

Abstract
Recent studies analysing immunogenetics and immune mechanisms controlling susceptibility to chronic bacterial infection in bronchiectasis implicate dysregulated immunity in conjunction with chronic bacterial infection. Bronchiectasis is a structural pathological end-point with many causes and disease associations. In about half of cases it is termed idiopathic, because it is of unknown aetiology. Bronchiectasis is proposed to result from a ‘vicious cycle’ of chronic bacterial infection and dysregulated inflammation. Paradoxically, both immune deficiency and excess immunity, either in the form of autoimmunity or excessive inflammatory activation, can predispose to disease. It appears to be a part of the spectrum of inflammatory, autoimmune and atopic conditions that have increased in prevalence through the 20th century, attributed variously to the hygiene hypothesis or the ‘missing microbiota’. Immunogenetic studies showing a strong association with human leucocyte antigen (HLA)-Cw*03 and HLA-C group 1 homozygosity and combinational analysis of HLA-C and killer immunoglobulin-like receptors (KIR) genes suggests a shift towards activation of natural killer (NK) cells leading to lung damage. The association with HLA-DR1, DQ5 implicates a role for CD4 T cells, possibly operating through influence on susceptibility to specific pathogens. We hypothesize that disruption of the lung microbial ecosystem, by infection, inflammation and/or antibiotic therapy, creates a disturbed, simplified, microbial community (‘disrupted microbiota’) with downstream consequences for immune function. These events, acting with excessive NK cell activation, create a highly inflammatory lung environment that, in turn, permits the further establishment and maintenance of chronic infection dominated by microbial pathogens. This review discusses the implication of these concepts for the development of therapeutic interventions.
Keywords: bronchiectasis, infection, lung immunology, microbiome, NK cell
Introduction
All mucosal surfaces pose complex challenges for immune protection, but the issues around effective, regulated immunity at the lung mucosa have posed particularly difficult questions for immunologists [1]. In the context of a massive surface area evolved for efficient gas exchange, constantly sampling the environment with all its pollutants, microbial pathogens and allergens the need is to harness an armamarium of physical and biochemical innate and adaptive mechanisms in order to retain optimal function. The potential for deviation from this, either through failure to properly clear microbial pathogens or through excessive inflammation and ensuing structural damage, is evident from the relatively common clinical end-points seen to result from these events. With the impact over the past 10 years of improved metagenomics methodologies for sequencing the microbiota has come the beginning of a greater appreciation that we must also grapple with the variable, environmental, competition within this niche between microbial pathogens and resident commensals [2].
In this review we summarize recent progress in analysing immune mechanisms controlling susceptibility to chronic bacterial infection in the lung, with particular attention to idiopathic bronchiectasis. Bronchiectasis is a disease defined by its pathological end-point that can be arrived at through many diverse aetiologies. Idiopathic bronchiectasis is defined as bilateral predominantly lower lobe bronichectasis and sinusitis of unknown aetiology [3]. Features of the clinical presentation and pathological picture indicate a fascinating interplay between immunogenetic susceptibility, immune dysregulation and chronic bacterial infection.
A very wide spectrum of overt or subtle immune deficiencies may predispose to recurrent bacterial and fungal infections. The term ‘idiopathic bronchiectasis’ describes a specific pathological profile, variably encompassing irreversible, abnormal, bronchial dilatation with chronic inflammation of the airways. Damage to elastic and muscle layers in bronchial airways causes abnormal dilatation, allowing accumulation of mucus and facilitating bacterial infection. The lack of ciliated cells in the damaged epithelium precludes effective removal of mucus, thus promoting chronic bacterial colonization. This is associated clinically with chronic cough and sputum production, recurrent chest infections and airflow obstruction. Bronchiectasis is often described in textbooks as being defined by ‘a common structural end-point with many causes’. These may range from viral, bacterial or mycobacterial post-infectious damage to cystic fibrosis, immune deficiencies or foreign body obstruction. According to a view first posited by Peter Cole, it has long been assumed that the disease results from a vicious cycle of chronic infection and dysregulated inflammation [4]. In its simplest form, a confounder for proponents of this view is that distinct strands of evidence variously implicate deficiencies in immunity and suboptimal immunity, or an excess of immunity in the form of either autoimmunity or excessive inflammatory activation. One of the immune deficiencies, transporter for antigen presentation (TAP) deficiency, is itself associated with aberrant and excessive immune activation as a consequence of impaired human leucocyte antigen (HLA) class I expression [5].The aim of this review is to summarize and synthesize the immunological, molecular and cellular pathways that may lead from initial insult to this common but distinctive pathological end-point.
Epidemiology and aetiology
The observation that there are very substantial differences in incidence of bronchiectasis across geographically and ethnically diverse populations is indicative of the aetiological mix of genetics and environment believed to underpin susceptibility to this disease. For example, in central Australian Aborigines, the incidence of bronchiectasis is 14 per 1000 (nearly 300 times the incidence reported from Finland), probably reflecting distinct aspects both of bacterial exposure and of immunogenetics [6]. In this population, human T lymphotrophic virus type 1 (HTLV-1) infection is associated with multi-focal bronchiectasis, increased morbidity and higher mortality, suggesting that HTLV-1 increases the risk of bronchiectasis and exacerbated outcome [7]. Bronchiectasis remains a significant cause of childhood morbidity in developing countries.
While bronchiectasis will often be diagnosed with no known underlying initiating cause (that is, idiopathic bronchiectasis), specific causes can include episodes of bacterial infection such as Bordetella pertussis, Mycobacterium tuberculosis, non-tuberculous mycobacteria (NTM), childhood measles or the immune response to fungal exposure with Aspergillus [allergic bronchopulmonary aspergillosis (ABPA)]. Bronchiectasis is a key feature of cystic fibrosis [3]. Some cases are due to mucociliary clearance defects, including primary ciliary dyskinesia (PCD) [8]. Presentation is typically with chronic cough and sputum production, recurrent bacterial infection and tiredness.
Genetic studies have focused thus far on candidate gene rather than genome-wide approaches. Analysis of HLA class II polymorphisms in a UK Caucasian disease cohort shows an association with HLA-DRB1*0101/HLA-DQB1*0501 [9]. In light of the diverse array of microbial pathogens that colonize the lungs during chronic infection, it seems most likely that genetic association with a single HLA class II haplotype implicates immunogenetic susceptibility to specific, initiating pathogens. However, no association has been found with polymorphisms in Toll-like receptor (TLR)-2 or TLR-4 [10].
It had been thought that the prevalence of bronchiectasis was decreasing in the developed world as a result of childhood immunizations, increased use of antibiotics and decreased tuberculosis. However, it is now clear that bronchiectasis is one of the spectrum of inflammatory, autoimmune and atopic conditions which shows an increasing disease burden: a recent US retrospective study covering the period 1993–2006 reported an average annual age-adjusted increase in the rate of bronchiectasis-associated hospitalizations of 2·4% in men and 3·0% in women [11].
Association with autoimmune disease
Any consideration of immunopathogenesis in bronchiectasis must account for the fact that, while there have been no data posited in support of this as an autoimmune disease, it is often seen as a part of the pathological spectrum in a subset of established autoimmune diseases [3]. It is seen most commonly with rheumatoid arthritis, ulcerative colitis and, more rarely, with Crohn's disease, systemic lupus erythematosus (SLE) and Sjögren's syndrome [12,13]. While rheumatoid arthritis (RA), ulcerative colitis (UC) and bronchiectasis may clearly all be considered under a common banner of ‘dysregulated immunity’, it is by no means clear what might be the mechanistic common denominators and why this respiratory pathology is associated more commonly with these autoimmune diseases than with others. Symptoms of RA may either precede or follow the diagnosed presence of bronchiectasis, and severe RA does not appear to confer any greater risk than mild RA. Any effort to identify common underlying features of these autoimmune pathologies as a predisposing risk for the diseases must also encompass the fact that up to 10% of cases are in individuals with common variable immunodeficiency (CVID) or other antibody deficiencies [3,14]. Interestingly, about 20% of patients with CVID also develop autoimmune complications, most commonly thrombocytopenia or haemolytic anaemia, but also inflammatory bowel disease, rheumatoid arthritis, primary biliary cirrhosis, thyroiditis, sicca syndrome and systemic lupus and a multi-system granulamatous disease. Human immunodeficiency virus (HIV) infection is a risk factor for bronchiectasis. Put simplistically, then, this is a pathological end-point that can sit alongside either deficient or excessive, but always dysregulated, immunity. Interestingly in this regard, while the interferon (IFN)-γ (+874 T/A) functional polymorphism and a CXCR-1 (+2607 G/C) polymorphism proposed to impact interleukin (IL)-8 binding and neutrophil trafficking were not associated with enhanced risk of bronchiectasis per se, they were associated strongly with bronchiectasis following colectomy in UC [15]. The observation that onset of bronchiectasis can often follow colectomy for UC raises the possibility that perturbation of the gut microbiota has had a systemic impact on immune regulation, leading to altered inflammatory control in the lung.
Both RA and UC are treated increasingly by tumour necrosis factor (TNF)-α antagonists, etanercept and infliximab, raising the issue of whether there is increased susceptibility to respiratory infection and bronchiectasis in this treatment group [16]. Some studies show greater susceptibility to respiratory bacterial and viral infections in these treatment groups. We are aware of one case report of bronchiectasis with non-tuberculous mycobacteria following etanercept treatment for RA [17]. However, when one considers the very large number of patient years' treatment accumulated thus far in UC and RA, one might conclude that there is no evidence that TNF blockade and any consequent increase in respiratory infection have resulted in an increased prevalence of bronchiectasis in this treatment group.
Lung infections in the context of the lung microbiome
The pathogens associated most commonly with infective exacerbations in bronchiectasis are Haemophilus influenzae, Haemophilus parainfluenzae and Pseudomonas aeruginosa. Other infections can include Streptococcus pneumoniae, Staphylococcus aureus, Stenotrophomonas, non-tuberculosis mycobacteria and Aspergillus. Across the range of chronic lung conditions, there has been growing interest in the use of a non-culture-based approach to a more comprehensive, metegenomics-based 16 s sequence analysis of the species present [18,19,20,21]. In general, inflammation is associated with reduced microbiome complexity [21]. Data sets are starting to emerge of 16 s sequences from the lung microbiota of healthy individuals, as well as those with asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis [18,19,20,21]. These data sets will become progressively more useful with the advent of more comprehensive RNAseq-based methodologies for sequencing, with greater momentum behind efforts to culture and manipulate the species, and with greater efforts to understand the functional correlates of microbiome changes. Certainly, the lung microbiota can be seen to change, between health and disease, with antibiotic use and between different parts of the lung. Increasing awareness of the lung microbiota has brought into question the distinction between those bacterial species that are pathogens and those commensals that comprise our physiological microbiome. Species within the nasopharyngeal microbiota such as S. pneumoniae will, to most people most of the time, be present as a commensal, existing in a state of asymptomatic, tolerated equilibrium with host immunity. A key aspect of this that remains relatively poorly understood is the activation of innate receptors, including TLRs by commensal organisms, and the downstream consequences of this activation for immune programming [22].
The fact that the bronchiectasis burden is increasing at a time when greater availability of antibiotics and improved hygiene might have been predicted to herald a decrease suggests that these or other environmental changes may contribute to a paradoxical rise. Falkow and Blaser have used the term ‘disappearing microbiota hypothesis’ to explain the rising burden of diseases, including those of inflammatory pathogenesis [23]. They argue that with changes in hygiene and clinical practice, perturbations in human macroecology have progressively modulated the composition of the indigenous microbiota, thus changing human physiology and disease risk. One possible mechanism for such influence is suggested by studies of immune repertoire changes in germ-free mice: it is clear that changes to host bacterial ecology can have a profound impact on immune subsets, including T helper type 1 (Th1)/Th2 balance and development of Th17, regulatory T cell (Treg) and invariant natural killer (iNK) T cells, with consequent downstream implications for disease susceptibility [24,25].
At minimum, a microbiome-based approach to lung disease offers a frame of reference for diseases such as bronchiectasis, in which neither the presence nor absence of any specific species can account for susceptibility [26]. Gordon and colleagues have used the term ‘community as pathogen’ to describe the situation such as that seen in inflammatory bowel disease, in which disease ensues from an inflammatory context imprinted by the microbial ecology [27]. It is likely that the relevance of this hypothesis extends to other diseases, including bronchiectasis.
At a time when academic departments of respiratory medicine increasingly embrace the new approaches offered by metegenomics, we need to be very clear why we are doing this and what rewards may be on offer. Certainly, to regard this as a high-technology route to identification and curation of ever more bacterial species that we can then ‘treat’ would be to miss the point entirely, and may exacerbate the pathologies we seek to alleviate. The enormous challenge, moving from the initial gains of 16 s sequence curation to annotating the functional genomics, will be to determine the rules and pathological correlates of lung bacterial ecology. Which are the ecological shifts that allow chronic colonization by pathogenic strains such as pseudomonas, how can these be manipulated, and how do natural or therapeutically induced changes in the microbiota contribute to reprogramming of local bacterial immunity?
Immune mechanisms
By Occam's razor, the HLA class II association in bronchiectasis would best be accounted for by a classic immune response gene effect, determining the magnitude of responsiveness to an underlying pathogen [9]. The immunocytochemical detection of T cells in bronchiectatic lung sections is compatible with a role of T cells in specific host defence or immunopathogenesis [28]. However, arguing against a simple model of this type, it appears unlikely that this disease is driven commonly by a single, dominant pathogen. Furthermore, lung histopathology shows infiltrating CD4 and CD8 cells, the latter predominating. T cell infiltrates are activated (HLA-DR+) and located mainly in the lamina propria [28]. There are also activated macrophages and dendritic cells. In a disease with chronic neutrophilia, one has to consider the possibility of excessive or dysregulated IL-17 release as a driver. While there is considerable focus on the role of Th17 cells in resistance to bacterial infection [29,30], it is likely that much IL-17 release may derive from other sources, including γδ T cells iNK T and NK cells. In line with this, the airway submucosa of non-cystic fibrosis (CF) bronchiectasis and CF patients show enhanced levels of IL-17+ cells, both innate and adaptive [31]. Furthermore, IL-17+ cells were correlated with neutrophilia, making them potential candidates in driving the vicious cycle of infection and inflammation in bronchiectasis.
In King's laboratory, Holdsworth and colleagues focused on baseline immune parameters in bronchiectasis patients compared to controls and the innate and adaptive immune response to non-typable H. influenzae [32,33]. Most aspects of baseline, systemic, cellular immunity were normal in this patient group, although minor subsets were outside the normal range either for CD4 numbers or for neutrophil oxidative burst.
Genotypic analysis of HLA-C and killer immunoglobulin-like receptors (KIR) has been used across a wide range of infectious, inflammatory and autoimmune disease phenotypes to elucidate the potential contribution of NK cell activation programmes in disease susceptibility [34,35]. From such studies has come the concept that, depending both on inheritance of predominantly activating or inhibitory KIR genomic repertoires and on differential inheritance of the cognate HLA class I ligands, humans may be considered to have an inherent potential for NK cell activation ranged across a spectrum of activation [36]. Susceptibility to viral infection, including hepatitis C virus (HCV) and HIV, has tended to be associated with inhibitory genotypes, while susceptibility to autoimmune and inflammatory pathology has often been associated with activating genotypes. Analysis of KIR and HLA class I genotypes appears informative with respect to susceptibility to idiopathic bronchiectasis: HLA-Cw*03 alleles and, in particular, HLA-C group 1 homozygosity were associated with increased risk of bronchiectasis [37,38]. Approximately half the bronchiectasis cohort were homozygous for HLA-C group 1 compared with a quarter of controls. Homozygosity effects of this type are reminiscent of several other NK cell disease studies, in which homozygotes missing ligands for inhibitory receptors are predicted to have fewer NK cells under inhibitory control [39,40]. Bronchiectasis patients encompass a significantly increased number of individuals expressing only HLA-C group 1 with 2DS1 and/or 2DS2 stimulatory KIRs – a combination that would be considered at the extreme end of the activation spectrum for NK cells. This model will acquire greater nuance and complexity as we gain the ability to factor in both polymorphic KIR variability and copy number variability [41,42]. Specific, functional data addressing the hypothesis based on the genotypic data are not yet available. Certainly, NK cells play a critical role at the innate–adaptive interface during lung infection, and are implicated in immunity to many of the key pathogens implicated in the respiratory infections of these patients [43,44].
Concluding remarks
Reductionist immunology has in the past demanded that specific respiratory infections tend to be studied experimentally as individual entities. Physiologically, however, the lung is a complex microbial ecosystem in which multiple, pathogenic bacterial, viral and fungal strains may all compete simultaneously for survival. In so doing, they alert the innate receptors of host immunity, eliciting innate and then adaptive responses or potentially diverse subset polarization (Fig. 1). This picture has become more complex in recent years, as it has been appreciated that these pathogenic strains are a minute part of a much larger and highly variable microbial ecosystem, comprising the non-pathogenic microbiota. Changes to the microbiota, whether through bacterial competition for niches, changes to the inflammatory milieu or anti-microbial therapy, are likely to impact upon T cell subset polarization, as is the case in the gut. Blaser and Falkow have made a highly articulate case for the ‘missing microbiota’ as a risk factor underlying the increase in inflammatory diseases. We consider bronchiectasis a probable example of such events. Certainly, inflammatory lung disease appears to be associated with a simplified microbiota. A more detailed, functional annotation of the lung microbiota in health and disease will be needed before we can really determine whether we should regard such diseases as arising out of a ‘missing’ or ‘disrupted’ microbiota. A prediction from the evidence in bronchiectasis is that the subset of patients in whom disease is identified following colectomy for ulcerative colitis are likely to show changes both in the lung microbiome and in lung immune subsets. If we are to break the vicious cycle of inflammation and infection that underpins chronic bacterial lung infection in bronchiectasis, we will need an improved understanding of how to modulate judiciously the lung ecosystem with anti-microbials, as well as of the consequences of infection and therapeutics on the specific patterns of respiratory innate and adaptive immune responses. Current advances in utilization of high-throughput sequencing for rapid and precise chracterization of respiratory pathogens will aid in the timely and appropriate prescribing of antibiotics. Some way further off is the potential for probiotic manipulation of the microbiota and for therapeutic intervention in KIR activation.
Fig. 1.
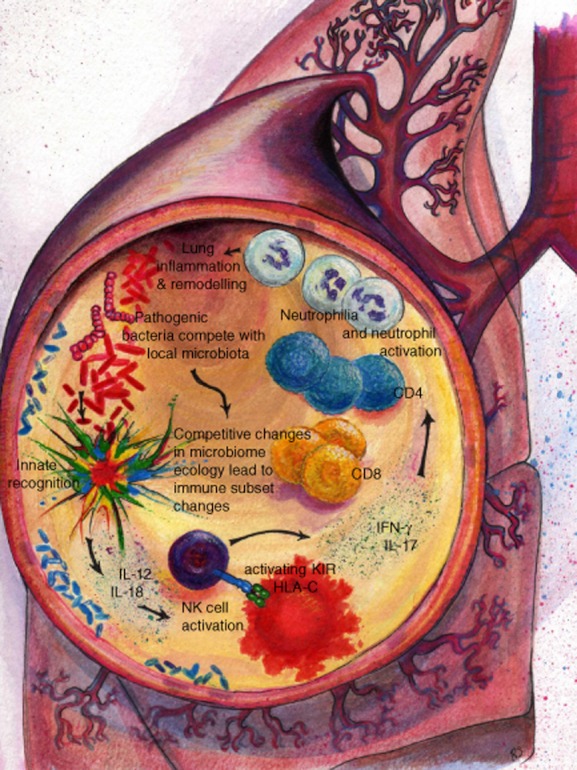
The disrupted microbiota in the vicious cycle of chronic bacterial infection and inflammation in the lung.
Acknowledgments
The authors would like to thank the Welton Foundation, Royal Brompton and Harefield NHS Trust Clinical Research Committee, BBSRC, NIH and MRC for funding and Trevor Lawley for discussions. R.B. is a Principle Investigator in the Centre for Respiratory Infection, Imperial College London. The authors are grateful for support from the NIHR Biomedical Research Centre funding scheme. The authors thank the illustrator, Rachel Scott.
Disclosure
The authors have declared that no competing interests exist.
References
- 1.Boyton RJ, Openshaw PJ. Pulmonary defences to acute respiratory infection. Br Med Bull. 2002;61:1–12. doi: 10.1093/bmb/61.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyton RJ. Bronchiectasis. Medicine. 2012;40:267–272. [Google Scholar]
- 4.Cole P. A new look at the pathogenesis and management of persistent bronchial sepsis: a ‘vicious circle’ hypothesis and its logical therapeutical connotations. In: Davies RJ, editor. Strategies for the management of chronic bronchial sepsis. Oxford: The Medicine Publishing Foundation; 1984. pp. 1–20. [Google Scholar]
- 5.Gadola SD, Moins-Teisserenc HT, Trowsdale J, Gross WL, Cerundolo V. TAP deficiency syndrome. Clin Exp Immunol. 2000;121:173–178. doi: 10.1046/j.1365-2249.2000.01264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang AB, Grimwood K, Mulholland EK, Torzillo PJ. Working group on indigenous paediatric respiratory health. Bronchiectasis in indigenous children in remote Australian communities. Med J Aust. 2002;177:200–204. doi: 10.5694/j.1326-5377.2002.tb04733.x. [DOI] [PubMed] [Google Scholar]
- 7.Einsiedel L, Fernandes L, Spelman T, Steinfort D, Gotuzzo E. Bronchiectasis is associated with human T-lymphotropic virus 1 infection in an Indigenous Australian population. Clin Infect Dis. 2012;54:43–50. doi: 10.1093/cid/cir766. [DOI] [PubMed] [Google Scholar]
- 8.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 9.Boyton RJ, Smith J, Jones M, et al. Human leucocyte antigen class II association in idiopathic bronchiectasis, a disease of chronic lung infection, implicates a role for adaptive immunity. Clin Exp Immunol. 2008;152:95–101. doi: 10.1111/j.1365-2249.2008.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds C, Ozerovitch L, Wilson R, Altmann D, Boyton R. Toll-like receptors 2 and 4 and innate immunity in neutrophilic asthma and idiopathic bronchiectasis. Thorax. 2007;62:279. [PMC free article] [PubMed] [Google Scholar]
- 11.Seitz AE, Olivier KN, Steiner CA, Montes de Oca R, Holland SM, Prevots DR. Trends and burden of bronchiectasis-associated hospitalizations in the United States, 1993–2006. Chest. 2010;138:944–949. doi: 10.1378/chest.10-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen M, Sahn SA. Bronchiectasis in systemic diseases. Chest. 1999;116:1063–1074. doi: 10.1378/chest.116.4.1063. [DOI] [PubMed] [Google Scholar]
- 13.Eaton TE, Lambie N, Wells AU. Bronchiectasis following colectomy for Crohn's disease. Thorax. 1998;53:529–531. doi: 10.1136/thx.53.6.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregersen S, Aaløkken TM, Mynarek G, et al. High resolution computed tomography and pulmonary function in common variable immunodeficiency. Respir Med. 2009;103:873–880. doi: 10.1016/j.rmed.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 15.Boyton RJ, Reynolds C, Wahid FN, et al. IFN gamma and CXCR-1 gene polymorphisms in idiopathic bronchiectasis. Tissue Antigens. 2006;68:325–330. doi: 10.1111/j.1399-0039.2006.00670.x. [DOI] [PubMed] [Google Scholar]
- 16.Lieberman-Maran L, Orzano IM, Passero MA, Lally EV. Bronchiectasis in rheumatoid arthritis: report of four cases and a review of the literature – implications for management with biologic response modifiers. Semin Arthritis Rheum. 2006;35:379–387. doi: 10.1016/j.semarthrit.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Esaki T, Sugimoto M, Mori S, Yamashita A, Matsumoto M, Kohrogi H. A case of pulmonary nontuberculous mycobacteriosis aggravated during treatment with etanercept for rheumatoid arthritis. Nihon Kokyuki Gakkai Zasshi. 2010;48:312–316. [PubMed] [Google Scholar]
- 18.Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. 2012;160:258–266. doi: 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Schloss PD, Kalikin LM, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sze MA, Dimitriu PA, Hayashi S, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han MK, Huang YJ, Lipuma JJ, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67:456–463. doi: 10.1136/thoraxjnl-2011-201183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Ann Rev Immunol. 2012;30:759–795. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov II, Littman DR. Modulation of immune homeostasis by commensal bacteria. Curr Opin Microbiol. 2011;14:106–114. doi: 10.1016/j.mib.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Relman DA. Microbial genomics and infectious diseases. N Engl J Med. 2011;365:347–357. doi: 10.1056/NEJMra1003071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva JR, Jones JA, Cole PJ, Poulter LW. The immunological component of the cellular inflammatory infiltrate in bronchiectasis. Thorax. 1989;44:668–673. doi: 10.1136/thx.44.8.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009;126:177–185. doi: 10.1111/j.1365-2567.2008.03017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol. 2007;19:377–382. doi: 10.1016/j.smim.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184:252–258. doi: 10.1164/rccm.201102-0236OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King PT, Ngui J, Gunawardena D, Holmes PW, Farmer MW, Holdsworth SR. Systemic humoral immunity to non-typeable Haemophilus influenzae. Clin Exp Immunol. 2008;153:376–384. doi: 10.1111/j.1365-2249.2008.03697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King PT, Ngui J, Farmer MW, Hutchinson P, Holmes PW, Holdsworth SR. Cytotoxic T lymphocyte and natural killer cell responses to non-typeable Haemophilus influenzae. Clin Exp Immunol. 2008;152:542–551. doi: 10.1111/j.1365-2249.2008.03667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyton RJ, Altmann DM. Natural killer cells, killer immunoglobulin-like receptors and human leucocyte antigen class I in disease. Clin Exp Immunol. 2007;149:1–8. doi: 10.1111/j.1365-2249.2007.03424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheent K, Khakoo SI. Natural killer cells: integrating diversity with function. Immunology. 2009;126:449–457. doi: 10.1111/j.1365-2567.2009.03045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khakoo SI, Carrington M. KIR and disease: a model system or system of models? Immunol Rev. 2006;214:186–201. doi: 10.1111/j.1600-065X.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- 37.Boyton RJ, Smith J, Ward R, et al. HLA-C and killer cell immunoglobulin-like receptor genes in idiopathic bronchiectasis. Am J Respir Crit Care Med. 2006;173:327–333. doi: 10.1164/rccm.200501-124OC. [DOI] [PubMed] [Google Scholar]
- 38.Boyton RJ. Regulation of immunity in bronchiectasis. Med Mycol. 2009;47(Suppl. 1):S175–182. doi: 10.1080/13693780802163370. [DOI] [PubMed] [Google Scholar]
- 39.Martin MP, Nelson G, Lee JH, et al. Cutting edge: susceptibility to psoriatic arthritis: influence of activating killer Ig-like receptor genes in the absence of specific HLA-C alleles. J Immunol. 2002;169:2818–2822. doi: 10.4049/jimmunol.169.6.2818. [DOI] [PubMed] [Google Scholar]
- 40.Nelson GW, Martin MP, Gladman D, Wade J, Trowsdale J, Carrington M. Cutting edge: heterozygote advantage in autoimmune disease: hierarchy of protection/susceptibility conferred by HLA and killer Ig-like receptor combinations in psoriatic arthritis. J Immunol. 2004;173:4273–4276. doi: 10.4049/jimmunol.173.7.4273. [DOI] [PubMed] [Google Scholar]
- 41.Middleton D, Gonzelez F. The extensive polymorphism of KIR genes. Immunology. 2010;129:8–19. doi: 10.1111/j.1365-2567.2009.03208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Traherne JA, Martin M, Ward R, et al. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum Mol Genet. 2010;19:737–751. doi: 10.1093/hmg/ddp538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology. 2009;128:151–163. doi: 10.1111/j.1365-2567.2009.03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. 2008;181:5481–5489. doi: 10.4049/jimmunol.181.8.5481. [DOI] [PMC free article] [PubMed] [Google Scholar]