

Abstract
The clinical efficacy of peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists in cell-mediated autoimmune diseases results from down-regulation of inflammatory cytokines and autoimmune effector cells. T cell islet autoimmunity has been demonstrated to be common in patients with phenotypic type 2 diabetes mellitus (T2DM) and islet-specific T cells (T+) to be correlated positively with more severe beta cell dysfunction. We hypothesized that the beneficial effects of the PPAR-γ agonist, rosiglitazone, therapy in autoimmune T2DM patients is due, in part, to the immunosuppressive properties on the islet-specific T cell responses. Twenty-six phenotypic T2DM patients positive for T cell islet autoimmunity (T+) were identified and randomized to rosiglitazone (n = 12) or glyburide (n = 14). Beta cell function, islet-specific T cell responses, interleukin (IL)-12 and interferon (IFN)-γ responses and islet autoantibodies were followed for 36 months. Patients treated with rosiglitazone demonstrated significant (P < 0·03) down-regulation of islet-specific T cell responses, although no change in response to tetanus, a significant decrease (P < 0·05) in IFN-γ production and significantly (P < 0·001) increased levels of adiponectin compared to glyburide-treated patients. Glucagon-stimulated beta cell function was observed to improve significantly (P < 0·05) in the rosiglitazone-treated T2DM patients coinciding with the down-regulation of the islet-specific T cell responses. In contrast, beta cell function in the glyburide-treated T2DM patients was observed to drop progressively throughout the study. Our results suggest that down-regulation of islet-specific T cell autoimmunity through anti-inflammatory therapy may help to improve beta cell function in autoimmune phenotypic T2DM patients.
Keywords: autoimmunity, PPAR-γ, rosiglitazone, T cells, type 2 diabetes
Introduction
Peroxisome proliferator-activated receptor-gamma (PPAR-γ) mediates important immune regulatory functions in conventional T cells, macrophages and dendritic cells [1–7]. The ability of ligand-activated PPAR-γ to inhibit interleukin (IL)-12 production by dendritic cells as well as its ability to inhibit interferon (IFN)-γ production by T cells indicates that this nuclear hormone receptor plays an important role during differentiation of naive T cells into their effector subsets [1]. PPAR-γ also plays a critical role in natural regulatory T cell (Treg) suppressive function and in the differentiation and stability of inducible Tregs [8–10]. In fact, PPAR-γ was shown recently to have a direct effect on visceral adipose tissue Treg accumulation, phenotype and function [11].
Consistent with the immunoregulatory effects of PPAR-γ, a number of PPAR-γ agonists have been used to treat effectively murine experimental autoimmune encephalomyelitis (EAE), colitis, asthma and allergic disease [12–19]. In humans, PPAR-γ agonists have demonstrated clinical efficacy in treating Crohn's disease, psoriasis and multiple sclerosis, reflecting a beneficial effect in cell-mediated autoimmune diseases [20–23].
During recent years, the relationship between inflammation, cytokine production, insulin resistance and subsequent development of type 2 diabetes mellitus (T2DM) has become apparent. Inflammation in the pancreatic islets of T2DM patients includes inflammatory cytokines [24,25] and proinflammatory immune cells [25,26]. The chronic systemic inflammation associated with T2DM patients has been hypothesized to contribute to the development of T cell islet-specific autoimmunity in some phenotypic T2DM patients [27–31]. Activation of islet-specific T cells (T+) in phenotypic T2DM patients has been found to be more common than appreciated previously [31], and correlated positively with a more severe β cell lesion [31,32].
Treatment of T2D patients with PPAR-γ agonists, such as rosiglitazone or pioglitazone, have been shown previously to have beneficial effects on glycaemic control, insulin sensitivity, insulin secretion and plasma adipokine levels [33]. Recently, the cumulative incidence of monotherapy failure at 5 years was shown to be significantly lower in phenotypic T2DM patients treated with the PPAR-γ agonist, rosiglitazone, compared to T2DM patients treated with metformin or glyburide. The clinical efficacy of rosiglitazone was believed to be due, in part, to a slower decline in beta cell function in rosiglitazone-treated patients [34]. We hypothesized that the beneficial effects of PPAR-γ agonists in T2DM patients might be due, in part, to the immunosuppressive properties on T cell islet autoimmunity and inflammatory cytokine production. In this study we compared the islet-specific T cell responses (T+), IL-12 production, IFN-γ production and glucagon-stimulated beta cell function in autoimmune phenotypic T2DM patients treated with the PPAR-γ agonist, rosiglitazone, to autoimmune T2DM patients treated with glyburide. Our results demonstrate that treatment of autoimmune phenotypic T2DM patients with an anti-inflammatory medication capable of suppressing the islet-specific T cell responses and inflammatory cytokine production may slow or stop the decline in beta cell function in T2DM patients.
Materials and methods
Subjects
Twenty-six phenotypic T2DM patients defined by obesity, age > 35 years, HbA1c levels (between 6–10%) and fasting C-peptide levels (> 0·8 ng/ml) positive for T cell responses to islet proteins (determined by cellular immunoblotting) were followed for 36 months. Patients on insulin were not eligible. Informed consent was obtained from all subjects. This study was approved by the Institutional Review Board at the University of Washington.
Study design
This was a randomized, open-label, multiple oral dose study. Randomization was achieved by the random number method with odd versus even indicating treatment group. T2DM patients meeting the inclusion criteria were randomized to either rosiglitazone or glyburide after 2 weeks off prestudy diabetes medications. Patients were scheduled for visits at 3-month intervals for 36 months of follow-up. Dosage for the rosiglitazone group was started at 4 mg once per day and increased to twice per day if glycaemic control (HbA1c ≤ 7·0%) was not achieved. Dosage for the glyburide group was started at 2·5 mg (or same dosage received prior to the study) and increased to twice per day up to a maximum of 10 mg twice per day if glycaemic control was not achieved. If monotherapy treatment did not achieve adequate overall control (HbA1c ≤7·0%), metformin was added and the dose increased gradually as needed up to 1000 mg ×2 per day. If necessary to achieve a HbA1c ≤ 7·0%, acarbose was added subsequently up to a maximum dose of 100 mg ×3 per day.
Autoantibody assays
Glutamic acid decarboxylase (GAD)-65 autoantibody assay
The determination of GAD-autoantibody levels were performed at the Northwest Lipid Metabolism and Diabetes Research Laboratories (NLMDRL) (Seattle, WA, USA). GAD-autoantibody was measured in a radiobinding immunoassay on coded serum samples, as described previously [31]. In the Immunology of Diabetes Society (IDS) Diabetes Antibody Standardization Program (DASP)-sponsored 2010 workshop, the sensitivity of the GAD assay was 82% and specificity was 93·3%. The NWLDRL is participating actively in the National Institutes of Health (NIH)-sponsored autoantibody harmonization programme.
Insulinoma-associated protein-2 autoantibody (IA-2) assay
The IA-2 autoantibodies were measured at the NLMDRL, as described previously [31]. Autoantibodies to IA-2 were measured under identical conditions to those described for GAD-autoantibody using the plasmid containing the cDNA coding for the cytoplasmic portion of IA-2. In the IDS-sponsored 2010 DASP workshop, the sensitivity of the IA-2 assay was 62% and specificity was 100%.
T cell assay: cellular immunoblotting (CI)
CI was performed on freshly isolated peripheral blood mononuclear cells (PBMCs) to test for the presence of islet reactive T cells, as described previously 35. Briefly, normal human islet cell preparations were subjected to preparative one-dimensional 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and electroblotted onto nitrocellulose, the nitrocellulose cut into molecular weight regions (blots) and then solubilized to form nitrocellulose particles. The nitrocellulose particles containing islet proteins were used to stimulate PBMCs at a concentration of 3·5 × 105 PBMCs per well. Positive T cell responses were determined to be a T cell stimulation index (SI) > 2·1, which corresponds to 3 standard deviations above the mean of T cell responses to islet proteins from normal control subjects 35. T1D patients have been shown to respond to 4–18 molecular weight proteins and normal controls (without diabetes) to 0–3 molecular weight regions [29,36]. Human pancreatic islets were obtained from the NIH-supported Islet Cell Resource Centers (ICR-ABCC). The tissue specificity of the T cell responses from diabetes patients to islet proteins has been demonstrated previously 35. Cellular immunoblotting has been validated in two distinct NIH-supported T cell validation studies designed to test the ability of several different assays, including CI, performed on masked specimens to distinguish T cell responses to islet proteins of T1D patients from control subjects [37,38]. In the first validation study, the sensitivity for detecting T1D patients from controls was 94% and specificity was 83% [37]. In the second validation study, the sensitivity was 74% and the specificity was 88% [38]. In 2009, the specificity and sensitivity of the CI assay were improved to 96% and 94%, respectively [39].
Tetanus toxoid
PBMC proliferative responses to tetanus toxoid (CalBioChem, La Jolla, CA, USA) were tested at each time-point for each patient as an antigen control response. Soluble tetanus toxoid was utilized in place of nitrocellulose-bound tetanus toxoid, as reported previously 35, for ease of use. No differences in responses have been observed between soluble and nitrocellulose-bound tetanus toxoid (data not shown). Furthermore, no differences in PBMC responses were noted for tetanus toxoid between rosiglitazone- and glyburide-treated patients (data not shown).
Enzyme-linked immunospot assay (ELISPOT)
IL-12 and IFN-γ production was measured using the Human Cytokine Elispot kit from U-CyTech (Utrecht, the Netherlands). PBMCs were isolated and added directly to a 96-well flat-bottom tissue culture plate at a concentration of 3 × 105 cells per well, coated previously with antibodies to either IFN-γ or IL-12. Cells were stimulated for 3 days with sonicated human islets at 37°C and 5% CO2. After 3 days cells were lysed, secondary antibodies added and the plates incubated overnight at 4°C. The plates were developed as per the manufacturer's instructions and results obtained using the BioSys BioReader-3000 (Austin, TX, USA). PBMC responses to tetanus toxoid were used as antigen control responses along with responses to concanavalin A (non-specific mitogenic responses).
ELISA
Adiponectin levels in serum of patients at baseline and every 12 months of follow-up were measured using the Quantikine Human Adiponectin Immunoassay from R&D Systems (Minneapolis, MN, USA) according to the manufacturer's instructions. The colour reaction was stopped after 30 min and optical density was measured at 450 nm using an MRX Revelation plate reader from Dynex Technologies (Chantilly, VA, USA).
Glucagon-stimulated C-peptide assay
C-peptide was measured at (NLMDRL) 6 min after stimulation with 1 mg glucagon administered intravenously, as described previously [32].
Statistics
All results for T cell and C-peptide are summarized as the mean, and measures of variability are reported as standard error (s.e.). Linear regression analysis was used to determine the best-fitted line, and an analysis of covariance was used to compare slopes between groups over the entire study. Two-tailed Mann–Whitney U-tests were used to compare results at individual time-points between the treatment groups. Two-tailed Wilcoxon matched-pairs signed-rank tests were used to compare results between individual time-points within the treatment groups.
Results
Demographic data, islet autoantibody and T cell responses to tetanus toxoid from patients treated with rosiglitazone and glyburide are shown in Table 1. No significant differences were observed in age, sex, race, body mass index (BMI), islet autoantibodies, tetanus responses or time since diagnosis between treatment groups at baseline or 36 months (Table 1). Islet-specific T cell responses in both patient groups increased during the first 12 months, becoming increased significantly (P < 0·05) compared to baseline at 9 months of treatment for both patient groups (Fig. 1). However, beginning at 15 months, T cell responses to islet proteins in the rosiglitazone-treated patients became suppressed significantly (P < 0·03). In fact, the T cell responses to islet proteins in the rosiglitazone-treated patients became negative at 15 months (fewer than four blot sections) and remained negative throughout follow-up (Fig. 1). In contrast, the T cell responses to islet proteins in the glyburide patients remained positive throughout the study (Fig. 1).
Table 1.
Demographic, islet autoantibody, and T cell responses to tetanus from patients treated with glyburide and rosiglitazone
| Demographic characteristics baseline and 36 months | Glyburide (n = 14) | Rosiglitazone (n = 12) |
|---|---|---|
| Age (years) (mean ± s.d.) | 54·5 ± 16·3 | 58 ± 9·8 |
| Sex, n (%) | ||
| Male | 12 (85·7%) | 8 (66·7%) |
| Female | 2 (14·3%) | 4 (33%) |
| BMI (mean ± s.d.) baseline | 30·9 ± 4·7 | 32·6 ± 4·5 |
| BMI (mean ± s.d.) 36 male | 30·1 ± 5·4 | 33·8 ± 6·8 |
| Race, n (%) | ||
| Caucasian | 8 (57·1%) | 9 (75·0%) |
| African American | 4 (28·6%) | 0 (0·0%) |
| Asian | 1 (7·1%) | 2 (16·7%) |
| Native American | 1 (7·1%) | 1 (8·3%) |
| HbA1c (mean ± s.d.) baseline | 6·8 ± 0·64 | 7·1 ± 0·51 |
| HbA1c (mean ± s.d.) 36 male | 7·22 ± 0·90 | 6·82 ± 0·61 |
| Length of disease (mean ± s.d.) | 2·92 ± 1·8 | 3·2 ± 1·7 |
| Autoantibodies baseline/36 male | ||
| ICA only | 4 / 2 | 2 / 0 |
| GADA only | 0 / 1 | 0 / 1 |
| IA-2 only | 0 / 1 | 0 / 0 |
| ICA/GADA | 0 / 0 | 2 / 1 |
| ICA/IA-2 | 1 /1 | 1 / 1 |
| IA-2/GADA | 0 / 0 | 0 / 0 |
| Antibody-negative | 9 / 9 | 7 / 9 |
| Tetanus toxoid response | ||
| SI at baseline (mean ± s.d.) | 18·9 ± 24·8 | 22·1 ± 34·6 |
| SI at 36 months (mean ± s.d.) | 11·6 ± 18·8 | 12·9 ± 11·3 |
| HLA | ||
| Risk associated for type 1 diabetes | ||
| 0201/0301 or 0302/04 | 7 (50%) | 4 (33%) |
| Protective for type 1 diabetes | ||
| 0602/1501 or 0303/0701 | 5 (35·7%) | 3 (25%) |
| Other | 2 (14·3%) | 5 (42%) |
BMI: body mass index; GADA: glutamic acid decarboxylase autoantibody; HLA: human leucocyte antigen; IA-2: insulinoma-associated protein-2; ICA: islet cell antibody; s.d.: standard deviation; SI: stimulation index.
Fig. 1.
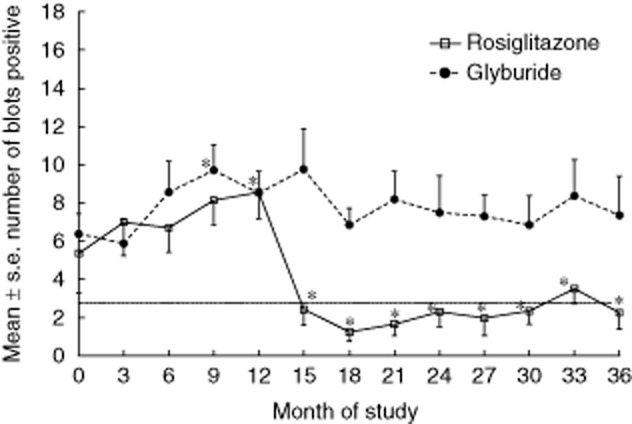
Mean ± standard error T cell responses to islet proteins for glyburide-treated (circles and dotted line) and rosiglitazone-treated (squares and solid line) phenotypic type 2 diabetes mellitus (T2DM) patients. The number of blot sections stimulatory to T cells [stimulation index (SI) > 2·0] is demonstrated on the y-axis. A horizontal line at three blot sections represents the cut-off for T cell positivity. Asterisk at 9 months identifies significant (P < 0·05) differences from baseline for both patient groups. Asterisks at other time-points identify significant (P < 0·03) differences between the groups.
Mean stimulated C-peptide responses for both glyburide- and rosiglitazone-treated patients are shown in Fig. 2. During the first 12 months of follow-up, at the time T cell proliferation increased, the C-peptide in the glyburide-treated patients remained stable, whereas the C-peptide responses in the rosiglitazone-treated patients declined significantly (P < 0·05). However, after 12 months of follow-up, when islet-reactive T cell responses were suppressed in rosiglitazone-treated patients (Fig. 1), the C-peptide responses in the rosiglitazone-treated patients improved. In contrast, the C-peptide in the glyburide patients was observed to continue to decline throughout the study, reaching significance (P < 0·05) from baseline at 36 months (Fig. 2). Comparison of the glucagon-stimulated C-peptide responses for the rosiglitazone- and glyburide-treated patients demonstrated significant differences (P < 0·05) beginning at 27 months (Fig. 2). Significant differences were also observed between the slopes (P < 0·01) of the responses from the two groups (data not shown).
Fig. 2.
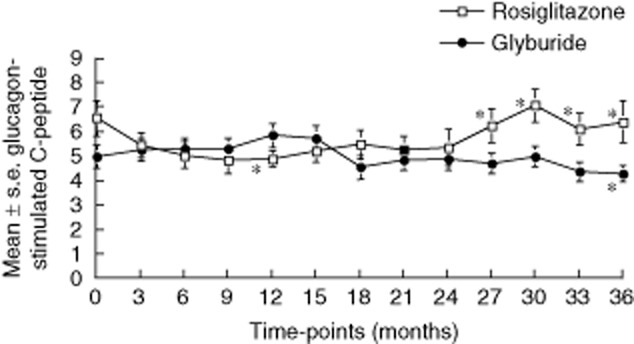
Mean ± standard error glucagon-stimulated C-peptide (ng/ml) responses for glyburide-treated (closed circles) and rosiglitazone-treated (open squares) autoimmune phenotypic phenotypic type 2 diabetes mellitus (T2DM) patients. Asterisks represent significant differences from baseline (P < 0·05) and between the groups. Significant differences between slopes (P < 0·01) are not shown on graphs.
The mean IFN-γ and IL-12 responses for the rosiglitazone- and glyburide-treated patients are shown in Fig. 3. For the glyburide-treated patients, the mean IFN-γ (Fig. 3a) and IL-12 (Fig. 3b) responses increased throughout the study and were elevated significantly (P ≤ 0·05) at 18 months for IFN-γ and 24 months for IL-12 compared to baseline. The IL-12 and IFN-γ responses in the rosiglitazone-treated patients increased during the first 12 months of follow-up and were increased significantly over baseline at 9 months for both IFN-γ and IL-12. However, after 12 months the responses to IFN-γ and IL-12 began to decrease. Significant (P < 0·05) differences were observed between the treatment groups for both IFN-γ and IL-12, beginning at 30 months of follow-up for IL-12 and 33 months for IFN-γ (Fig. 3a and b). IFN-γ and IL-12 responses to tetanus toxoid and concanavalin A were similar between rosiglitazone- and glyburide-treated patients (data not shown).
Fig. 3.
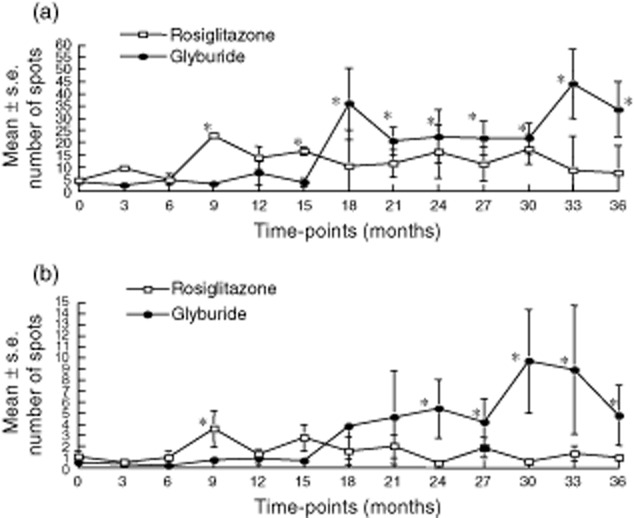
Mean ± standard error of number of spots for the enzyme-linked immunospot (ELISPOT) cytokine response (a) interferon (IFN)-γ and (b) interleukin (IL)-12 production in rosiglitazone-treated (open squares) and glyburide-treated (closed circles) during follow-up. Asterisks identify significant differences (P < 0·05) from baseline and between the groups.
Previously, other researchers have identified increases in serum adiponectin levels in patients treated with rosiglitazone. We also observed that adiponectin levels increased significantly (P < 0·001) in rosiglitazone-treated patients compared to baseline, whereas adiponectin levels in glyburide-treated patients remained stable. Significant differences in overall plasma concentrations of adiponectin were also significantly (P < 0·03) higher in patients treated with rosiglitazone compared to patients treated with glyburide (Fig. 4).
Fig. 4.
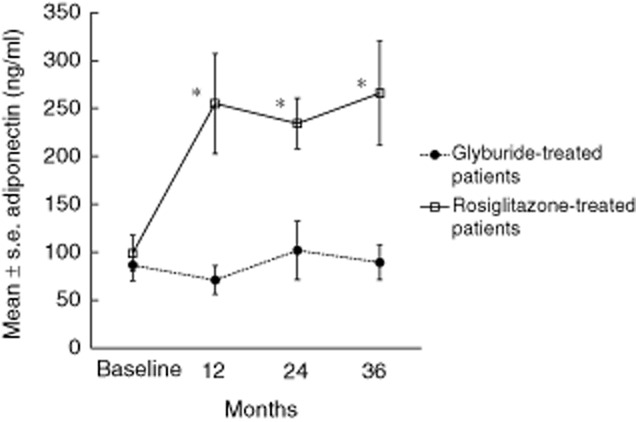
Mean ± standard error of adiponectin responses in the serum of glyburide-treated (closed circles and dotted line) and rosiglitazone-treated (open squares and solid line) patients at baseline and every 12 months of follow-up. Asterisks identify significant differences from baseline and between groups (P < 0·001).
Discussion
Systemic inflammation has been demonstrated to be involved in the development of T2DM. Over the years, we have used the validated cellular immunoblotting assay to study islet-specific T cell autoimmunity in both T1DM and T2DM patients [29,31,32,35–39]. The presence of the islet-specific T cells in T2DM patients has also been linked to a more severe beta cell dysfunction [32]. We therefore postulated that suppression of the islet-specific T cells in T2DM patients might benefit these patients by slowing or reversing beta cell function. Although the beneficial effect of PPAR-γ agonists in T2DM immunotherapy was believed originally to be due to an increase in insulin sensitivity, PPAR-γ agonists have also been reported to have anti-inflammatory properties and may be useful in suppressing autoimmune responses [21]. We propose yet another possible mechanism for the protection offered by PPAR-γ agonists such as rosiglitazone against T2DM disease progression; namely, the suppression of islet-specific T cell autoimmunity.
In this study, we observed that rosiglitazone was able to down-regulate significantly islet-specific T cell proliferative responses compared to patients treated with glyburide, but not affect T cell reactivity to a recall antigen (tetanus toxoid) or non-specific responses (concanavalin A). Islet autoantibody responses were also not affected by either treatment. We postulate that the length of time from initiation of treatment to observation of diminished islet-specific T cell proliferative responses (12 months) is due, in part, to the time needed for the immune system to establish regulatory cells or other regulatory mechanisms needed to suppress the autoreactive T cell responses. Because T cell responses to tetanus toxoid or concanavalin A were not suppressed, it is unlikely that rosiglitazone has a toxic effect on the islet-reacting T cells but, rather, instills regulation of the autoimmune T cell response.
Other markers of inflammation and autoimmunity were also down-regulated in the rosiglitazone-treated patients (IFN-γ and IL-12) compared to the glyburide-treated patients. Additionally, the anti-inflammatory cytokine, adiponectin, was significantly (P < 0·001) higher at 12 months of follow-up in the plasma of the rosiglitazone-treated patients coinciding with down-regulation of the islet-specific T cell responses. In contrast, the adiponectin levels in the plasma of the glyburide-treated patients were not different from baseline during follow-up. In other autoimmune diseases, rosiglitazone has been shown to be effective in reducing the development of inflammation and autoimmunity by increasing levels of regulatory cytokines such as IL-4 and IL-10, increasing adiponectin, inhibiting T helper cell proliferative responses and decreasing IL-12 production [2,40–45]. We hypothesized that the beneficial effects of thiazolidinediones (TZDs) in treating type 2 diabetes may be explained partly by the down-regulation of islet autoimmunity in these patients. Our data suggest that this may indeed be one mechanism of action of the TZDs in type 2 diabetes.
We therefore propose that part of the clinical efficacy of rosiglitazone therapy on beta cell function in autoimmune T2DM patients results from the immunosuppressive effects on the islet-specific autoreactive T cell responses and cytokine (IL-12 and IFN-γ) production and the up-regulation of adiponectin. Thus, assessment of islet T cell autoimmunity may be important to determine whether phenotypic T2DM patients might benefit from treatment with rosiglitazone or other anti-inflammatory medications capable of suppressing islet-specific T cell autoimmunity.
Acknowledgments
This work was supported (in part) by the Medical Research Service of the Department of Veterans Affairs and GlaxoSmithKline. In addition, the following National Institutes of Health grants provided partial support: P01-DK053004, P30-DK017047. We would also like to thank Mrs Jessica Reichow for help in preparation of this manuscript.
Disclosure
This study was supported in part by an investigator-initiated grant from Glaxo-SmithKline. Dr Jerry Palmer has been a consultant for and been on the speakers’ bureau for Glaxo-SmithKline.
References
- 1.Cunard R, Ricote M, DiCampli D, et al. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol. 2002;1689:2795–2802. doi: 10.4049/jimmunol.168.6.2795. [DOI] [PubMed] [Google Scholar]
- 2.Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol. 2000;164:1364–1371. doi: 10.4049/jimmunol.164.3.1364. [DOI] [PubMed] [Google Scholar]
- 3.Faveeuw C, Fougeray S, Angeli V, et al. Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 2000;486:261–266. doi: 10.1016/s0014-5793(00)02319-x. [DOI] [PubMed] [Google Scholar]
- 4.Harris SG, Phipps RP. The nuclear receptor PPAR gamma is expressed by mouse T lymphocytes and PPAR gamma agonists induce apoptosis. Eur J Immunol. 2001;31:1098–1105. doi: 10.1002/1521-4141(200104)31:4<1098::aid-immu1098>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 5.Housley WJ, O'Conor CA, Nichols F, et al. PPAR gamma regulates retinoic acid-mediated DC induction of Tregs. J Leukoc Biol. 2009;86:293–301. doi: 10.1189/jlb.1208733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung SW, Kang BY, Kim TS. Inhibition of interleukin-4 production in CD4+ T cells by peroxisome proliferator-activated receptor gamma (PPAR-gamma) ligands: involvement of physical association between PPAR-gamma and the nuclear factor of activated T cells transcription factor. Mol Pharmacol. 2003;64:1169–1179. doi: 10.1124/mol.64.5.1169. [DOI] [PubMed] [Google Scholar]
- 7.Yang XY, Wang LH, Chen T, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4544. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]
- 8.Lei J, Hasegawa H, Matsumoto T, Yasukawa M. Peroxisome proliferator-activated receptor α and γ agonists together with TGFβ convert human CD4+CD25– T cells into functional Foxp3+ regulatory T cells. J Immunol. 2010;185:7186–7198. doi: 10.4049/jimmunol.1001437. [DOI] [PubMed] [Google Scholar]
- 9.Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and independent mechanisms. J Immunol. 2007;178:4129–4135. doi: 10.4049/jimmunol.178.7.4129. [DOI] [PubMed] [Google Scholar]
- 10.Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol. 2007;178:2940–2949. doi: 10.4049/jimmunol.178.5.2940. [DOI] [PubMed] [Google Scholar]
- 11.Cipolletta D, Feuerer M, Am L, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bassaganya-Riera J, Reynolds K, Martino-Catt S, et al. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gasteroenterology. 2004;127:777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 13.Beales PE, Liddi R, Giorgini AE, et al. Troglitazone prevents insulin dependent diabetes in the non-obese diabetic mouse. Eur J Pharmacol. 1998;357:221–225. doi: 10.1016/s0014-2999(98)00574-3. [DOI] [PubMed] [Google Scholar]
- 14.Diab A, Deng C, Smith JD, et al. Peroxisome proliferator-activated receptor-γ agonist 15 –deoxy-Δ12,14-prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- 15.Feinstein DL, Galea E, Gavrilyuk V, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 16.Mueller C, Weaver V, Vanden Heuvel JP, August A, Cantorna MT. Peroxisome proliferator-activated receptor gamma ligands attenuate immunological symptoms of experimental allergic asthma. Arch Biochem Biophys. 2003;418:186–196. doi: 10.1016/j.abb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Woerly G, Honda K, Loyens M, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med. 2003;198:411–421. doi: 10.1084/jem.20021384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nino M, Iwabuchi K, Kikuchi S, et al. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor γ. J Neuroimmunol. 2001;116:40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 19.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-γ agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 20.Dubuquoy L, Rousseaux C, Thuru X, et al. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55:1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pershadsingh HA. Peroxisome proliferator-activated receptor-gamma: therapeutic target for diseases beyond diabetes: quo vadis? Exp Opin Investig Drugs. 2004;13:215–228. doi: 10.1517/13543784.13.3.215. [DOI] [PubMed] [Google Scholar]
- 22.Drew PD, Xu J, Racke MK. PPAR-gamma: therapeutic potential for multiple sclerosis. PPAR Res. 2008;2008:627463–627472. doi: 10.1155/2008/627463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt S, Moric E, Schmidt M, Sastre M, Feinstein DL, Heneka MT. Anti-inflammatory and anti-proliferative actions of PPAR-gamma agonists on T lymphocyte derived from MS patients. J Leukoc Biol. 2004;75:478–485. doi: 10.1189/jlb.0803402. [DOI] [PubMed] [Google Scholar]
- 24.Ehses JA, Ellingsgaard H, Boni-Schnetzler M, Donath MY. Pancreatic islet inflammation in type 2 diabetes: from α and β cell compensation to dysfunction. Arch Physiol Biochem. 2009;115:240–247. doi: 10.1080/13813450903025879. [DOI] [PubMed] [Google Scholar]
- 25.Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes. Diabetes Care. 2008;31:S161–164. doi: 10.2337/dc08-s243. [DOI] [PubMed] [Google Scholar]
- 26.Richardson SJ, Wilcox A, Bone AJ, Foulis AK, Morgan NG. Islet-associated macrophages in type 2 diabetes. Diabetologia. 2009;52:1686–1688. doi: 10.1007/s00125-009-1410-z. [DOI] [PubMed] [Google Scholar]
- 27.Brooks-Worrell B, Palmer JP. Immunology in the clinic review series; focus on metabolic diseases: development of islet autoimmune disease in type 2 diabetes patients: potential sequelae of chronic inflammation. Clin Exp Immunol. 2012;167:40–46. doi: 10.1111/j.1365-2249.2011.04501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks-Worrell B, Narla R, Palmer JP. Biomarkers and immune-modulating therapies for Type 2 diabetes. Trends Immunol. 2012;33:546–553. doi: 10.1016/j.it.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Brooks-Worrell B, Juneja R, Minokadeh A, Greenbaum CJ, Palmer JP. Cellular immune response to human islet proteins in antibody-positive type 2 diabetic patients. Diabetes. 1999;48:983–988. doi: 10.2337/diabetes.48.5.983. [DOI] [PubMed] [Google Scholar]
- 30.Mayer A, Fabien N, Gutowski MC, et al. Contrasting cellular and humoral autoimmunity associated with latent autoimmune diabetes in adults. Eur J Endocrin. 2007;157:53–61. doi: 10.1530/EJE-07-0060. [DOI] [PubMed] [Google Scholar]
- 31.Brooks-Worrell B, Reichow JL, Goel A, Ismail H, Palmer JP. Identification of autoantibody negative autoimmune type 2 diabetes patients. Diabetes Care. 2011;34:168–173. doi: 10.2337/dc10-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goel A, Chiu H, Felton J, Palmer JP, Brooks-Worrell B. T cell responses to islet antigens improves detection of autoimmune diabetes and identifies patients with more severe β-cell lesions in phenotypic type 2 diabetes. Diabetes. 2007;56:2110–2115. doi: 10.2337/db06-0552. [DOI] [PubMed] [Google Scholar]
- 33.Miyaaki Y, DeFronzo RA. Rosiglitazone and pioglitazone similarly improve insulin sensitivity and secretion, glucose tolerance and adipocytokines in type 2 diabetic patients. Diabetes Obes Metab. 2008;10:1204–1211. doi: 10.1111/j.1463-1326.2008.00880.x. [DOI] [PubMed] [Google Scholar]
- 34.Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 35.Brooks-Worrell B, Starkebaum GA, Greenbaum C, Palmer JP. Peripheral blood mononuclear cells of insulin-dependent diabetic patients respond to multiple islet cell proteins. J Immunol. 1996;157:5668–5674. [PubMed] [Google Scholar]
- 36.Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the pre-clinical period of human type 1 diabetes. J Immunol. 2001;166:5265–5270. doi: 10.4049/jimmunol.166.8.5265. [DOI] [PubMed] [Google Scholar]
- 37.Seyfert-Margolis V, Gisler TD, Asare AL, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes. 2006;55:2588–2594. doi: 10.2337/db05-1378. [DOI] [PubMed] [Google Scholar]
- 38.Herold KC, Brooks-Worrell B, Palmer J, et al. Validity and reproducibility of measurement of islet autoreactivity by T cell assays in subjects with early type 1 diabetes. Diabetes Care. 2009;58:2588–2595. doi: 10.2337/db09-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks-Worrell B, Warsen A, Palmer JP. Improved T cell assay for identification of Type 1 diabetes patients. J Immunol Methods. 2009;344:79–83. doi: 10.1016/j.jim.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, Drew PD. Peroxisome proliferator-activated receptor-g agonists suppress the production of IL-12 family cytokines by activated glia. J Immunol. 2007;178:1904–1913. doi: 10.4049/jimmunol.178.3.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 42.Manetti R, Parronchi P, Giudizi MG, et al. Natural killer cell stimulatory factor (interleukin-12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4 producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Windhagen A, Newcombe J, Dangond F, et al. Expression of co-stimulatory molecules B7-1 (CD80), B-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995;182:1985–1996. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Comabella M, Balashov K, Issaadeh S, Smith D, Weiner HL, Khoury SJ. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest. 1998;102:671–678. doi: 10.1172/JCI3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicoletti F, Patti F, Cocuzza C, et al. Elevated serum levels of interleukin-12 in chronic progressive multiple sclerosis. J Neuroimmunol. 1996;70:87–90. doi: 10.1016/s0165-5728(96)00101-4. [DOI] [PubMed] [Google Scholar]