

Summary
The tumour microenvironment is complex containing not only neoplastic cells but also a variety of host cells. The heterogeneous infiltrating immune cells include subsets of cells with opposing functions, whose activities are mediated either directly or through the cytokines they produce. Systemic delivery of cytokines such as interleukin‐2 ( IL‐2) has been used clinically to enhance anti‐tumour responses, but these molecules are generally thought to have evolved to act locally in a paracrine fashion. In this study we examined the effect of local production of IL‐2 on the growth and the immune response to B16 melanoma cells. We found that the local production of IL‐2 enhances the number of interferon‐γ‐expressing CD8 T and natural killer cells in the tumour, as well as inducing expression of vascular cell adhesion molecule 1 on tumour vessels. These responses were largely absent in interferon‐γ knockout mice. The expression of IL‐2 in the tumour microenvironment decreases tumour growth despite also enhancing Foxp3+ CD4+ regulatory T cells and anti‐inflammatory cytokines such as IL‐10. Higher levels of IL‐2 in the tumour microenvironment eliminated the progressive growth of the B16 cells in vivo, and this inhibition was dependent on the presence of either T cells or, to a lesser extent, natural killer cells. Surprisingly however, the B16 tumours were not completely eliminated but instead were controlled for an extended period of time, suggesting that a form of tumour dormancy was established.
Keywords: dormancy, interleukin‐2, interferon‐γ, tumour microenvironment
Introduction
A growing tumour is a mixture of transformed cells and a variety of host cells and factors that both enhance and suppress the immune response.1–3 This may reflect aspects of normal homeostatic mechanisms that help to tightly regulate an immune response.1–6 At the cellular level, this dichotomy is reflected by finding that tumours contain effector cells, such as cytotoxic T cells, as well as suppressive cell populations, such as regulatory T (Treg) cells and myeloid‐derived suppressor cells.1,3,7–9 Similarly, at the molecular level, tumours contain cytokines such as interferon‐γ (IFN‐γ), as well as interleukin‐10 (IL‐10) and transforming growth factor‐β, which can oppose one another in regulating an immune response.1 One prominent feature thought to dramatically impact the growth of tumours is the host immune response, which can affect tumour growth at many points during its evolution from initiation to metastasis.8,10 Although anti‐tumour responses can be clearly demonstrated in humans and in mice,1,3,8 it has become evident that the immune response to tumours is extremely complex and that many factors influence its efficacy. To grow and progress, the transformed cells require additional nutrients and oxygen, which involves an intricate process of vessel development and formation that in turn may influence the nature and number of infiltrating host cells.11–13 Vessel development as well as immune cell infiltration can also be influenced by factors that the transformed cells produce themselves, such as vascular endothelial growth factor, by cytokines such as IL‐6 produced by stromal cells14 or by cytokines such as IFN‐γ produced by the host immune cells.15,16 The integration of all these interactions ultimately determines the outcome; tumour growth, tumour control, or elimination. We hypothesize that it is the relative balance of these factors in the tumour microenvironment that is critical in determining the fate of the tumour and that altering this balance may affect the immune response and tumour growth.
Cytokines comprise a key set of molecules that can dramatically affect immune responses. Given their crucial role in immune responses, they have been used in a variety of systems and in clinical trials in an effort to enhance immune responses to tumours.17–19 One of the first cytokines discovered and characterized, IL‐2, has been approved by the US Food and Drug Administration to treat melanoma and renal cancer.17,20,21 Strikingly, systemic IL‐2 treatment has led to remarkable and durable remissions in a fraction of patients with melanoma (7% in one study17), although the adverse effects of systemic treatment limit its utility. Interleukin‐2, originally described as a T‐cell growth factor (indeed initially called T‐cell growth factor), has a multifaceted role in the immune system.22,23 Studies have suggested that IL‐2 has a paradoxical role in both promoting and down‐regulating immune responses. For example, IL‐2 is a potent growth and differentiation factor for T cells and natural killer (NK) cells.22,24,25 In contrast, IL‐2 is important in the development of Treg cells,26 and for activation‐induced cell death, both of which act to suppress immune responses.22,23,27,28
The dual role of IL‐2 in promoting as well as inhibiting the immune response has led to a renewed interest in IL‐2 as an immunomodulating agent that might either enhance or suppress immune responses.29–31 In the current study, we examine the effect of local IL‐2 expression on the host response and tumour growth in the B16 tumour model, an aggressive mouse model of melanoma that is relatively resistant to systemic treatment with IL‐2.32 Here we show that local expression of IL‐2 at low levels within the tumour microenvironment of B16 tumours results in an increased frequency of NK cells and effector T cells and slower tumour growth despite an increase in infiltrating Foxp3+ T regulatory CD4 T cells. Surprisingly, higher levels of IL‐2 at the tumour site result in tumour control that is dependent upon NK cells or T cells, but does not result in total tumour clearance, indicating that a long‐term form of tumour dormancy was established.
Materials and methods
Mice, cell lines and generation of transfectants
C57BL/6J and B6.129S7‐Ifngtm1Ts (IFN‐γ−/−) were purchased from the Jackson Laboratory (Bar Harbor, ME). Nude mice were purchased from Taconic Farms (Hudson, NY). All mice were treated in accordance with guidelines approved by the University Committee on Animal Resources. The B16‐F0 (B16) cell line, a spontaneously arising C57BL/6‐derived melanoma, was obtained from the American Type Culture Collection (Manassas, VA; CRL 6322) and maintained in MAT/P medium supplemented with 100 units/ml penicillin, 100 mg/ml streptomycin and 2% fetal calf serum. B16 tumour cells were transfected to express IL‐2 as previously described.33,34 This paper used two clones of B16/IL‐2. The first, referred to as B16/IL‐2.19, secretes much lower levels of IL‐2 (< 10 pg/ml/48 hr), when compared with the second clone, B16/IL‐2.4 (9000 pg/ml/48 hr), when assayed as described below.
Analysis of IL‐2 and IFN‐γ protein levels
To determine the levels of IL‐2 produced by the transfected tumour cell lines, 2 × 105 cells were plated in 2 ml medium and supernatants were harvested after 48 hr. These supernatants were then assayed using the Luminex assay (Luminex, Austin, TX) or the CTLL‐2 assay according to the manufacturer's directions or as previously described.35 Intratumoral levels of IFN‐γ were examined by homogenizing tumour pieces in 500 μl lysis buffer as described.36 Samples were centrifuged at 4° for 8 min and supernatant was tested for total protein using a bicinchoninic acid kit (Pierce, Rockford, IL) and IFN‐γ by ELISA (PeproTech, Rocky Hill, NJ) per the manufacturer's protocol. All samples were normalized to total protein.
Tumour growth and whole mount analysis
Mice were injected with either 2 × 105 or 1 × 106 (indicated in figure legend) tumour cells intramuscularly into the left thigh and mean thigh diameter was measured as previously described.34 Mice were killed when the mean thigh diameter reached 10–13 mm. Tumours were analysed by whole mount histology as previously described.37 Briefly, tumour pieces were removed and stained using fluorescently conjugated antibodies anti‐CD31 (clone MEC13.3), anti‐vascular cell adhesion molecule 1 (anti‐VCAM‐1; clone 429), anti‐CD45 (clone 30‐F11), anti‐CD8 (clone 53‐6.7) and anti‐NK1.1 (clone PK136) (BD Pharmingen, San Jose, CA). All monochrome images were pseudo‐coloured and overlays were performed using ImagePro Plus software 5.0 (Media Cybernetics, Rockville, MD). Ten‐micrometre‐thick sections were obtained from paraffin‐embedded tissue consisting of normal muscle containing tumour foci and stained with haematoxylin & eosin before being examined by a pathologist at the Research Animal Diagnostic Laboratory (RADIL) at the University of Missouri.
Antibody depletion of immune cells
The NK cells in C57BL/6J mice were depleted by intraperitoneal antibody injection twice a week of 100 µg/mouse and 300 µg/mouse once weekly of rat anti‐mouse NK1.1 (clone PK136) or balanced salt solution control. In nude mice, NK and extrathymically derived T cells were depleted using a rat anti‐mouse IL‐2Rβ monoclonal antibody (TM‐β1) 16 on days 1 and 7 after tumour injection and then three times a week for the duration of the experiment.
Flow cytometry
Tumour samples were dissociated and single cell suspensions were stained as previously described.38 Cells were stained using the following antibodies: anti‐CD45 (clone 30‐F11; BD Pharmingen), anti‐NK1.1 (clone PK136; BD Pharmingen), anti‐CD4 (clone GK1.5; BD Pharmingen), anti‐CD8 (clone 53‐6.7; eBioscience, San Diego, CA), anti‐F4/80 (clone BMB; eBioscience), anti‐CD11b (clone M170; eBioscience), anti‐Gr‐1 (clone RB6‐8C5; BD Pharmingen), anti‐Foxp3 (clone FJK‐16S; eBioscience), anti‐CD25 (clone PC61; BD Pharmingen). Intracellular IFN‐γ staining was performed on single cell suspensions directly out of the tumour (with no antigen re‐stimulation). Cells were fixed, permeabilized and stained using anti‐IFN‐γ (Clone XMG1.2; BD Pharmingen) as described previously.38 Foxp3 staining was performed using the eBioscience anti‐mouse/rat Foxp3 Staining Set following the manufacturer's protocol. All samples were analysed using a FACSCanto II flow cytometer (BD Biosciences) and FlowJo Software (Tree Star Inc., Ashland, OR).
Quantitative real‐time PCR analysis
Tumours were excised and pieces were snap‐frozen in buffer RLT (QIAGEN, Valencia, CA). Samples were processed into homogenates and total RNA was isolated using an RNeasy fibrous tissue mini kit (QIAGEN) according to the manufacturer's protocol. Reverse transcription reaction was performed using iScript™ cDNA synthesis kit (Bio‐Rad, Hercules, CA) and the resulting cDNA was used for SYBR® Green‐based quantitative RT‐PCR analysis. Cycle thresholds were normalized to GAPDH RNA within each sample, before each value was expressed as fold increase over a single control sample that is used for all the samples. Comparing all controls and experimental samples to a single data‐point allows us to show the variation of mRNA levels among individual mice within each group and also highlights the relevant differences in mRNA between the B16/IL‐2.19 tumours and the parental B16 cell lines for given genes. The primers used were obtained from Eurofins MWG Operon, and their sequences are listed in the Supplementary material, Fig. S1. To specifically detect endogenously produced IL‐2 we used a primer that was in the 3′ untranslated region of the naturally produced host IL‐2 mRNA but was not present in the IL‐2 derived from the β‐actin expression vector. The specificity of this primer set was validated using cell lines and activated spleen cells.
Results
In vivo effects of local expression of IL‐2 on B16 cells
To examine the effects of local expression of IL‐2, we transfected the B16 tumour cell line with an IL‐2 expression plasmid, and isolated B16 clones that express IL‐2. One clone, B16/IL‐2.19, expressing a low amount of cytokine that is near the limit of detection of the assays (< 10 pg/ml assayed as described in the Materials and methods section) was used to examine the effects of local expression of IL‐2 on tumour growth and the host immune response. As expected, in mice injected with the parental B16 cells, tumours grew progressively and rapidly. However, tumours formed from the B16/IL‐2.19 cells expressing IL‐2, while still growing progressively, were significantly smaller than tumours from parental B16 cells (Fig. 1a). This was not a result of different intrinsic growth rates, as their in vitro growth rate was very similar (data not shown). Interestingly, when we examined the tumours using whole mount histology there was a dramatically increased infiltration of CD45‐positive immune cells in the IL‐2‐expressing tumours compared with parental B16 tumours (Fig. 1b,c).
Figure 1.
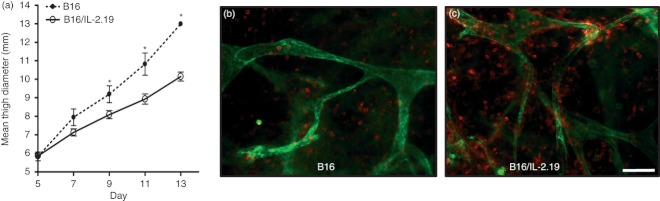
B16/IL‐2.19 tumours exhibit delayed tumour growth and increased infiltration of CD45+ immune cells. (a) 1 × 106 tumour cells were injected intramuscularly and monitored for growth over time [B16 (dotted lines) or B16/IL‐2.19 (solid lines)]. Bars indicate standard error of the mean (n = 6 for the B16/IL‐2.19 tumours, n = 3 for B16 tumours). A second independent experiment showed the same pattern of tumour growth. B16 (b) or B16/IL‐2.19 (c) were excised on day 13 following injection, stained simultaneously for blood vessels (anti‐CD31, green) and immune cells (anti‐CD45, red), and visualized by whole mount histology. *Significant difference (P < 0·05 by Student's t‐test). Scale bar indicates 100 μm.
To quantify the increase and further characterize the infiltrating cells, we performed flow cytometry using a panel of markers to delineate CD8, CD4 and regulatory T cells, NK cells, myeloid‐derived suppressor cells (Gr1+, CD11b+), and macrophages (Fig. 2a–f). The presence of IL‐2 significantly increased the density of effector CD8 T cells (Fig. 2a) and NK cells (Fig. 2b) as well as Treg cells (defined as Foxp3+ CD4+ T cells) as shown in (Fig. 2d). Interestingly, the presence of IL‐2 did not increase all regulatory cell populations, as IL‐2‐expressing tumours tended to have a lower density of myeloid‐derived suppressor cells (Fig. 2e). Additionally, IL‐2‐expressing tumours tended to contain more CD4 T cells (Fig. 2c) as well as macrophages (Fig. 2f), although these differences did not reach statistical significance. Further characterization of the Treg‐cell population (Fig. 2g), revealed differences in the expression of CD25 (Fig. 2h), the α chain of the IL‐2 receptor, which is often used to help delineate Treg cells. Interestingly, the expression of CD25 was lower on Treg cells in the B16 tumour (light coloured line) compared with the same population of cells in the B16/IL‐2.19 tumour (darker line) (average mean fluorescence intensity 850 compared with 298) (Fig. 2h). Additionally, we examined the functional activity of the CD8 T cells and NK cells isolated directly from the tumour by assessing their expression of IFN‐γ without re‐stimulation in vitro. We found there was an increased percentage of IFN‐γ‐producing CD8 T cells and NK cells within the IL‐2‐producing tumours, consistent with increased functional activity (Fig. 2i,j).
Figure 2.
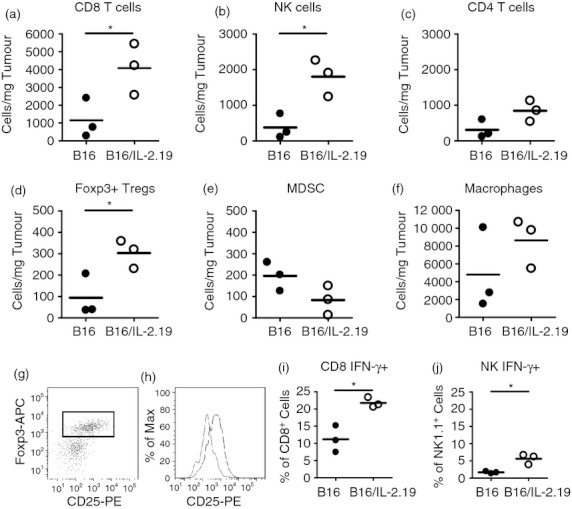
Local interleukin‐2 (IL‐2) increases the frequency of tumour infiltrating effector and regulatory cells. Flow cytometry analyses were performed on day 13 as described in the Materials and methods on either B16 or B16/IL‐2.19 tumours (as described in Fig. 1). The number of particular immune cell subsets is shown per mg of tumour to reflect the density of the cell type. Closed circles indicate B16 tumours, open circles indicate B16/IL‐2.19 and each symbol represents the results from an individual tumour. (a) CD8 T cells, (b) natural killer (NK) cells, (c) CD4 T cells, (d) Foxp3+ CD4 (Treg) cells, (e) myeloid‐derived suppressor cells (MDSC), (f) macrophages, (g) delineation of the CD4+, Foxp3+ population which was analysed for CD25 expression shown in (h) in which Treg cells from B16 are shown in grey and B16/IL‐2.19 are indicated in black. Representative histograms for each group are shown (three tumours of each type analysed). (i,j) Frequency of intracellular interferon‐γ (IFN‐γ) staining in (i) CD8 T cells and (j) NK cells. (Each symbol represents a single mouse). *Significant difference (P < 0·05 by Student's t‐test).
To further characterize the effect of local IL‐2 expression on the tumour microenvironment, we performed a series of quantitative real‐time PCR analyses (Fig. 3). As expected, the IL‐2‐expressing B16/IL‐2.19 tumours grown in vivo expressed higher levels of IL‐2 message compared with B16 tumours (Fig. 3a). Both T‐bet, a critical transcription factor for cell mediated responses, and Eomesodermin (Eomes), a T‐cell‐specific T‐box transcription factor important for CD8 T‐cell function,39 were elevated in the B16/IL‐2.19 tumours, consistent with the recruitment of more T cells into the tumour in the presence of IL‐2 (Fig. 3b,c). In addition, Granzyme B and Fas ligand, effector molecules required for CD8+ cytotoxic T lymphocytes to mediate tumour cell killing were up‐regulated (about 40‐fold and 58‐fold respectively), supporting the hypothesis that IL‐2 can enhance T‐cell responses (Fig. 3d,e). There was also an increase in the inflammatory cytokines tumour necrosis factor and IFN‐γ (approximately 40‐fold), as well as a corresponding increase in IFN‐inducible gene IP‐10 (Fig. 3f–h). However, IL‐12p35, IL‐5 and IL‐17 (Fig. 3i–k) were not substantially increased in B16/IL‐2.19 tumours, indicating that the observed increase in tumour necrosis factor and IFN‐γ transcript levels was not a generalized pattern for all cytokines. Furthermore, these data suggest that neither T helper type 17 nor classic T helper type 2 responses were induced. At the same time, Foxp3 and cytotoxic T lymphocyte antigen‐4 mRNA, transcripts most often associated with counter‐regulation of immune responses, were increased 5‐fold and 11‐fold, respectively (Fig. 3l,m). Increases of Foxp3 mRNA are consistent with the flow cytometry data that indicated an increased infiltration of Treg cells. Message for other inhibitory or anti‐inflammatory factors including programmed death‐1, and IL‐10 were also up‐regulated (Fig. 3n,o). Nevertheless, despite the increase of the Treg‐cell population, as well as other inhibitory factors, the IL‐2‐expressing tumours grew more slowly than the parental B16 tumours (Fig. 1a). Interestingly, using primers that only amplified endogenously derived IL‐2, production of IL‐2 by the tumour also increased the expression of IL‐2 by host cells (Fig. 3p).
Figure 3.
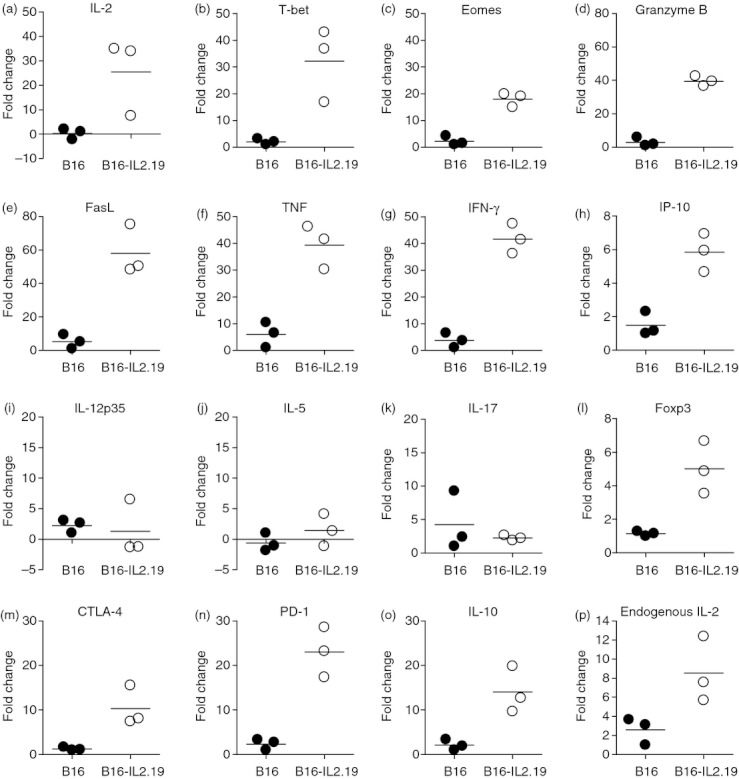
Effects of local expression of interleukin‐2 (IL‐2) on the tumour microenvironment examined by real‐time PCR. Quantitative real‐time PCR was performed for the indicated targets as described in the Materials and methods section on tumours described in Fig. 1. Data are expressed as fold increases relative to the mRNA levels in one selected sample within the B16 tumour group. This sample was used for all the fold induction calculations allowing the range of values for the parental B16 as well as the B16/IL‐2.19 to be seen. Closed circles indicate B16 tumour homogenates, open circles represent B16/IL‐2.19 tumour homogenates. Bars indicate the average of three tumours.
Vascular VCAM‐1 expression and infiltration of CD8 T cells and NK cells are increased in IL‐2‐expressing tumours in an IFN‐γ‐dependent manner
Flow cytometry and real‐time PCR analyses indicated that CD8 T cells and NK cells were increased in the IL‐2‐expressing B16 tumours. One of the key effector cytokines produced by these cells is IFN‐γ and the level of mRNA encoding IFN‐γ was highly up‐regulated in the IL‐2‐expressing tumours. The level of IFN‐γ protein was also significantly elevated in the IL‐2‐expressing tumour microenvironment (Fig. 4a). As IFN‐γ can increase the expression of VCAM‐1, an adhesion molecule involved in lymphocyte trafficking, we also examined the expression of VCAM‐1 on B16 and B16/IL‐2.19 tumour vessels. The expression of VCAM‐1 is induced in the IL‐2‐expressing B16/IL‐2.19 tumours. Further, this expression is dependent upon IFN‐γ, because this induction is lost in tumours grown in the IFN‐γ knockout mice (Fig. 4b). Interestingly, the increased number of CD8 T cells and NK cells appeared largely dependent on IFN‐γ. As shown in Figure 4(c,d), the IL‐2‐expressing tumours show an increased frequency of CD8 T cells and NK cells compared with parental B16 tumours, however these cell populations are decreased in IL‐2‐expressing tumours grown in IFN‐γ knockout mice.
Figure 4.
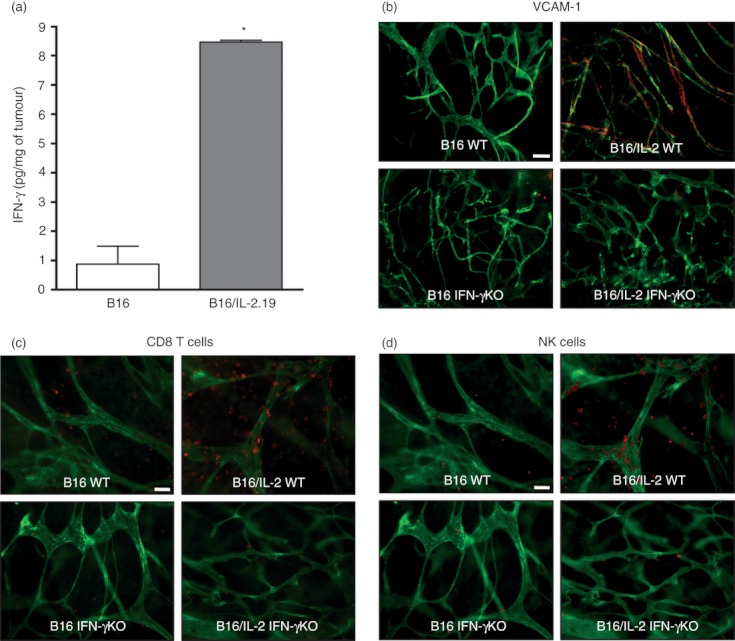
Local expression of interleukin‐2 (IL‐2) results in the induction of vascular cell adhesion molecule 1 (VCAM‐1) on vessel endothelial cells and increased infiltration of CD8 T cells and natural killer (NK) cells in an IFN‐γ‐dependent manner. (a) Levels of interferon‐γ (IFN‐γ) protein as determined by ELISA in homogenates from B16 and B16/IL‐2.19 tumours (as described in Fig. 1). The amount of IFN‐γ is expressed per mg of total protein in tumour homogenates (n = 3 tumours). *Significance determined by Student's t‐test (P < 0·05). (b–d) Whole mount analyses of either B16 or B16/IL‐2.19 grown in C57BL/6J or IFN‐γ−/− mice. (b) Tumour samples were stained simultaneously with anti‐CD31 to visualize blood vessels (green) and anti‐VCAM‐1 (red). CD31‐labelled vessels that express VCAM‐1 appear yellow. White measurement bar indicates 100 μm. (c, d) Tumour samples were stained simultaneously with anti‐CD8‐phycoerythrin (pseudocoloured red in c), anti‐NK1.1‐allophycocyanin (pseudocoloured red in d), anti‐CD31‐FITC (pseudocoloured green in c, d) and examined by whole mount histology. Collecting images from three separate fluorescent channels allowed for the visualization of both CD8+ and NK1.1+ cells in the same field of view. White measurement bar indicates 50 μm.
Higher local levels of IL‐2 can result in long‐term tumour control
The data above suggested that a very low level of IL‐2 could induce an increase in T cells and NK cells and a concomitant reduction in tumour growth. We hypothesized that a higher local level of IL‐2 might have more dramatic effects on tumour growth and the host immune response. We therefore examined the effect of a higher level of local IL‐2 using the IL‐2‐transfected line B16/IL‐2.4, which expresses approximately 9000 pg/ml assayed as described in the Materials and methods section. The parental B16 and the B16/IL‐2.4 cell lines grew similarly in vitro (data not shown). In Figure 5, we compared the growth of B16/IL‐2.4 with parental B16 controls in vivo. As expected, the B16 tumours grew rapidly and progressively and mice were killed at about 2 weeks post injection (Fig. 5a). In contrast, the expression of IL‐2 abrogated the growth of B16/IL‐2.4 tumours (Fig. 5b). Hence, local expression of high levels of IL‐2 by the B16/IL‐2.4 tumour cells dramatically decreased their ability to grow in vivo. To investigate which cells were required for the inhibition of tumour growth, we performed a series of experiments examining the ability of the IL‐2‐transfected tumours to grow in mice in which particular immune cell subsets were absent as a result of antibody depletion or mutation. We depleted C57BL/6J mice of NK cells using anti‐NK1.1 and examined the growth of the B16 and B16/IL‐2.4 tumours (Fig. 5c,d) and determined that the growth of the B16/IL‐2.4 tumours remained inhibited, indicating that NK cells were not absolutely essential for controlling tumour growth. To determine whether T cells were essential, we inoculated nude mice that are deficient in T cells but retain NK cells, with B16 or B16/IL‐2.4 cells (Fig. 5e,f). In these mice, the B16/IL‐2.4 tumour growth was delayed, although tumours eventually grew out sporadically. These data suggest that T cells, although not absolutely required to delay tumour growth, play an important role in controlling tumour progression in wild‐type mice. Further, we examined tumour growth in nude mice treated with anti‐IL‐2Rβ antibody to remove NK cells and any residual T cells that developed extra‐thymically (Fig. 5g,h). As expected, the B16 tumours grew rapidly and progressively. Importantly, in a majority of the mice, the B16/IL‐2.4 tumours also grew progressively in a fashion very similar to parental B16 tumours. Because NK cells and T cells are major producers of IFN‐γ, a key cytokine that is important in many of their effector functions, we examined the growth of the B16/IL‐2.4 tumours in IFN‐γ knockout mice. As can be seen in Fig. 5(i), parental B16 tumours grew progressively in the IFN‐γ knockout mice as expected; however, B16/IL‐2.4 tumours demonstrated an initial period of growth followed by regression (Fig. 5j). Interestingly, whereas B16/IL‐2.4 tumour growth in wild‐type mice remained inhibited (Fig. 5b), several tumours eventually grew out in IFN‐γ knockout mice (Fig. 5j). Collectively, these data suggest that T cells, and to a lesser extent NK cells, mediate the inhibition of tumour growth and that IFN‐γ can play an important role in the maintenance of tumour control.
Figure 5.
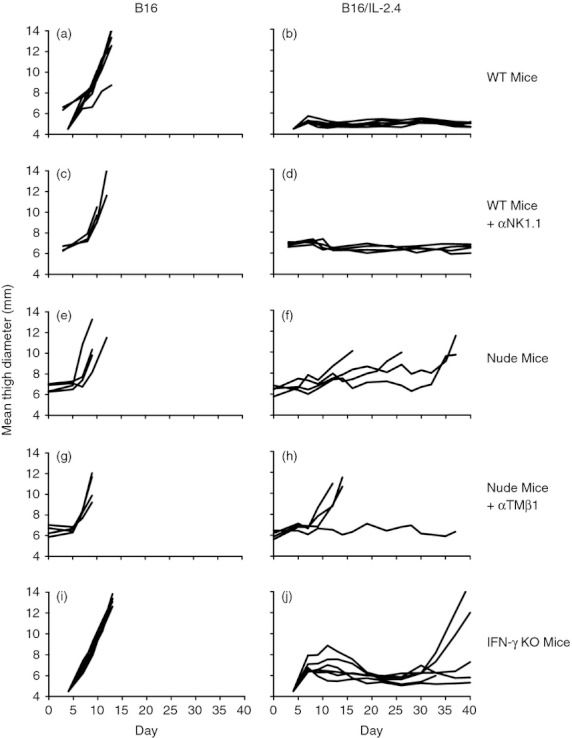
Control of tumour growth of B16 and B16/IL‐2.4 cells in mice lacking specific immune cells or interferon‐γ (IFN‐γ). Growth of B16 tumours or B16/IL‐2.4 tumours in the indicated mice. (a, b) C57BL/6J mice, (c,d) C57BL/6J mice treated with anti‐NK1.1, (e,f) nude mice, (g,h) nude mice treated with anti‐TMβ1 (an antibody to the IL‐2 receptor β chain) to deplete natural killer (NK) cells, and (i,j) IFN‐γ−/− mice. Mice were injected with 2 × 105 B16 or B16/IL‐2.4 cells. Lines indicate the growth of tumours in individual mice. Note that the end of the line indicates that the mice were killed or had died during the course of the experiment.
Initial host response to B16/IL‐2.4 tumours and establishment of tumour dormancy
To gain further insight as to the mechanism of tumour growth inhibition, we examined the B16 or B16/IL‐2.4 tumour site at very early time‐points after tumour inoculation (days 4, 6 and 8) and analysed the tumour immune infiltrate by flow cytometry. These early time‐points were chosen because this cell line did not grow progressively in wild‐type B6 mice, in marked contrast to the B16/IL‐2.19 cell line used previously in Fig. 1–4 that expressed lower levels of IL‐2. As can be seen in Fig. 6, we found that even though both B16 and B16/IL‐2.4 tumours were infiltrated by immune cells, the IL‐2‐expressing tumours had a greater density of NK cells compared with the B16 tumours, suggesting an initial rapid host response to IL‐2‐expressing tumours. However, one of the unexpected findings in the case of B16/IL‐2.4 tumours was that tumours would grow out after an extended period of time as seen in the T‐cell‐deficient mice (Fig. 5f) and in a few cases in the wild‐type mice (data not shown). This infrequent outgrowth of tumours suggested a mechanism in which the tumours were controlled but not eliminated. We therefore examined the animals that had been inoculated with the B16/IL‐2.4 tumour after 14 or 28 days even though no tumour was detectable by external measurement (Fig. 5b). Interestingly, we found that in mice with no palpable or clearly measurable tumour, melanated tumour cells were still clearly present by gross examination at day 14 (Fig. 7a). At this time‐point, mice inoculated with the same number of parental B16 tumour cells would often need to be killed because of progressive tumour growth. The B16/IL‐2.4 tumours were also analysed by whole mount histology on day 28, where melanated tumour cells (black) as well as infiltrated CD45‐positive cells (red) were clearly visible (Fig. 7b). Further, despite being relatively near many vessels, these tumour cells did not appear to induce new vasculature at this time‐point, because they were largely devoid of new vessels, which exhibit sprouting (Fig. 7c). Even after 100 days, there was evidence of a small number of pigmented cells seen by conventional haematoxylin & eosin histology (Fig. 7d). Slides from these samples were read by a pathologist at the Research Animal Diagnostic Laboratory (RADIL – University of Missouri) who reported that the histopathology was consistent with an experimentally induced malignant melanoma. To examine whether these cells were truly viable, at varying times after tumour inoculation we surgically excised tissues containing the tumour foci from mice implanted with the B16/IL‐2.4 cells. These excised tissue fragments were digested with collagenase and then plated and cultured in vitro. From these explants, we were able to isolate viable clonogenic cells in one out of two animals from explants isolated at day 24 and from two out of three animals taken at day 42. Interestingly, we found that the cells from the explants expressed IL‐2 in all cases (data not shown). These data are consistent with the concept that the tumours are not completely eliminated but rather are being controlled by the immune effectors or their products and that a form of tumour dormancy is established.
Figure 6.
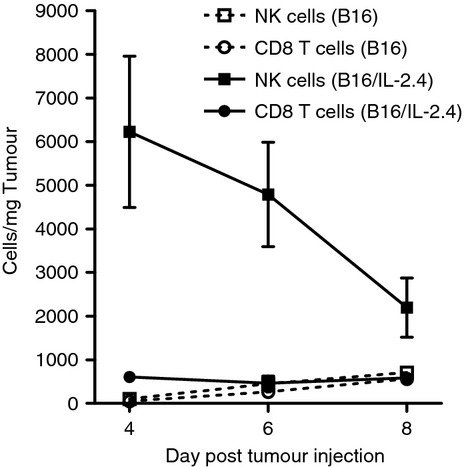
B16/IL‐2.4 induces a rapid natural killer (NK) cell infiltrate. Mice were injected with 1 × 106 B16 or B16/IL‐2.4 cells. The tumours were processed into single cell suspensions and analysed by flow cytometry at the indicated days. The frequency of T cells or NK cells was determined and normalized to the weight of the tumour to correct for differences in tumour size (n = 4 tumours examined in each condition). Squares indicate NK cells, circles indicate CD8 T cells. Dotted lines indicate cells from B16 tumours, solid lines those from B16/IL‐2.4 tumours.
Figure 7.
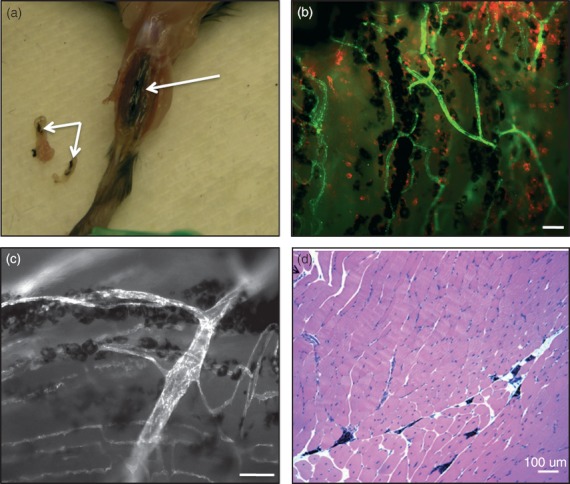
Expression of interleukin‐2 (IL‐2) can result in long‐term tumour control and dormancy. 1 × 106 B16/IL‐2.4 tumour cells were injected and the injection site was analysed at various time‐points. (a) Fourteen days following injection, B16/IL‐2.4 cells can still be seen at the tumour site as well as portions of tissue that were excised (arrows indicated melanin‐expressing tumour cells). (b) The injection site was excised 28 days after tumour injection and stained simultaneously for blood vessels using anti‐CD31 (green) and immune cells (anti‐CD45, red), and examined by whole mount histology. White measurement bar indicates 100 μm. (c) Higher magnification of B16/IL‐2.4 tumours at day 28 showing the tumours growing along side vessels. White measurement bar indicates 50 μm. (d) Haematoxylin & eosin staining of a paraffin section of the thigh muscle 100 days following injection of B16/IL‐2.4. Melanin‐expressing tumour cells (black) can be seen between muscle fibres. White measurement bar indicates 100 μm.
We further investigated whether dormant tumours engendered a systemic immune response. Dormant B16/IL‐2.4 tumours were established as in previous experiments (Fig. 8a). At day 21 these mice were challenged with parental B16 tumours in the opposite leg. Naive mice were inoculated with the same dose of parental B16 for comparison and the original dormant tumours as well as tumours arising from the challenge were measured over time (Fig. 8a,b). In eight out of nine of the mice with dormant tumours, the original tumours remained dormant for the duration of the experiment (Fig. 8a). The one dormant tumour that grew out had lost expression of IL‐2 (data not shown). As expected, naive mice challenged with B16 cells rapidly grew tumours (Fig. 8b). In the mice with dormant tumours, there was a statistically significant delay of approximately 2 days in the growth of the B16 challenge tumours compared with naive animals (Fig. 8c). Interestingly, in one of the mice with a dormant tumour, the challenge tumour did not grow during this time period, although it did grow out after an extended period of time (30 days after challenge). These data indicate that a systemic immune response was present, although in most cases it did not provide complete protection from challenge with parental B16.
Figure 8.
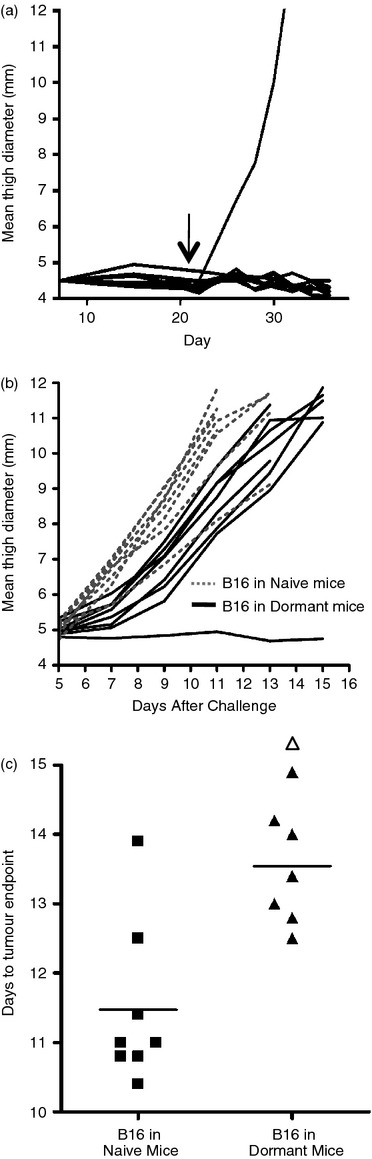
Challenge of mice with an established but dormant tumour. (a) To establish dormant tumours, mice were inoculated with 1 × 106 B16/IL2.4 tumours and growth was followed over time. The arrow indicates the time of challenge with parental B16 in the opposite leg. The one dormant tumour that grew out had lost expression of interleukin‐2 (IL‐2) (data not shown). (b) The animals with dormant tumours depicted in (a), were inoculated with 2 × 105 parental B16 cells in the opposite leg at day 21 and the tumour growth was measured. As controls, naive animals were also challenged with the same dose of parental B16 cells. Dotted lines indicate growth of challenge tumours in naive mice and solid lines indicate growth of tumours in mice harbouring dormant tumours. (c) For ease of comparison, the time required for the parental B16 tumour challenge to reach a tumour endpoint (a defined size of 11‐mm leg diameter) for naive mice or for mice with dormant tumours is indicated. Symbols represent individual mice; filled squares represent tumours in naive mice, triangles represent growth of tumours in mice with dormant tumours. The open triangle represents the tumour that did not grow out during the course of the experiment shown in Fig. 8(b) although it eventually grew out 30 days after challenge and is plotted at the maximum day. The groups are significantly different from one another P = 0·0019 using the Mann–Whitney U‐test.
Discussion
IL‐2 has striking effects on number and types of immune cells in the tumour
The presence of even extremely low levels of IL‐2 in the context of viable B16 tumours results in increased numbers of T cells and NK cells in the tumour (Fig. 2 and 4). Staining of these cells by flow cytometry also revealed a greater number of IFN‐γ‐producing cells (Fig. 2). The IFN‐γ‐producing‐cells would be expected to exert anti‐tumour effects in vivo. This increase in cellular effectors could result from increased infiltration of cells into the tumour as well as proliferation of effectors within the tumour microenvironment. The increased expression of chemokines such as IP‐10 (Fig. 3) in the IL‐2‐expressing tumours and the enhanced expression of VCAM‐1 on vessels (Fig. 4) would be expected to aid in the infiltration of T cells and NK cells that express very late antigen‐4, the receptor for VCAM‐1.38 Further, IL‐2 can not only enhance the proliferation and differentiation of NK and T cells22,23 but in naive mice even in the absence of antigen it can expand CD8 memory phentoype T cells in vivo that can provide protection against Listeria or vaccinia virus in infection models,40 providing a potential mechanism for the anti‐tumour effects observed here.
Finding that the presence of IL‐2 at the tumour site can have dramatic effects on B16 tumour growth is noteworthy. Early reports indicated that IL‐2 can have beneficial anti‐tumour effects in the B16 model,41 but it is thought that this aggressive tumour is relatively resistant to systemic IL‐2 treatment.32 The levels of intratumoral IL‐2 appear to play an important role, as lower levels of IL‐2 resulted in slower tumour growth whereas higher levels resulted in long‐term tumour control. The current study highlights the potential differences in local expression versus systemic delivery. Importantly, as seen in Fig. 2 and 3, these studies reveal that altering the level of one key cytokine locally at the tumour site can have profound effects on the cellular infiltration that, in turn, can alter the cytokine milieu within the tumour microenvironment.
Treg cells are enhanced in IL‐2‐expressing tumours
Interleukin‐2 is a particularly interesting cytokine because it has been shown to exert opposing effects upon the immune system. It can enhance the proliferation and differentiation of T cells and NK cells yet at the same time is required for Treg‐cell development and survival.5,22,23 In the experiments presented here, we found that, as in other models, the B16 tumours are infiltrated by Treg cells.8,42,43,44 An increased number of Treg cells are found in the IL‐2‐expressing tumours but conventional CD4 T cells, as well as CD8 T cells and NK cells, are also increased (Fig. 2). Interestingly, immunotherapy with IL‐2 in patients can also augment Treg cells, illustrating the complexity of IL‐2 action in vivo.45,46 It is possible that this reflects the action of IL‐2 directly on Treg cells, but may also reflect the result of a normal homeostatic counter‐regulatory response of the immune system that has evolved to control vigorous immune responses.5,6,45
In the data presented here, the Foxp3+ cells in the parental B16 tumours exhibit a relatively low level of CD25, a marker often used to help delineate Treg cells, whereas this expression is higher in the B16/IL‐2.19 tumours (Fig. 2). This may reflect a limiting amount of IL‐2 in the B16 tumour and might suggest that the Treg cells expressing the higher levels of CD25 in the B16/IL‐2.19 tumours might be more functional. Nevertheless, in contrast to a previously reported autoimmune model of type I diabetes30 in which additional IL‐2 dampened the T‐cell response and enhanced CD25 expression, the presence of IL‐2 in the B16/IL‐2.19 tumours does not result in dominance of the Treg‐cell activity. This may reflect differences in the levels of IL‐2 at the actual tissue site as well as the levels of IL‐2 required in various physiological contexts.47
Despite the presence of Treg cells, the B16/IL‐2.19 tumours exhibit slower growth compared with parental B16 tumours. How can one explain this apparently paradoxical finding? One possibility is that it is the relative ratio of NK cells or effector CD4 or CD8 cells to Treg cells that is critical. Experimental manipulations that alter this ratio by enhancing T effector cells or inhibiting Treg cells can result in beneficial anti‐tumour responses.4,6,7,44,48,49 Interestingly, an extensive review correlating clinical responses in a diverse panel of human cancers highlighted the importance of CD8 T cells and T helper type 1 responses whereas the correlation with Treg cells was much more variable and complex.3 In this light, finding that IL‐10 was up‐regulated in the B16/IL‐2.19 tumours would be consistent with an increase in IL‐10‐producing Treg cells and increased suppression. However, recent work has suggested that IL‐10, while often immunosuppressive, could also have beneficial immunologically relevant anti‐viral and anti‐tumour effects.50,51 Interestingly, it has also been suggested that at different times during an immune response, effectors might be differentially susceptible to the influence of Treg cells, and not as sensitive to inhibition when inflammatory signals are present or when antigen or cytokines such as IL‐2 that act through the common γ chain are plentiful.52 Finally, the levels of IL‐2 may be sufficient to overcome the suppressive effects of the Treg cells. In several in vitro systems used to measure Treg‐cell function, many, although not all, of the effects of Treg cells can be overcome by the addition of exogenous IL‐2 to the cultures.5 The IL‐2‐expressing tumours might represent an analogous in vivo situation.
Tumour control but not total elimination of IL‐2‐expressing tumours
Our experiments using the B16/IL‐2.4 tumour cell line showed a profound growth inhibition and did not result in measurable tumour growth. However, occasionally, a tumour would begin to grow out after an extended period of time (such as seen in Fig. 8a). This was striking because the B16 melanoma line is an aggressive tumour, and in typical experiments with the B16 model, the recipient mice would need to be killed after 12–15 days because of progressive tumour growth (Fig. 1 and 5). Further, in the case of B16/IL‐2.4, even when there was no visible tumour by external measurements, tumour was apparent when the inoculation site was opened and examined grossly and by whole mount histology (Fig. 7) at day 28. Importantly, it was possible to recover viable tumour cells capable of growing in culture even 42 days after tumour inoculation. Strikingly, tumour cells were still detectable after 100 days as detected by haematoxylin & eosin staining (Fig. 7). Data from immune‐deficient mice or mice depleted of immune cells using antibodies suggested that tumour control could be mediated either by T cells or to a lesser extent by NK cells (Fig. 5). Notably, these data also revealed that the tumours, although not totally eliminated, could be controlled, and this was dependent upon immune cells and IFN‐γ. How this tumour dormancy is established and maintained remains to be determined. The dormancy could be maintained by the lymphocytes themselves through continuously eliminating tumour cells and so controlling tumour growth, or more indirectly, by factors such as the IFN‐γ that the immune cells produce. Several lines of evidence presented here, including studies with immune‐deficient or immune‐cell‐depleted mice, strongly indicate that the immune system and cytokines such as IFN‐γ, are important in maintaining dormancy. Interestingly, although we see evidence of a systemic anti‐tumour immune response in challenge experiments in mice with ‘dormant’ tumours (Fig. 8), it is not fully protective, and this finding may help explain why the tumour is not completely eliminated at the dormant site. Taken together, these experiments suggest that the local production of IL‐2 probably plays an important role in establishing or maintaining dormancy in addition to direct cytotoxic effects via immune cells on the tumour cells themselves. For example, IFN‐γ can limit vessel growth through the induction of anti‐angiogenic factors such as IP‐10. Interestingly, a recent report in which the antigens on vasculature were targeted directly by cellular immune responses also resulted in tumour dormancy that required CD8 T cells.53 Interleukin‐2 could also have direct effects on vessels because it was recently reported that pulmonary vessels express the IL‐2 receptor.54 The apparent dormancy of the B16/IL‐2.4 tumour is also reminiscent of some human tumours,55,56 although whether the same processes underlie this phenomenon is an important, and as yet, unanswered question.
Local expression of IL‐2 in B16 tumours alters the tumour microenvironment and enhances tumour effectors
Paracrine delivery of cytokines as a means of enhancing the initiation of immunity to tumours has been extensively studied.57 In contrast, here we focused on the effects of local IL‐2 expression on the immune effectors within the tumour microenvironment of viable tumours. Surprisingly, given that B16 cells grown in vivo are relatively insensitive to systemic treatment with IL‐232,58 the presence of IL‐2 in the tumour microenvironment affected tumour growth and the anti‐tumour immune response. Even the extremely low levels of IL‐2 produced by the B16/IL‐2.19 clone dramatically changed the composition of the infiltrating cells, the cytokines produced, and slowed tumour growth. Unexpectedly, the higher levels of IL‐2 produced by the B16/IL‐2.4 cell line led to extended tumour control but not complete tumour elimination in a process dependent upon immune cells and mediated in part by IFN‐γ. In vivo, expression of IL‐2 may drive differentiation toward T effector cells rather than toward the development of memory T cells24 which would result in strong local effects but more limited systemic effects. Interestingly, even in systemic treatment of patients, it has been speculated that the success of IL‐2 treatment is a result of IL‐2‐mediated changes at the tumour site.59 To maintain the efficacy of IL‐2 while minimizing systemic side effects, strategies are now being explored to increase IL‐2 expression in tumours in ways that could be applied to disseminated tumour sites. These approaches include coupling a cytokine such as IL‐2 to a tumour‐reactive antibody60–64 or by developing an IL‐2 fusion protein that could be activated by proteases expressed preferentially in the tumour microenvironment.35 By further dissecting the in vivo mechanism of action of IL‐2 and other immunomodulators, it should also be possible to combine immunotherapy with new molecularly targeted therapies as well as conventional therapies, such as radiation and chemotherapy, in clinically significant ways.1,2,65
Acknowledgments
This work was supported by National Institutes of Health Grant CA28332. E.W.S., A.L.S. and D.S. were supported in part by the National Institutes of Health Training Grant AI07285. The authors thank Mr Ryan Cummings who provided helpful comments on the manuscript.
Glossary
- Eomes
eomesodermin
- FoxP3
forkhead box P3
- IFN‐γ
interferon‐γ
- IL‐2
interleukin 2
- IP‐10
interferon‐inducible protein 10
- NK
natural killer
- Treg cells
regulatory T cells
- VCAM‐1
vascular cell adhesion molecule‐1
Disclosure
The authors declare no competing financial interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Primers.
References
- 1.Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 2.Weiner LM. Cancer immunotherapy – the endgame begins. N Engl J Med. 2008;358:2664–5. doi: 10.1056/NEJMp0803663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fridman WH, Pages F, Sautes‐Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 4.Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. 2011;71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shevach EM. Mechanisms of foxp3+ T regulatory cell‐mediated suppression. Immunity. 2009;30:636–45. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Rowswell‐Turner RB, Harden JL, Nair RE, Gu T, Kilinc MO, Egilmez NK. Chronic chemoimmunotherapy achieves cure of spontaneous murine mammary tumors via persistent blockade of posttherapy counter‐regulation. J Immunol. 2011;187:4109–18. doi: 10.4049/jimmunol.1101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baecher‐Allan C, Anderson DE. Immune regulation in tumor‐bearing hosts. Curr Opin Immunol. 2006;18:214–9. doi: 10.1016/j.coi.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104–18. doi: 10.1111/j.1600-065X.2011.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 11.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnem T, Al‐Shibli K, Andersen S, Al‐Saad S, Busund LT, Bremnes RM. Combination of low vascular endothelial growth factor A (VEGF‐A)/VEGF receptor 2 expression, high lymphocyte infiltration is a strong and independent favorable prognostic factor in patients with non‐small cell lung cancer. Cancer. 2010;116:4318–25. doi: 10.1002/cncr.25333. [DOI] [PubMed] [Google Scholar]
- 13.Dings RP, Vang KB, Castermans K. Enhancement of T‐cell‐mediated antitumor response: angiostatic adjuvant to immunotherapy against cancer. Clin Cancer Res. 2011;17:3134–45. doi: 10.1158/1078-0432.CCR-10-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher DT, Chen Q, Skitzki JJ. IL‐6 trans‐signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. 2011;121:3846–59. doi: 10.1172/JCI44952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen EW, Gerber SA, Frelinger JG, Lord EM. Suppresses vascular endothelial growth factor receptor 3 expression on tumor vessels by two distinct IFN‐γ‐dependent mechanisms. J Immunol. 2010;184:1858–66. doi: 10.4049/jimmunol.0903210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Yang JC, Topalian SL. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high‐dose bolus interleukin 2. JAMA. 1994;271:907–13. [PubMed] [Google Scholar]
- 18.Donskov F. Interleukin‐2 based immunotherapy in patients with metastatic renal cell carcinoma. Dan Med Bull. 2007;54:249–65. [PubMed] [Google Scholar]
- 19.Reang P, Gupta M, Kohli K. Biological response modifiers in cancer. MedGenMed. 2006;8:33. [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 21.Den Otter W, Jacobs JJ, Battermann JJ. Local therapy of cancer with free IL‐2. Cancer Immunol Immunother. 2008;57:931–50. doi: 10.1007/s00262-008-0455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malek TR. The biology of interleukin‐2. Annu Rev Immunol. 2008;26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 23.Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. 2007;8:1142–8. doi: 10.1038/sj.embor.7401099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang N, Bevan MJ. CD8+ T cells: foot soldiers of the immune system. Immunity. 2011;35:161–8. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyman O, Sprent J. The role of interleukin‐2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12:180–90. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 26.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–90. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 27.Boyman O, Purton JF, Surh CD, Sprent J. Cytokines and T‐cell homeostasis. Curr Opin Immunol. 2007;19:320–6. doi: 10.1016/j.coi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Waldmann TA. Effective cancer therapy through immunomodulation. Annu Rev Med. 2006;57:65–81. doi: 10.1146/annurev.med.56.082103.104549. [DOI] [PubMed] [Google Scholar]
- 29.Overwijk WW, Schluns KS. Functions of γC cytokines in immune homeostasis: current and potential clinical applications. Clin Immunol. 2009;132:153–65. doi: 10.1016/j.clim.2009.03.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang Q, Adams JY, Penaranda C. Central role of defective interleukin‐2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–97. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koreth J, Matsuoka K, Kim HT. Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med. 2011;365:2055–66. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im2001s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAdam AJ, Pulaski BA, Harkins SS, Hutter EK, Lord EM, Frelinger JG. Synergistic effects of co‐expression of the TH1 cytokines IL‐2 and IFN‐ γ on generation of murine tumor‐reactive cytotoxic cells. Int J Cancer. 1995;61:628–34. doi: 10.1002/ijc.2910610508. [DOI] [PubMed] [Google Scholar]
- 34.McAdam AJ, Pulaski BA, Storozynsky E, Yeh KY, Sickel JZ, Frelinger JG, Lord EM. Analysis. of the effect of cytokines (interleukins 2, 3, 4 and 6, granulocyte–monocyte colony‐stimulating factor and interferon‐ γ) on generation of primary cytotoxic T lymphocytes against a weakly immunogenic tumor. Cell Immunol. 1995;165:183–92. doi: 10.1006/cimm.1995.1204. [DOI] [PubMed] [Google Scholar]
- 35.Puskas J, Skrombolas D, Sedlacek A, Lord E, Sullivan M, Frelinger J. Development of an attenuated interleukin‐2 fusion protein that can be activated by tumour‐expressed proteases. Immunology. 2011;133:206–20. doi: 10.1111/j.1365-2567.2011.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerber SA, Pober JS. IFN‐α induces transcription of hypoxia‐inducible factor‐1α to inhibit proliferation of human endothelial cells. J Immunol. 2008;181:1052–62. doi: 10.4049/jimmunol.181.2.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber SA, Rybalko VY, Bigelow CE, Lugade AA, Foster TH, Frelinger JG, Lord EM. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am J Pathol. 2006;169:1739–52. doi: 10.2353/ajpath.2006.051222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation‐induced IFN‐γ production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180:3132–9. doi: 10.4049/jimmunol.180.5.3132. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Y, Ju S, Chen E. T‐bet and eomesodermin are required for T cell‐mediated antitumor immune responses. J Immunol. 2010;185:3174–83. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton SE, Schenkel JM, Akue AD, Jameson SC. IL‐2 complex treatment can protect naive mice from bacterial and viral infection. J Immunol. 2010;185:6584–90. doi: 10.4049/jimmunol.1001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenberg SA, Mule JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high‐dose recombinant interleukin 2. J Exp Med. 1985;161:1169–88. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kline J, Brown IE, Zha YY. Homeostatic proliferation plus regulatory T‐cell depletion promotes potent rejection of B16 melanoma. Clin Cancer Res. 2008;14:3156–67. doi: 10.1158/1078-0432.CCR-07-4696. [DOI] [PubMed] [Google Scholar]
- 43.Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–6. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM‐CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–45. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartzentruber DJ, Lawson DH, Richards JM. gp100 peptide vaccine and interleukin‐2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Vliet HJ, Koon HB, Yue SC. Effects of the administration of high‐dose interleukin‐2 on immunoregulatory cell subsets in patients with advanced melanoma and renal cell cancer. Clin Cancer Res. 2007;13:2100–8. doi: 10.1158/1078-0432.CCR-06-1662. [DOI] [PubMed] [Google Scholar]
- 47.Hofer T, Krichevsky O, Altan‐Bonnet G. Competition for IL‐2 between regulatory and effector T cells to chisel immune responses. Front Immunol. 2012;3:268. doi: 10.3389/fimmu.2012.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL‐10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol. 2011;41:3615–26. doi: 10.1002/eji.201141700. [DOI] [PubMed] [Google Scholar]
- 49.Colombo MP, Piconese S. Regulatory‐T‐cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7:880–7. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 50.Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL‐10 at the peak of coronavirus‐induced encephalitis. J Immunol. 2011;186:3642–52. doi: 10.4049/jimmunol.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mumm JB, Emmerich J, Zhang X. IL‐10 elicits IFNγ‐dependent tumor immune surveillance. Cancer Cell. 2011;20:781–96. doi: 10.1016/j.ccr.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Walker LS. Regulatory T cells overturned: the effectors fight back. Immunology. 2009;126:466–74. doi: 10.1111/j.1365-2567.2009.03053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X, Bose A, Komita H. Vaccines targeting tumor blood vessel antigens promote CD8+ T cell‐dependent tumor eradication or dormancy in HLA‐A2 transgenic mice. J Immunol. 2012;188:1782–8. doi: 10.4049/jimmunol.1101644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL‐2 immunotherapy by selective stimulation of IL‐2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107:11906–11. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brackstone M, Townson JL, Chambers AF. Tumour dormancy in breast cancer: an update. Breast Cancer Res. 2007;9:208. doi: 10.1186/bcr1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ossowski L, Aguirre‐Ghiso JA. Dormancy of metastatic melanoma. Pigment Cell Melanoma Res. 2010;23:41–56. doi: 10.1111/j.1755-148X.2009.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pardoll DM. Paracrine cytokine adjuvants in cancer immunotherapy. Annu Rev Immunol. 1995;13:399–415. doi: 10.1146/annurev.iy.13.040195.002151. [DOI] [PubMed] [Google Scholar]
- 58.Levin AM, Bates DL, Ring AM. Exploiting a natural conformational switch to engineer an interleukin‐2 ‘superkine’. Nature. 2012;484:529–33. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Panelli MC, White R, Foster M, Martin B, Wang E, Smith K, Marincola FM. Forecasting the cytokine storm following systemic interleukin (IL)‐2 administration. J Transl Med. 2004;2:17. doi: 10.1186/1479-5876-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang G, Li W, Holle L, Chen N, Chen WY. A novel design of targeted endocrine, cytokine therapy for breast cancer. Clin Cancer Res. 2002;8:1196–205. [PubMed] [Google Scholar]
- 61.Tomblyn S, Springs AE, Langenheim JF, Chen WY. Combination therapy using three novel prolactin receptor antagonist‐based fusion proteins effectively inhibits tumor recurrence and metastasis in HER2/neu transgenic mice. Int J Oncol. 2009;34:1139–46. doi: 10.3892/ijo_00000242. [DOI] [PubMed] [Google Scholar]
- 62.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–59. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 63.Hirsch B, Brauer J, Fischdick M, Loddenkemper C, Bulfone‐Paus S, Stein H, Durkop H. Anti‐CD30 human IL‐2 fusion proteins display strong and specific cytotoxicity in vivo. Curr Drug Targets. 2009;10:110–7. doi: 10.2174/138945009787354566. [DOI] [PubMed] [Google Scholar]
- 64.Marlind J, Kaspar M, Trachsel E. Antibody‐mediated delivery of interleukin‐2 to the stroma of breast cancer strongly enhances the potency of chemotherapy. Clin Cancer Res. 2008;14:6515–24. doi: 10.1158/1078-0432.CCR-07-5041. [DOI] [PubMed] [Google Scholar]
- 65.Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011;364:985–7. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers.