

Abstract
Common variable immunodeficiency (CVID) is the most common severe primary immunodeficiency, but the pathology of this condition is poorly understood. CVID involves a defect in the production of immunoglobulin from B cells, with a subsequent predisposition to infections. Approximately 10–20% of cases are inherited, but even in families with a genetic defect the penetrance is far from complete. A classification system for CVID has been suggested (EUROclass) based on B cell immunophenotyping, but it has not been shown that altered B cell immunophenotype is not a consequence of the complications and treatment of CVID. This study compares the EUROclass B cell immunophenotype of CVID patients (n = 30) with suitable disease controls with bronchiectasis (n = 11), granulomatous disease (Crohn's disease) (n = 9) and neurological patients on immunoglobulin treatment (n = 6). The results of this study correlate with previous literature, that alterations in B cell immunophenotype are associated strongly with CVID. Interestingly, three of the 11 bronchiectasis patients without known immunodeficiency had an altered B cell immunophenotype, suggesting the possibility of undiagnosed immunodeficiency, or that bronchiectasis may cause a secondary alteration in B cell immunophenotype. This study showed a significant difference in B cell immunophenotype between CVID patients compared to disease control groups of granulomatous disease and immunoglobulin treatment. This suggests that granulomatous disease (in Crohn's disease) and immunoglobulin treatment (for chronic neurological conditions) are not causal of an altered B cell immunophenotype in these control populations.
Keywords: B cell immunophenotyping, bronchiectasis, common variable immunodeficiency, granulomatous disease, immunoglobulin
Introduction
Common variable immunodeficiency (CVID) is the most common severe primary immunodeficiency [1], with an incidence of between one in 10 000 and one in 50 000 [2]. It is characterized by low immunoglobulins (IgG, IgA and IgM) and recurrent infections. Intrinsic or extrinsic defects in the early and late stages of B cell differentiation may account for the impaired immunoglobulin synthesis in CVID. B cells mature and migrate from the bone marrow to the periphery and acquire different surface markers, such as CD19, IgM, IgD, CD27, CD21 and CD38. T cell abnormalities and defects in monocytes and plasmacytoid dendritic cells also play a part in B cell development [3–11]. B cell surface markers can be used as a classification system for CVID. The EUROclass B cell immunophenotypical classification 12 measures the percentage of B cells (CD19+), the percentage of B cells of class-switched memory phenotype (CD19+, CD27+, IgM−, IgD−), the percentage B cells of transitional type (CD19+, CD38++, IgM+) and the percentage of B cells expressing low levels of CD21 in peripheral blood.
The EUROclass classification of the B cell phenotype provides a basis for defining and comparing CVID patients. The reduced class-switched memory B cell percentage in CVID patients is associated with complications [13,14], and a CD21 low B cell expansion in CVID patients is associated with autoimmunity [15–17]. Published trials have compared CVID patients with and without complications, but have not included non-CVID disease controls. Thus, despite the routine use of the EUROclass B cell phenotype in the evaluation of antibody deficiency, it is unclear if alterations in B cell marker expression are associated with CVID, its complications (e.g. bronchiectasis, granulomatous disease) and treatment (e.g. immunoglobulin replacement), or whether bronchiectasis, immunoglobulin treatment or granulomatous disease may cause abnormalities independently of the presence of CVID. Intravenous immunoglobulin has been shown to reduce the numbers of monocytes in peripheral blood 4 h after administration [18], indicating that immunoglobulins do have effects on immune cells, and raising the possibility that immunoglobulin could have direct effects on B cell immunophenotype.
B cell types
Class-switched memory B cells (CD19+CD27+IgM−IgD−) are a sensitive marker for sufficient germinal centre function, and their numbers are often reduced in CVID [19]. In CVID there is a correlation between IgG antibody response to pneumococcal polysaccharide and percentage of class-switched memory B cells [13]. Some studies suggest a clinical correlation in CVID patients between persistent infection, requirement for antibiotic treatment and reduced class-switched memory B cells [20]. Reduced class-switched memory B cells are correlated with complications of CVID, such as bronchiectasis, granulomas, autoimmunity and splenomegaly [13,14]. The gating and staining pattern by the EUROclass method unfortunately includes plasmablasts within the class-switched memory B cell gate (which are also CD19+CD27+IgM−IgD−) [normal range (including plasmablasts) = 6·5–29·1% of B cells [14], but normal levels of plasmablasts in blood are low (0·4–3·6% of B cells [21]), so the majority of counted cells in this gate are switched memory B cells.
Materials and methods
We conducted a case–control study comparing CVID patient's B cell immunophenotype with that of disease controls without CVID (bronchiectasis without known hypogammaglobulinaemia, Crohn's (granulomatous) disease or neurological patients receiving current long-term immunoglobulin treatment for immune-mediated neuropathies). Primary end-points were differences in percentages of switched memory B cells between patient groups.
Definitions
CVID patients were defined according to ESID criteria [22]: patients with an IgG level two standard deviations below the mean, evidence of increased susceptibility to infection (recurrent infection and/or poor response to test vaccination) and exclusion of secondary causes of hypogammaglobulinaemia. Bronchiectasis was diagnosed on high-resolution computed tomography by consultant radiologists at Barts Health National Health Service (NHS) Trust. Granulomatous disease control patients had a diagnosis of Crohn's disease and activity was scored using the Harvey Bradshaw Index [23].
Patients
Patients were recruited from immunology, respiratory and gastroenterology specialist clinics within Barts Health NHS Trust. Patients were recruited sequentially from clinics attended by members of the research team and gave written consent for inclusion in the study.
The groups within the study were as follows: healthy controls; CVID patients: with no bronchiectasis or granulomatous disease and not on immunoglobulin (CVID without complications), with bronchiectasis (CVID and bronchiectasis), on immunoglobulin (CVID on immunoglobulin), with granulomatous disease (CVID and granulomatous disease), CVID patients could be a member of more than one subgroup; bronchiectasis patients without CVID (bronchiectasis controls); neurology patients on long-term immunoglobulin without CVID, e.g. diagnoses: chronic inflammatory demyelinating polyneuropathy, multi-focal acquired demyelinating sensory and motor neuropathy (MADSAM) (immunoglobulin controls); and Crohn's disease patients without CVID (granulomatous controls).
Analysis of samples
Samples were processed according to our laboratory standard EUROclass protocol for B cell immunophenotyping and analysed by flow cytometry (Appendix 1) [24]. Samples were collected at routine medical visits, or at attendance for in-hospital immunoglobulin treatment. For neurology and CVID patients receiving immunoglobulin treatment the timing of blood collection varied from immediately before the infusion to several days after.
Statistics
Switched memory B cell numbers were compared between all CVID groups and disease control groups using the Wilcoxon–Mann–Whitney test.
Ethics
Ethical approval was attained through the Moorfields and Whittington Research Ethics Committee (REC number 10/H0721/3) and was approved within Barts and the London NHS Trust. The trial was registered with clinicaltrials.gov; registration number NCT01196702.
Results
Numbers of patients and matched disease/healthy controls are shown in Table 1. Age range (21–78 versus 28–87 years) and sex (male/female; 13/17 and 18/19) of participants was similar for CVID and control groups.
Table 1.
Patient [common variable immunodeficiency (CVID)] and control (non-CVID) group numbers and percentage in each group of studied subjects with less than 2% class-switched memory B cells (CD19+CD27+IgM–IgD− B cells)
| Number of subjects studied (in brackets: percentage of studied subjects with switched memory B cells < 2% total B cells) | ||
|---|---|---|
| CVID | Controls | |
| No complication or immunoglobulin treatment | 6 (17%) | 11* (0%) |
| Bronchiectasis | 11 (54%) | 11† (27%) |
| Immunoglobulin treatment | 23 (56%) | 6‡ (0%) |
| Granulomatous disease | 7 (86%) | 9§ (0%) |
| Total | 30 (47%) | 37 (8%) |
Healthy normal controls.
Bronchiectasis controls.
Neurology patients on long-term immunoglobulin – immunoglobulin controls.
Crohn's disease patients – granulomatous controls.
Three CVID patients on immunoglobulin (which included two with bronchiectasis and one with granulomatous disease) were excluded from analysis due to a lack of B cells (i.e. < 1% lymphocytes were B cells).
Comparison of switched memory B cell percentages between CVID and control patients
No healthy controls, patients on immunoglobulin treatment for neurological reasons (immunoglobulin controls) or Crohn's disease patients (granulomatous controls) had abnormal switched memory B cells, whereas 47% (14 of 30) of CVID patients had low switched memory B cell numbers. Three of 11 (27%) of bronchiectasis patients had low switched memory B cells.
None of the disease control groups (bronchiectasis without CVID, Crohn's disease or immunoglobulin treatment for neurological reasons) had statistically significant differences in switched memory B cell percentages from normal controls [bronchiectasis without CVID (P = 0·123), neurological immunoglobulin treatment (P = 0·725) and Crohn's disease (P = 0·648)]. Similarly, uncomplicated, untreated CVID patients (‘CVID without complications’) showed a trend to reduction in switched memory B cell numbers, but this did not reach significance (P = 0·087). In contrast, patient groups with CVID on immunoglobulin or with complications from CVID (granulomatous disease or bronchiectasis) had significantly reduced switched memory B cell numbers (see Fig. 1).
Fig. 1.
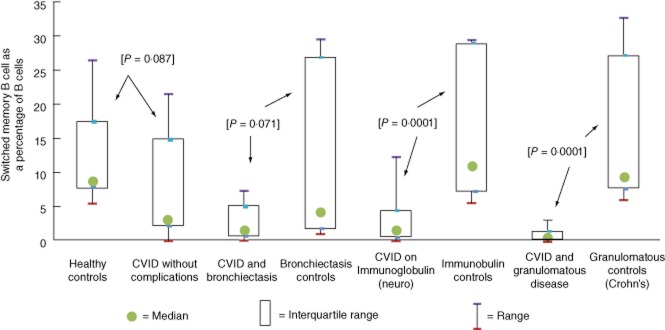
Comparison of switched memory B cells as a percentage of B cells between cases and controls, with P-value in square brackets comparing cases and control groups (arrowed) by Wilcoxon–Mann–Whitney test.
There was a significant reduction in switched memory B cell numbers for CVID patients on immunoglobulin or with granulomatous disease compared with individual disease control groups. The apparent reduction in switched memory B cell numbers for bronchiectasis patients with CVID did not reach significance on this analysis due to three of 11 bronchiectasis controls having unexpectedly low switched memory B cell numbers.
Similarly, when each factor was analysed by an adjusted linear regression model, CVID and immunoglobulin [which was associated closely with CVID, owing to low numbers in the immunoglobulin disease control group (n = 6)] were associated with low switched memory B cell numbers, while granulomatous disease and bronchiectasis were not (data not shown).
Discussion
Switched memory B cell percentages
In keeping with the EUROclass study 12 there is a reduction in switched memory B cell percentage in patients with CVID, compared with controls. In contrast to the other CVID subgroups, this reduction did not reach significance for uncomplicated CVID patients not on immunoglobulin (one of six CVID versus none of 11 controls), probably because of low numbers. Comparisons with relevant disease control groups suggested that the abnormalities in B cell immunophenotype are CVID-associated, rather than associated with immunoglobulin treatment or granulomatous disease. The choice of Crohn's disease as a granulomatous control may not be ideal, especially as only one of nine Crohn's patients had active disease. Other granulomatous disorders, such as newly diagnosed tuberculosis or active sarcoid, may be a better choice. Of note, five of nine Crohn's patients were taking long-term azathioprine, without discernable effect on the B cell phenotype. Mouse data suggest that azathioprine does alter B cell number and function [25]. If the lack of effect of azathioprine on B cell phenotype in humans is confirmed, this could be useful in distinguishing primary from secondary antibody deficiencies in patients who have received azathioprine for autoimmunity prior to the diagnosis of hypogammaglobulinaemia.
Of the bronchiectasis patients without CVID, 27% had an abnormally reduced switched memory B cell population on EUROclass (i.e. < 2% with normal levels being above approximately 6–7% of B cells [14]). This profound reduction was not found in any of the other control populations. The incidence of hypogammaglobulinaemia in patients with bronchiectasis is thought to be low although some, but not all, series suggest that undiagnosed, usually subtle, antibody deficiency may be present. In one report, 13 of 103 adult patients with bronchiectasis had reduced IgG3 levels without panhypogammaglobulinaemia [26]. Another study found that four of 56 bronchiectasis patients had either a low IgG or poor response to test vaccination [27]. None of the studies have measured switched memory B cell numbers. The incidence of hypogammaglobulinaemia is believed to be low in the other disease control groups, although published data are lacking.
The low levels of switched memory B cells in bronchiectasis control patients could be attributable either to alterations in switched memory B cells secondary to bronchiectasis, or undiagnosed immunodeficiency in some bronchiectasis patients. IgG levels on these three bronchiectasis patients were within the normal range, and one of them had specific antibody levels to pneumococcus, Haemophilus influenzae B and tetanus measured, which were normal. Test vaccinations have not been administered. Of note, one bronchiectasis control patient in this study was shown subsequently to have low total IgG, probably secondary to treatment for an anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis and ongoing immunosuppression. Another bronchiectasis patient had a normal total IgG, but low pneumococcal-specific antibody levels (< 0·35ug/ml) [28] to seven of 13 serotypes. Both of these patients had a normal EUROclass B cell immunophenotype. It will be necessary to test more bronchiectasis patients and exclude antibody deficiency more rigorously than was possible in this study to understand fully the relevance of these findings.
Conclusions
Patients with bronchiectasis should be investigated for antibody deficiency. Neither the granulomatous control group nor the immunoglobulin control group had abnormal B cell immunophenotypes, providing preliminary support for the hypothesis that granulomatous disease in Crohn's disease and immunoglobulin for neurological conditions do not alter the B cell immunophenotype. Azathioprine did not significantly alter EUROclass B cell immunophenotype for the five patients studied.
Acknowledgments
We would like to acknowledge the patients who participated, the clinicians at Barts and The London who helped me in recruiting patients who included Dr J. Lindsay, Dr L. Langmead (Consultant Gastroenterologists), Dr V. White, Dr L. Kuitert (Consultant Respiratory Physicians) and Dr B. Turner (Consultant Neurologist). We would also like to acknowledge Mr Tianqiang Zhang for statistical advice. Finally, Dr A. Manson and Dr A. Polycarpou were extremely helpful in assisting with some clinical data on patients. Bio Products Laboratory funded the project, with a grant made to the Autoantibodies Fund of the Special Trustees of Barts and The London.
Appendix 1
Flow plots of normal and abnormal immunophenotypes
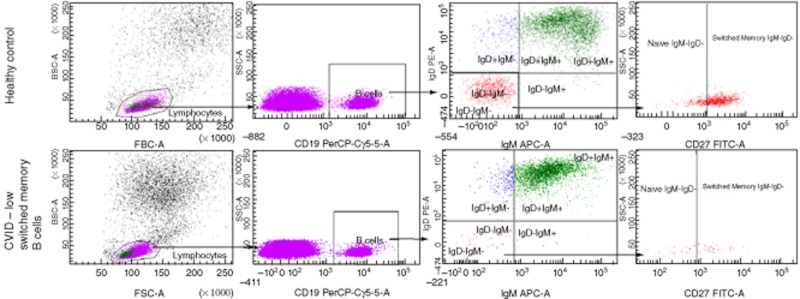
Disclosure
Dr Longhurst reports that she and members of her department have received funding to attend conferences and other educational events, donations to her departmental fund and/or have participated in clinical trials with the following immunoglobulin manufacturers: BPL, CSL Behring, Octapharma, Baxter and Grifols. She has been a member of the medical advisory panel for Baxter and CSL. This project was funded by Bio Products Laboratory with an educational grant made to the Autoantibodies Fund of the Special Trustees of Barts and The London.
References
- 1.Conley ME, Notarangelo LD, Etzioni A representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Diagnostic criteria for primary immunodeficiencies. Clin Immunol. 1999;93:190–197. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 2.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 3.Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30:2516–2520. doi: 10.1002/1521-4141(200009)30:9<2516::AID-IMMU2516>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Taubenheim N, von Hornung M, Durandy A, et al. Defined blocks in terminal plasma cell differentiation of common variable immunodeficiency patients. J Immunol. 2005;175:5498–5503. doi: 10.4049/jimmunol.175.8.5498. [DOI] [PubMed] [Google Scholar]
- 5.Stagg AJ, Funauchi M, Knight SC, Webster AD, Farrant J. Failure in antigen responses by T cells from patients with common variable immunodeficiency (CVID) Clin Exp Immunol. 1994;96:48–53. doi: 10.1111/j.1365-2249.1994.tb06228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.North ME, Webster AD, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28− (‘suppressor’) CD8+ subsets. Clin Exp Immunol. 1998;111:70–75. doi: 10.1046/j.1365-2249.1998.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aspalter RM, Sewell WA, Dolman K, Farrant J, Webster AD. Deficiency in circulating natural killer (NK) cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia. Clin Exp Immunol. 2000;121:506–514. doi: 10.1046/j.1365-2249.2000.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giovannetti A, Pierdominici M, Mazzetta F, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- 9.Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol. 2000;164:488–494. doi: 10.4049/jimmunol.164.1.488. [DOI] [PubMed] [Google Scholar]
- 10.Bayry J, Lacroix-Desmazes S, Kazatchkine MD, et al. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood. 2004;104:2441–2443. doi: 10.1182/blood-2004-04-1325. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin Immunol. 2005;115:147–153. doi: 10.1016/j.clim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–321. doi: 10.1016/j.clim.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alachkar H, Taubenheim N, Haeney MR, Durandy A, Arkwright PD. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency. Clin Immunol. 2006;120:310–318. doi: 10.1016/j.clim.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(–)IgD(–)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–1551. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 16.Warnatz K, Wehr C, Drager R, et al. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;206:502–513. doi: 10.1078/0171-2985-00198. [DOI] [PubMed] [Google Scholar]
- 17.Boileau J, Mouillot G, Gérard L, et al. Autoimmunity in common variable immunodeficiency: correlation with lymphocyte phenotype in the French DEFI study. J Autoimmun. 2011;36:25–32. doi: 10.1016/j.jaut.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Siedlar M, et al. Preparations of intravenous immunoglobulins diminish the number and proinflammatory response of CD14+CD16++ monocytes in common variable immunodeficiency (CVID) patients. Clin Immunol. 2011;139:122–132. doi: 10.1016/j.clim.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 19.MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 20.Oksenhendler E, Gerard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- 21.Warnatz K, Schlesier M. Flow cytometric phenotyping of common variable immunodeficiency. Cytometry B Clin Cytom. 2008;74:261–271. doi: 10.1002/cyto.b.20432. [DOI] [PubMed] [Google Scholar]
- 22.European Society for Immunodeficiencies (ESID) Diagnostic criteria: common variable immunodeficiencyLast updated September 2005. Available at: http://www.esid.org/clinical-diagnostic-criteria-for-pid-73-0#Q3 (accessed August 2012)
- 23.Harvey RF, Bradshaw JM. A simple index of Crohn's disease activity. Lancet. 1980;1:514. doi: 10.1016/s0140-6736(80)92767-1. [DOI] [PubMed] [Google Scholar]
- 24.Ferry BL, Jones J, Bateman EA, et al. Measurement of peripheral B cell subpopulations in common variable immunodeficiency (CVID) using a whole blood method. Clin Exp Immunol. 2005;140:532–539. doi: 10.1111/j.1365-2249.2005.02793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salinas-Carmona MC, Perez LI, Galan K, Vazquez AV. Immunosuppressive drugs have different effect on B lymphocyte subsets and IgM antibody production in immunized BALB/c mice. Autoimmunity. 2009;42:537–544. doi: 10.1080/08916930903019119. [DOI] [PubMed] [Google Scholar]
- 26.King PT, Hutchinson P, et al. Assessing immune function in adult bronchiectasis. Clin Exp Immunol. 2006;144:440–446. doi: 10.1111/j.1365-2249.2006.03091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stead A, Douglas JG, Broadfoot CJ, Kaminski ER, Herriot R. Humoral immunity and bronchiectasis. Clin Exp Immunol. 2002;130:325–330. doi: 10.1046/j.1365-2249.2002.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jodar L, et al. Serological criteria for evaluation and licensure of new pneumococcal conjugate vaccine formulations for use in infants. Vaccine. 2003;21:3265–3272. doi: 10.1016/s0264-410x(03)00230-5. [DOI] [PubMed] [Google Scholar]