disease course, the mechanisms that regulate the appearance of discrete lesions within the diverse tracts of CNS white matter that could be targeted by anti‐myelin responses remain largely uncharacterized.
Previous work within the EAE model revealed that disruption of the encephalitogenic T cell's capacity to produce interferon‐γ (IFN‐γ) resulted in significant changes in clinical outcome, with the extent and exact type of changes determined by the EAE system studied.12–18 In some of these studies differential lesion development in the spinal cord and cerebellum of mice following T‐cell recognition of myelin antigen revealed that disease development in both tissues is critically regulated by T‐cell cytokine production and host cytokine recognition.17,18 In these models, the capacity of a fraction of pathogenic T cells to produce IFN‐γ was sufficient to induce lesion development within the spinal cord and prevent lesion development within the cerebellum and brainstem. Interestingly, one of these studies revealed that IFN‐γ had dual functions as both a pro‐inflammatory and anti‐inflammatory agent and simultaneously acted to block EAE disease in the cerebellum while potentiating EAE in the spinal cord. These functions were the result of separable interactions with the different host tissues and were found to have different requirements for numbers of T cells capable of producing IFN‐γ.18 Because of the differential requirements for IFN‐γ‐producing cells, administration of mixed populations of IFN‐γ‐deficient and IFN‐γ‐sufficient cells at specific ratios resulted in the development of EAE in both the spinal cord and cerebellum within the same host. The study also demonstrated that sub‐optimal numbers of IFN‐γ‐producing T cells that were insufficient to induce disease alone were nonetheless sufficient to determine the site of lesion development.
These data, combined with earlier studies, allowed the development of two EAE models, classical EAE (cEAE) and non‐classical/atypical EAE (nEAE), induced by adoptive transfer of IFN‐γ‐sufficient or IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells, respectively (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein). Further, by using MOG35–55‐specific EAE induction in C57BL/6 mice we were able to examine the effects of IFN‐γ on inflammation during both acute and chronic phases of EAE. Although these studies clearly delineated a vital role for IFN‐γ signalling in T‐cell invasion and inflammation during the initiation of EAE disease, the role of IFN‐γ in secondary T‐cell recruitment to established CNS inflammatory sites and the subsequent exacerbation of established disease were not addressed.
Here, we used serial adoptive transfer of polyclonal IFN‐γ‐sufficient or IFN‐γ‐deficient MOG35–55‐specific CD4+ T‐cell lines to examine the role of IFN‐γ production in secondarily recruited T‐cell activity within established sites of inflammation in both classical spinal‐cord‐targeted EAE (cEAE) and non‐classical cerebellar‐targeted EAE (nEAE). We used these CNS‐region‐specific diseases to examine the activities of IFN‐γ following disease initiation within each tissue. We made use of a serial adoptive transfer methodology to dissect the effects of IFN‐γ on different stages of disease development including the initiation of lesions, secondary T‐cell recruitment to established sites of inflammation, and exacerbation of chronic disease. Using these techniques, we found that previous formation of lesions, even clinically silent lesions, abrogates the effect of IFN‐γ production on future inflammation of both the spinal cord and cerebellum. These data suggest that initiation of EAE within a tissue makes that tissue more permissive to future inflammation.
Materials and methods
Mice
C57BL/6J and congenic CD45.1 (Ly5.2) mice were purchased from the National Cancer Institute (Bethesda, MD) and IFN‐γ‐deficient B6.129S7‐ifngtm1Ts/J mice were purchased from Jackson Laboratories (Bar Harbor, ME). All experiments were approved by the University of Maryland, Baltimore Animal Care Committee and all mice were housed in an AAALAC approved facility.
T‐cell lines
To generate MOG‐specific cell lines, C57BL/6J or IFN‐γ‐deficient B6.129S7‐ifngtm1Ts/J mice were immunized subcutaneously with 50 μg MOG35–55 peptide (Sigma Genosys, Woodlands, TX) emulsified in incomplete Freund's adjuvant and supplemented with 500 μg/ml Mycobacterium tuberculosis. CD4+ cells were isolated from the spleen as previously described.19 Cells were then stimulated at a concentration of 1 × 106 cells/ml in the presence of 5 × 106 cells/ml irradiated C57BL/6 splenocytes, 10 μg/ml MOG35–55, 10 units/ml interleukin‐12 (IL‐12) and 10 units/ml IL‐2, in RPMI‐1640 complete medium containing 10% fetal calf serum. Cells were isolated 7 days later by density centrifugation then restimulated in the absence of IL‐12 under all other conditions outlined above. Cells underwent two to four rounds of stimulation then were tested for cytokine production capacity as previously described. Cells were then stored at −70° in 10% DMSO containing fetal calf serum. Before use, cells were thawed and restimulated for 7 days with 10 μg/ml MOG35–55 and 10 units/ml IL‐2. Before injection, cells were again restimulated for 4 days with MOG35–55 and IL‐2, separated by density centrifugation, then washed and resuspended in Hanks’ balanced salt solution. The cytokine profile of each line was determined as previously described.20 Ovalbumin (OVA) ‐specific T‐cell lines were produced using the same protocol with 900 μg whole OVA substituted for MOG35–55. To determine the appropriate T‐cell dose to induce EAE, variable numbers of cells in 300 μl Hanks’ balanced salt solution were transferred by intravenous injection into the tails of 6‐ to 8‐week‐old mice, and animals were monitored for the development of clinical scores.
Two distinct wild‐type and two distinct IFN‐γ‐deficient T‐cell lines were used for the experiments presented here. Each table includes data generated using both sets of cell lines to insure reproducibility among individual T‐cell lines.
Clinical scoring
Mice were examined daily for symptoms for at least 30 days after adoptive transfer. Symptoms of classical EAE were scored based on area(s) affected (1 = partial or total flaccid paralysis of tail; 2 = hind limb weakness/disrupted righting reflex; 3 = flaccid paralysis in one hind limb; 4 = flaccid paralysis in both hind limbs; 5 = moribund/dead). Symptoms of non‐classical EAE were scored based on a non‐progressive analysis of anatomical area(s) affected, signs of rigor, signs of ataxia/dysequilibrium, and the severity of the effects (1 = rigor in the tail; 2 = widened stance, leaning or wobbling to one side, mild ataxia; 3 = constant turning to one side, rigor in one limb, moderate ataxia; 4 = rolling of animal, rigor in two or more limbs, severe ataxia; 5 = moribund/dead).
Statistics
All statistics were performed using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA).
Results
We wished to examine the role of IFN‐γ in secondary T‐cell‐mediated inflammation. A previous report indicated that IFN‐γ‐producing encephalitogenic T cells blocked the development of EAE inflammation of the cerebellum.18 Given this information, we wanted to determine if IFN‐γ could act therapeutically on established cerebellar nEAE disease. We have previously reported that while cells begin to accumulate within preparations made from the CNS within 3 days of adoptive transfer, reliable invasion of the cells into perivasculature and parenchyma occurred ~5 days after transfer21. Hence, to determine if production of IFN‐γ signalling after initial T‐cell invasion was sufficient to protect the cerebellum from fulminant EAE development, we induced non‐classical EAE by administration of 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells then, either immediately following the initial injection or 5 days later, we injected a pathogenic dose (5 × 106) of an IFN‐γ‐sufficient MOG35–55‐specific cell line. Very early administration of IFN‐γ‐producing cells relative to the nEAE initiating IFN‐γ‐deficient T‐cell administration resulted in blockade of nEAE and induction of cEAE, whereas later administration of IFN‐γ‐producing cells had no effect on the number of mice displaying nEAE (Table 1).
Table 1.
Interferon‐γ production after disease initiation does not protect the cerebellum from non‐classical (cerebellar) experimental autoimmune encephalomyelitis (nEAE)
| Transferred cells1 | Incidence of nEAE (Peak CS ± SD)2 | Incidence of cEAE (Peak CS ± SD)2 | Mortality3 | |
|---|---|---|---|---|
| Group A | Day 0 IFN‐γ−/− | 19/20 (2·7 ± 1·0) | 0/20 | 1/20 |
| Group B | >Day 0 IFN‐γ−/− Day 0 WT |
0/10 | 10/10 (3·0 ± 1·6) | 2/10 |
| Group C | Day 0 IFN‐γ−/− Day 5 WT |
25/25 (4·7 ± 0·5)4 | 5/25 (2·2 ± 1·6) | 21/255 |
| Group D | Day 0 IFN‐γ−/− Day 5 IFN‐γ−/− |
14/14 (3·5 ± 0·9) | 0/14 | 5/146 |
| Group E | Day 0 IFN‐γ−/− Day 5 OVA |
10/10 (3·2 ± 1·0) | 0/10 | 1/10 |
| Group F | Day 0 WT Day 5 IFN‐γ−/− |
0/8 | 8/8 (2·9 ± 0·6) | 0/8 |
| Group G | Day 5 IFN‐γ−/− | 20/20 (2·1 ± 0·7) | 0/20 | 0/20 |
| Group H | Day 5 WT | 0/20 | 18/20 (2·6 ± 1·1) | 1/20 |
Transferred cells: 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells (IFN‐γ−/−) and/or 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (WT) were administered at the start of the experiment (Day 0) or 5 days later (Day 5) (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein).
(Peak CS ± SD): The mean peak clincial score and standard deviations for each group are shown.
Mortality: includes euthanasia due to acquisition of a nEAE or cEAE clinical score of 5 (the majority of mice) or spontaneous death (three mice in Group C, one mouse in Group D).
Results of Group C peak clinical score were significantly different compared with all other groups by a Kruskal–Wallis test with Dunn's multiple comparison test (P < 0·05).
Group C: mortality was associated with severe nEAE. Only one of the fatalities showed symptoms of classical disease. Results of group C were found to be significantly different from those of all other groups by one‐way analysis of variance using Tukey's multiple comparison test (P < 0·05).
Results of Group D were were found to be significantly different from those of Group G by a Kruskal–Wallis test with Dunn's multiple comparison test (P < 0·05).
Interestingly, mice that received IFN‐γ‐producing cells after initiation of cerebellar inflammation appeared to develop exacerbated disease symptoms, when compared with mice given equivalent numbers of an activated IFN‐γ‐sufficient cell line specific for OVA or additional numbers of IFN‐γ‐deficient MOG35–55‐specific T cells (Table 1). Indeed we found, using a Kruskal–Wallis test with Dunn's multiple comparison test, that the nEAE scores associated with a secondary dose of IFN‐γ‐sufficient cells were significantly increased over the nEAE scores of mice that received IFN‐γ‐deficient T cells or cells that lack CNS antigen specificity (P < 0·05 for both). Further, using a one‐way analysis of variance with Tukey's multiple comparison test we found that the addition of IFN‐γ‐sufficient T cells after initial invasion resulted in a significant increase in EAE mortality (P < 0·05 compared with all other conditions) (Fig. 1). These data suggested that while production of IFN‐γ blocked the development of cerebellar lesions during initiation of neuroinflammation, subsequent production of IFN‐γ exacerbated established cerebellar EAE. These findings may help to explain many of the seemingly contradictory reports regarding the role of IFN‐γ in EAE and MS pathogenesis by highlighting differential functions of IFN‐γ at different stages of neuroinflammation.12,13,22–24
Figure 1.
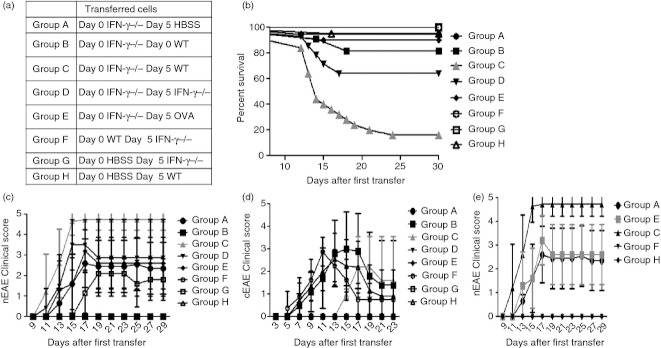
Addition of freshly activated encephalitogenic interferon‐γ (IFN‐γ) ‐sufficient cells shortly after induction of non‐classical experimental autoimmune encephalomyelitis (EAE) exacerbates disease. Mice received 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells (Groups A, C, D, E), 5 × 106 IFN‐γ‐sufficient MOG35–55‐specific CD4+ T cells (Group F), or Hanks’ balanced salt solution (HBSS) (Groups G, H) (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein). In addition, one group (Group B) received both 5 × 106 wild‐type and 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells concurrently. Five days later mice received 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (Groups C, H), 5 × 106 wild‐type ovalbumin‐specific CD4+ T cells (Group E), 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells (Groups D, F, G), or an equivalent amount of HBSS (Group A). Group composition is summarized in (a). Mouse survival was then followed over time (b). Mice were examined for the development of both non‐classical (c) and classical (d) EAE symptoms. Results shown were collected from two separate experiments using five mice in each group.
We performed complementary experiments to determine if a secondary injection of IFN‐γ‐deficient MOG35–55‐specific T cells was sufficient to exacerbate established cEAE. To induce cEAE we injected 5 × 106 IFN‐γ‐sufficient MOG35–55‐specific CD4+ T cells. At 5 days after the initial injection, we injected 5 × 106 of an IFN‐γ‐deficient MOG35–55‐specific cell line, an IFN‐γ‐sufficient OVA‐specific cell line, or the initially injected IFN‐γ‐sufficient MOG35–55‐specific T‐cell line, and examined mice over time for clinical symptoms of EAE (Fig. 2). Interestingly, both MOG35–55‐specific cell lines increased peak classical EAE clinical scores, apparently without regard for IFN‐γ production capacity, resulting in a statistically significant difference in clinical scores compared with mice that received OVA‐specific cells or diluent (Fig. 2). These data demonstrate that following lesion formation within the spinal cord, newly recruited MOG35–55‐specific T cells are not required to produce IFN‐γ to exacerbate disease.
Figure 2.
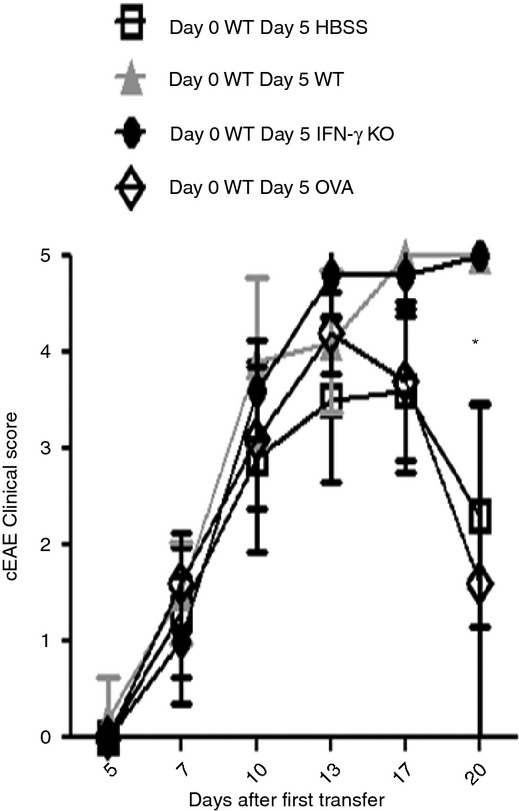
Addition of freshly activated encephalitogenic cells exacerbates ongoing classical experimental autoimmune encephalomyelitis (cEAE) regardless of the capacity of new cells to produce interferon‐γ (IFN‐γ). Mice received 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein). Five days later mice received either another 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (▲), 5 × 106 wild‐type ovalbumin‐specific CD4+ T cells (◊), 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells (●), or an equivalent amount of Hanks’ balanced salt solution (HBSS) (□). Mice were then followed for the clinical symptoms of cEAE over time. The * indicates a statistically significant difference (P < 0·05) at the 20‐day time‐point for both HBSS and ovalbumin‐treated groups when compared with both wild‐type (WT) and IFN‐γ knockout (KO) groups by a Kruskal–Wallis test with Dunn's multiple comparison tests. Results shown were collected from two separate experiments using five mice in each group.
Given the capacity of both IFN‐γ‐sufficient and IFN‐γ‐deficient cell lines to exacerbate established acute cEAE we wished to determine what effect, if any, IFN‐γ production would have on exacerbation of chronic EAE. To address this we examined the capacity of newly activated IFN‐γ‐sufficient and IFN‐γ‐deficient cells to exacerbate chronic cEAE symptoms.
To determine the role of IFN‐γ in exacerbation of previously resolved EAE we induced cEAE by injecting 5 × 106 IFN‐γ‐sufficient MOG35–55‐specific CD4+ T cells and examined mice over time for clinical symptoms of EAE. On day 23 (during or immediately after acute disease resolution) or day 90 (≥ 60 days after acute EAE resolution in all mice) after the initial injection, we injected 5 × 106 of an IFN‐γ‐deficient MOG35–55‐specific cell line, an IFN‐γ‐sufficient OVA‐specific cell line, or the initially injected IFN‐γ‐sufficient MOG35–55‐specific T‐cell line. At both time points, re‐administration of the initially injected IFN‐γ‐sufficient MOG35–55‐specific T‐cell line was sufficient to re‐induce acute clinical symptoms similar in severity to those seen following initial EAE development (Table 2). This exacerbation of previously resolved disease was antigen dependent as adoptive transfer of equal numbers of an OVA‐specific T‐cell line had little to no effect on clinical manifestation (Table 2). Interestingly, a second injection of an IFN‐γ‐deficient MOG35–55‐specific cell line was also sufficient to re‐induce acute cEAE symptoms, at both time‐points (Table 2, Fig. 3). At day 23 most mice developed only classical disease, suggesting that sufficient numbers of the initial IFN‐γ‐sufficient encephalitogenic T cells remain active at this time to determine lesion localization. In contrast, at day 90 mice developed both cerebellar and spinal cord forms of disease, suggesting that in the absence of continued IFN‐γ signalling prior inflammation still allowed secondary inflammation and reactivation of resolved disease. Together, these data suggest that the presence of established lesions is sufficient to allow exacerbation of chronic cEAE symptoms regardless of the IFN‐γ‐producing status of secondarily invasive encephalitogenic T cells. These findings also correspond with the conclusions of previous works that described enhanced T‐cell CNS invasion in the context of previous inflammation due to exogenous mediators.4,21,25,26
Table 2.
Addition of freshly activated encephalitogenic cells exacerbates chronic classical experimental autoimmune encephalomyelitis (cEAE) disease regardless of the capacity of new cells to produce interferon‐γ (IFN‐γ)
| Transferred cells1 | Incidence of nEAE (Peak CS ± SD)2 | Incidence of 2nd cEAE (Peak CS ± SD)2 |
|---|---|---|
| Day 0 WT Day 90 WT |
0/12 | 12/12 (3·3 ± 1·0) |
| Day 0 WT Day 90 IFN‐γ−/− |
11/12 (2·4 ± 1·1) | 12/12 (3·6 ± 0·8) |
| Day 0 HBSS Day 90 IFN‐γ−/− |
10/10 (2·8 ± 1·3) | 0/10 |
| Day 0 WT Day 90 OVA |
0/10 | 0/10 |
Transferred cells: Either 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (WT) or diluent (Hanks’ balanced salt solution; HBSS) was administered at the start of the experiment (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein). After 90 days mice received 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (WT), 5 × 106 IFN‐γ‐deficient MOG35–55‐specific CD4+ T cells (IFN‐γ−/−), or 5 × 106 wild‐type ovalbumin‐specific CD4+ T cells (OVA).
(Peak CS ± SD): The mean peak clincial score and standard deviations for each group are shown.
Figure 3.
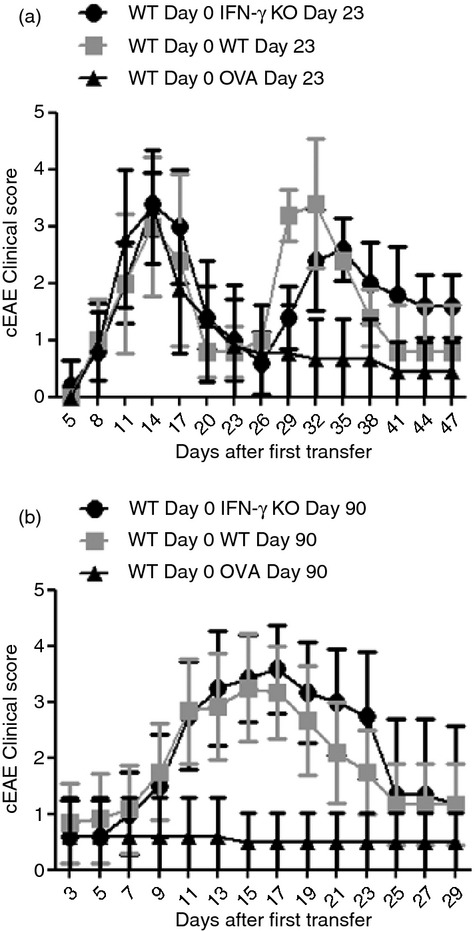
Addition of freshly activated encephalitogenic cells exacerbates chronic classical experimental autoimmune encephalomyelitis (cEAE) disease regardless of the capacity of new cells to produce interferon‐γ (IFN‐γ). Mice received 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein). After 23 days (a) or 90 days (b) mice received 5 × 106 of an IFN‐γ‐deficient MOG35–55‐specific cell line (●), an IFN‐γ‐sufficient ovalbumin‐specific cell line (▲), or the initially injected IFN‐γ‐sufficient MOG35–55‐specific T‐cell line (■). Mice were then followed for the clinical symptoms of cEAE over time. Results shown were collected from eight mice (a) or 10–12 mice (b) in each group examined over two experiments.
We wished to further examine the effects of established lesions on regional disease exacerbation by examination of the nEAE model. Unfortunately, the non‐classical model of EAE does not follow the predictable and closely timed resolution of acute clinical symptoms observed in the cEAE model, instead displaying characteristics of a severe chronic disease state. As such, rather than examining the capacity of cells to exacerbate resolved disease, we instead examined whether the formation of clinically silent lesions within the cerebellum and brainstem would allow future IFN‐γ‐sufficient MOG35–55‐specific cells to initiate nEAE. To address this issue we adoptively transferred a suboptimal dose (1 × 106) of an IFN‐γ‐deficient MOG35–55‐specific cell line, and then either 5 or 23 days later gave a secondary injection containing 5 × 106 of an IFN‐γ‐sufficient MOG35–55‐specific T‐cell line. Interestingly, pretreatment with the IFN‐γ‐deficient line allowed induction of nEAE in a substantial proportion of mice following transfer of a pathogenic dose of IFN‐γ‐sufficient encephalitogenic cells (Table 3). These data demonstrated that the existence of clinically silent lesions within the cerebellum or brainstem can allow future inflammation at those sites regardless of IFN‐γ production by secondary pathogenic T cells.
Table 3.
Incidence and severity experimental autoimmune encephalomyelitis (EAE) induced following clinically silent inflammation of the cerebellum
| Transferred cells1 | Incidence of nEAE2 (peak CS ± SD) | Incidence of cEAE2 (peak CS ± SD) |
|---|---|---|
| Day 0: 1 × 106 IFN‐γ−/− | 1/20 (2) | 0/20 |
| Day 0: 1 × 106 IFN‐γ−/− Day 5: WT |
16/20 (3·4 ± 1·1) | 20/20 (2·8 ± 1·0) |
| Day 0: 1 × 106 IFN‐γ−/− Day 5: OVA |
5/20 (1·8 ± 0·4) | 0/20 |
| Day 0: 1 × 106 IFN‐γ−/− Day 23: WT |
6/20 (2·6 ± 0·8) | 20/20 (3·0 ± 1·0) |
| Day 0: 1 × 106 IFN‐γ−/− Day 23: OVA |
1/20 (1) | 0/20 |
Transferred cells: 1 × 106 interferon‐γ (IFN‐γ) ‐deficient MOG35–55‐specific CD4+ T cells (IFN‐γ−/−) were administered at the start of the experiment then either 5 or 23 days later mice received 5 × 106 wild‐type MOG35–55‐specific CD4+ T cells (WT) or 5 × 106 wild‐type ovalbumin‐specific CD4+ T cells (OVA) (where MOG35–55 is a peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein).
(Peak CS ± SD): The mean peak clincial score and standard deviations for each group are shown.
Discussion
Overall, the data presented here demonstrate that the molecular requirements for T‐cell invasion of the CNS may differ depending on the underlying inflammatory condition of the CNS tissue. Indeed, these data suggest that previous T‐cell invasion of CNS tissue, even in the absence of clinically visible disease, is sufficient to make that tissue more permissive to future inflammation. Ultimately, these data suggest that regarding pre‐clinical investigation of new therapeutics, the most clinically applicable results may be provided by examination of therapeutic function in the context of ongoing CNS inflammation.
These studies made extensive use of a serial adoptive transfer technique to examine sequential instances of disease activity. Several studies have suggested that resolution of acute EAE is associated with active regulation of ongoing T‐cell responses.27,28 These findings would seem to suggest that exacerbation of chronic EAE by introduction of fresh activated encephalitogenic T cells could be inhibited by ongoing suppression of myelin antigen‐specific T cells within the CNS, an idea that would fit well with the emerging models suggesting that tissue‐specific inflammation can result in enhanced activity of local regulatory T cells.29,30 However, we previously reported that adoptive transfer of a second dose of MOG35–55‐specific encephalitogenic T cells following acute EAE resolution was sufficient to induce a second instance of acute inflammation, suggesting that active regulation was not occurring in our system.21 This apparent contradiction could result from the differences involved in timing, induction method, or method of re‐challenge.
There is a wealth of data suggesting that conditioning of tissue within the CNS is vital to the development of EAE.4,21,25,26 Current models suggest that under homeostatic conditions the unique cellular architecture of the CNS system prevents CNS antigen‐specific effectors from invading the parenchyma. Instead, EAE initiation is thought to begin with infiltration of small numbers of autoreactive T cells into the subarachnoid space of the CNS, with some evidence to suggest that this infiltration occurs through the choroid plexus or directly into the subarchnoid space following introduction of exogenous inflammatory mediators.2,5,31,32,33 This primary infiltration of the CNS is followed by restimulation of the CNS‐specific T cells, induction of chemokine and adhesion molecule expression in distal regions of the CNS (conditioning), and subsequent large‐scale recruitment of inflammatory cells to the CNS parenchyma, resulting in lesion formation.5,34
Regional recognition of cytokines has previously been shown to regulate lesion development in both the cerebellum and spinal cord.17,18,35 Here we examined the effect of previously established CNS inflammation on induction of new neuroinflammatory symptoms and found that previous regional cerebellar inflammation was sufficient to allow new disease induction in the presence of the normally suppressive cytokine IFN‐γ. Additionally, previous inflammation of the spinal cord was sufficient to allow IFN‐γ‐deficient T cells to induce new EAE symptoms associated with the spinal cord, an area that these cells are typically unable to invade. These data suggest either that IFN‐γ functions early in the process of spinal cord lesion formation or that the signal generated by IFN‐γ during spinal cord disease induction is extremely long‐lived.
It is clear that many factors contribute to localized development of lesions, including responding T‐cell repertoire and functional capacity, CNS regional chemokine production and immune cell chemokine expression, and variable T‐cell/endothelial cell adhesion molecule recognition.11,17,31,36–39 However, the study presented here examined only CNS tissue intrinsic changes following inflammation induction. As such, the effect of previous inflammation on other mechanisms of lesion localization remains an important question.
In conclusion, the data presented here suggest that previous inflammatory insults predispose CNS tissue to further inflammation long after the clinical effects of the primary insult have been resolved. These findings are vital to consider during the experimental investigation of new therapeutics, studies often performed by examining the effects of treatment on the induction of a primary inflammatory insult. Further, these data clearly suggest that examinations of new therapeutic agents should account for the apparent contribution of previously established CNS inflammation in the development of new or exacerbated symptoms.
Acknowledgments
This work was supported in part by research grants from the National Multiple Sclerosis Society (RG 4305A1/T and RG 4492‐A‐2). We would like to acknowledge Dr John H. Russell, Dr Kamal D. Moudgil, Dr Jonathan S. Bromberg and Ms Julia Sim for invaluable assistance with reagents and with the editing of this manuscript.
Glossary
- EAE
experimental autoimmune encephalomyelitis
- nEAE
non‐classical (cerebellar) EAE
- cEAE
classical (spinal cord) EAE
- IFN‐γ
interferon‐γ
- CNS
central nervous system
- MS
multiple sclerosis
- MOG35–55
peptide comprising amino acids 35 to 55 of the myelin oligodendrocyte glycoprotein
Disclosures
The authors have no conflict of interest to declare.
References
- 1.Engelhardt B, Ransohoff RM. The ins and outs of T‐lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–95. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–81. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 3.Constantin G. Chemokine signaling and integrin activation in lymphocyte migration into the inflamed brain. J Neuroimmunol. 2008;198:20–6. doi: 10.1016/j.jneuroim.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 4.Bartholomaus I, Kawakami N, Odoardi F. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462:94–8. doi: 10.1038/nature08478. et al. [DOI] [PubMed] [Google Scholar]
- 5.Kivisakk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, Khoury SJ. Localizing central nervous system immune surveillance: meningeal antigen‐presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol. 2009;65:457–69. doi: 10.1002/ana.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flugel A, Berkowicz T, Ritter T. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001;14:547–60. doi: 10.1016/s1074-7613(01)00143-1. et al. [DOI] [PubMed] [Google Scholar]
- 7.Langdon DW, Thompson AJ. Multiple sclerosis: a preliminary study of selected variables affecting rehabilitation outcome. Mult Scler. 1999;5:94–100. doi: 10.1177/135245859900500205. [DOI] [PubMed] [Google Scholar]
- 8.Riise T, Gronning M, Aarli JA, Nyland H, Larsen JP, Edland A. Prognostic factors for life expectancy in multiple sclerosis analysed by Cox‐models. J Clin Epidemiol. 1988;41:1031–6. doi: 10.1016/0895-4356(88)90041-8. [DOI] [PubMed] [Google Scholar]
- 9.Riise T, Gronning M, Fernandez O. Early prognostic factors for disability in multiple sclerosis, a European multicenter study. Acta Neurol Scand. 1992;85:212–8. doi: 10.1111/j.1600-0404.1992.tb04031.x. et al. [DOI] [PubMed] [Google Scholar]
- 10.Naismith RT, Trinkaus K, Cross AH. Phenotype and prognosis in African‐Americans with multiple sclerosis: a retrospective chart review. Mult Scler. 2006;12:775–81. doi: 10.1177/1352458506070923. [DOI] [PubMed] [Google Scholar]
- 11.Sobel RA. Genetic and epigenetic influence on EAE phenotypes induced with different encephalitogenic peptides. J Neuroimmunol. 2000;108:45–52. doi: 10.1016/s0165-5728(99)00270-2. [DOI] [PubMed] [Google Scholar]
- 12.Furlan R, Brambilla E, Ruffini F. Intrathecal delivery of IFN‐γ protects C57BL/6 mice from chronic‐progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system‐infiltrating lymphocytes. J Immunol. 2001;167:1821–9. doi: 10.4049/jimmunol.167.3.1821. et al. [DOI] [PubMed] [Google Scholar]
- 13.Krakowski M, Owens T. Interferon‐γ confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–6. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 14.Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon‐γ inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129(Pt 5):1306–18. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- 15.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN‐γ plays a critical down‐regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–7. [PubMed] [Google Scholar]
- 16.Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D, Lira SA, Zagzag D, Lafaille JJ. IFN‐γ determines distinct clinical outcomes in autoimmune encephalomyelitis. J Immunol. 2005;174:1416–23. doi: 10.4049/jimmunol.174.3.1416. [DOI] [PubMed] [Google Scholar]
- 17.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by TH1 and TH17 cells. Nat Med. 2008;14:337–42. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN‐γ determine lesion localization patterns during EAE pathogenesis. J Exp Med. 2008;205:2633–42. doi: 10.1084/jem.20080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archambault AS, Sim J, Gimenez MA, Russell JH. Defining antigen‐dependent stages of T cell migration from the blood to the central nervous system parenchyma. Eur J Immunol. 2005;35:1076–85. doi: 10.1002/eji.200425864. [DOI] [PubMed] [Google Scholar]
- 20.Lees JR, Iwakura Y, Russell JH. Host T cells are the main producers of IL‐17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J Immunol. 2008;180:8066–72. doi: 10.4049/jimmunol.180.12.8066. [DOI] [PubMed] [Google Scholar]
- 21.Lees JR, Archambault AS, Russell JH. T‐cell trafficking competence is required for CNS invasion. J Neuroimmunol. 2006;177:1–10. doi: 10.1016/j.jneuroim.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with γ interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 23.Renno T, Taupin V, Bourbonniere L. Interferon‐γ in progression to chronic demyelination and neurological deficit following acute EAE. Mol Cell Neurosci. 1998;12:376–89. doi: 10.1006/mcne.1998.0725. et al. [DOI] [PubMed] [Google Scholar]
- 24.Ferber IA, Brocke S, Taylor‐Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN‐γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 25.Piccio L, Rossi B, Colantonio L. Efficient recruitment of lymphocytes in inflamed brain venules requires expression of cutaneous lymphocyte antigen and fucosyltransferase‐VII. J Immunol. 2005;174:5805–13. doi: 10.4049/jimmunol.174.9.5805. et al. [DOI] [PubMed] [Google Scholar]
- 26.Piccio L, Rossi B, Scarpini E. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P‐selectin glycoprotein ligand‐1 and heterotrimeric G(i)‐linked receptors. J Immunol. 2002;168:1940–9. doi: 10.4049/jimmunol.168.4.1940. et al. [DOI] [PubMed] [Google Scholar]
- 27.McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+ CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–32. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- 28.Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+ CD25+ T regulatory cells and IL‐10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007;178:3447–56. doi: 10.4049/jimmunol.178.6.3447. [DOI] [PubMed] [Google Scholar]
- 29.Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak‐Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480:538–42. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seneschal J, Clark RA, Gehad A, Baecher‐Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873–84. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reboldi A, Coisne C, Baumjohann D. C‐C chemokine receptor 6‐regulated entry of TH‐17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–23. doi: 10.1038/ni.1716. et al. [DOI] [PubMed] [Google Scholar]
- 32.Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM‐1, VCAM‐1, and MAdCAM‐1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am J Pathol. 1996;148:1819–38. [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda M, Tsukada N, Miyagi K, Yanagisawa N. Adhesion of lymphocytes to endothelial cells in experimental allergic encephalomyelitis before and after treatment with endotoxin lipopolysaccharide. Int Arch Allergy Immunol. 1995;106:335–44. doi: 10.1159/000236863. [DOI] [PubMed] [Google Scholar]
- 34.Brown DA, Sawchenko PE. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. J Comp Neurol. 2007;502:236–60. doi: 10.1002/cne.21307. [DOI] [PubMed] [Google Scholar]
- 35.Gimenez MA, Sim J, Archambault AS, Klein RS, Russell JH. A tumor necrosis factor receptor 1‐dependent conversation between central nervous system‐specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am J Pathol. 2006;168:1200–9. doi: 10.2353/ajpath.2006.050332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B, Korn T. Th17 lymphocytes traffic to the central nervous system independently of α4 integrin expression during EAE. J Exp Med. 2011;208:2465–76. doi: 10.1084/jem.20110434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroenke MA, Chensue SW, Segal BM. EAE mediated by a non‐IFN‐γ/non‐IL‐17 pathway. Eur J Immunol. 2010;40:2340–8. doi: 10.1002/eji.201040489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL‐12‐ and IL‐23‐modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–41. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abromson‐Leeman S, Bronson R, Luo Y, Berman M, Leeman R, Leeman J, Dorf M. T‐cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165:1519–33. doi: 10.1016/S0002-9440(10)63410-4. [DOI] [PMC free article] [PubMed] [Google Scholar]