

Abstract
Cholesterol gallstones are associated with slow intestinal transit in humans as well as in animal models, but the molecular mechanism is unknown. We investigated in C57L/J mice whether the components of a lithogenic diet (LD; 1.0% cholesterol, 0.5% cholic acid and 17% triglycerides), as well as distal intestinal infection with Helicobacter hepaticus, influence small intestinal transit time. By quantifying the distribution of 3H-sitostanol along the length of the small intestine following intraduodenal instillation, we observed that, in both sexes, the geometric centre (dimensionless) was retarded significantly (P < 0.05) by LD but not slowed further by helicobacter infection (males, 9.4 ± 0.5 (uninfected), 9.6 ± 0.5 (infected) on LD compared with 12.5 ± 0.4 and 11.4 ± 0.5 on chow). The effect of the LD was reproduced only by the binary combination of cholesterol and cholic acid. We inferred that the LD-induced cholesterol enrichment of the sarcolemmae of intestinal smooth muscle cells produced hypomotility from signal-transduction decoupling of cholecystokinin (CCK), a physiological agonist for small intestinal propulsion in mice. Treatment with ezetimibe in an amount sufficient to block intestinal cholesterol absorption caused small intestinal transit time to return to normal. In most cholesterol gallstone-prone humans, lithogenic bile carries large quantities of hepatic cholesterol into the upper small intestine continuously, thereby reproducing this dietary effect in mice. Intestinal hypomotility promotes cholelithogenesis by augmenting formation of deoxycholate, a pro-lithogenic secondary bile salt, and increasing the fraction of intestinal cholesterol absorbed.
Key points
Peristaltic function of the small intestine is compromised in cholelithogenic humans and in animal models of cholesterol gallstones.
In a mouse gallstone model fed a cholesterol- and cholic acid-enriched diet, we show that slowing of small intestinal transit is due to absorption of excess cholesterol molecules from the upper small intestine followed by their incorporation into sarcolemmal membranes of smooth muscle cells.
Blocking cholesterol absorption with ezetimibe (Zetia), an inhibitor of intestinal sterol transport, prevents cholesterol enrichment of sarcolemmal membranes and normalizes the motility disorder.
Although the primary source of intestinal cholesterol in mice is the lithogenic diet, in most cholesterol gallstone-prone humans, the small intestine is flooded continuously with an abundance of liver-secreted cholesterol molecules via bile.
Our findings imply that small intestinal hypomotility will amplify the cholelithogenic state because of hyperabsorption of cholesterol and ‘secondary’ bile salt synthesis by the gut's anaerobic microbiota.
Introduction
Cholesterol cholelithogenesis is characterized by cholesterol supersaturation of bile, principally from hepatic hypersecretion of cholesterol, which results in gallbladder hypomotility and rapid cholesterol nucleation and phase transitions in gallbladder bile (Paigen & Carey, 2002). Hypomotility of the small and possibly large intestines is also part of this ‘syndrome’ and may augment the lithogenic process by production of additional secondary bile salts, especially deoxycholate, and increasing the fraction of intestinal cholesterol absorbed (van Erpecum & van Berge-Henegouwen, 1999; Dowling, 2000; Heaton, 2000). Two independent groups (Heaton et al. 1993; Azzaroli et al. 1999) estimated that whole gut transit times are nearly 30% longer in normal-weight women with cholesterol gallstones compared with non-stone formers. In addition, acromegalics treated with octreotide display prolonged intestinal transit times, elevated deoxycholate conjugates and higher cholesterol saturation indices (CSIs) in gallbladder bile plus rapid cholesterol crystallization (Dowling et al. 1992; Veysey et al. 2001; Thomas et al. 2005); however, cisapride, a prokinetic agent, counteracted this effect on intestinal transit (Veysey et al. 2001). Furthermore, cholesterol-rich diets prolong small intestinal transit times in hamsters (Fan et al. 2007) and ground squirrels (Xu et al. 1996). These diets lead to a two-fold increase in the cholesterol saturation of bile in these models, which promotes cholesterol gallstone formation, a trend reversible with the prokinetic agent erythromycin (Xu et al. 1998). However, neither in humans nor in animal models has the molecular mechanism for prolongation of small intestinal transit in the lithogenic state been investigated.
Most recent research on pathogenesis of cholesterol gallstones has utilized inbred mice (Wang et al. 1997, 1999b; Lammert et al. 2001; Paigen & Carey, 2002; Lyons & Wittenburg, 2006). The gallstone-prone C57L mouse (Khanuja et al. 1995; Lammert et al. 2001; Paigen & Carey, 2002; Lyons & Wittenburg, 2006) develops gallstones when fed a lithogenic diet (LD) and does so much more frequently when infected with certain strains of enterohepatic Helicobacter species (Maurer et al. 2005), but not the gastrotropic H. pylori (Maurer et al. 2006). The standard LD contains 1.0% cholesterol, 0.5% cholic acid and 17% triglycerides and induces a significant increase in the proportion of biliary deoxycholate conjugates (from cholic acid), as an enlarged pool of hepatic conjugates of dietary cholate replaces the endogenous muricholates. This alteration in bile acid composition promotes cholesterol hyperabsorption, increases biliary cholesterol secretion and, concomitantly with cholesterol supersaturation, induces a solid crystalline phase change in bile and eventually cholesterol gallstone formation (Wang et al. 1999a, 2009).
In this study we reasoned that small intestinal transit time may be affected by one or more components of the LD or, alternatively, by enterohepatic helicobacter infection. Since it was established earlier that increased cholesterol incorporation by the sarcolemmae of smooth muscle cells in the gallbladder impaired its contractile response to a CCK agonist (Yu et al. 1996, 1998; Xiao et al. 1999), we hypothesised that cholesterol molecules absorbed from the LD and similarly integrated by small intestinal sarcolemmae might slow intestinal transit in C57L mice. We demonstrate here that the LD, and not H. hepaticus infection, significantly contributes to slowing of small intestinal transit and that increased absorption of cholesterol molecules, with their incorporation into the lipid structure of sarcolemmae, is the critical factor responsible for this observation.
Moreover, by ablating intestinal cholesterol absorption with ezetimibe (EZ; Zetia), a drug that inhibits the intestinal NPC1L1 sterol transporter, LD-induced slowing of small intestinal transit was abolished, and sarcolemmal lipid composition returned to normal. Analogous to studies of human gallbladders (Xiao et al. 1999), cholesterol enrichment of upper small intestinal sarcolemmae blocks signal-transduction coupling with CCK, which, in addition to inducing gallbladder contraction in health, is a physiological agonist for the caudad propulsive activity of the small intestine in vivo (Wang et al. 2004).
Methods
Animal sources and husbandry
Animal protocols were reviewed and approved by the Massachusetts Institute of Technology (MIT) and Harvard Medical School Standing Committees on Animals. The authors have also read, and the experiments comply with, the policies and regulations of The Journal of Physiology described by Drummond (2009). Three- to five-week-old male and female C57L mice, free of Helicobacter spp. infection, were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in polycarbonate micro-isolator cages under specific pathogen-free conditions (free of Helicobacter spp., Citrobacter rodentium, Salmonella spp., endo-parasites, ectoparasites, and all known murine viral pathogens) in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. Mouse rooms were maintained at constant temperature (22°C) and humidity (40–60%) on a 12:12 h regular light–dark cycle. Mice were fed food and water ad libitum.
Experimental diets
Male C57L/J mice were fed a standard chow diet (Purina 5001; PMI Feeds, Richmond, IN, USA) until 8 weeks of age, after which they were either maintained on chow or changed to an experimental diet. The latter included a standard LD (a balanced mouse diet replete with vitamins and minerals plus 1.0% cholesterol, 0.5% cholic acid and 17% triglycerides (Khanuja et al. 1995) prepared in the ‘diet kitchen’ of The Jackson Laboratory), a high-cholesterol diet (Purina 5001 containing 1.0% cholesterol; Harlan Teklad, Madison, WI, USA), a high-cholic acid diet (Purina 5001 containing 0.5% cholic acid; Harlan Teklad), and a high-cholesterol + high-cholic acid diet (Purina 5001 containing 1.0% cholesterol plus 0.5% cholic acid; Harlan Teklad). In addition, certain experimental diets (chow, high cholesterol and lithogenic) were supplemented with 60 mg kg−1 of EZ (Schering-Plough, Kenilworth, NJ, USA), a specific inhibitor of NPC1L1, the intestinal transporter responsible for sterol absorption (Altmann et al. 2004), to yield a dose of approximately 8 mg (kg body weight (BW))−1 day−1. This level of drug causes at least a tenfold decrease in intestinal cholesterol absorption in mice (Wang et al. 2008). Mice were fed LD for 8 days, 14 days or 8 weeks; all other diets were fed for 8 weeks.
Helicobacter hepaticus infection
Designated mice were infected with Helicobacter hepaticus (strain 3BI) with appropriate uninfected controls. H. hepaticus was grown under microaerobic conditions on blood agar plates at 37°C, as described previously (Maurer et al. 2005, 2006). Four-week-old mice were gavaged orally with the organisms three times on alternate days during the first week, and re-dosed once at 8 and 12 weeks. The dose employed per inoculum was approximately 1.5 × 108 to 3.0 × 108 organisms.
Measurement of small intestinal transit times
Small intestinal transit times were measured by a validated method for mice (Wang et al. 2001, 2003, 2004), with a single modification that the latency period between instillation of the 3H-sitostanol dissolved in medium-chain triglycerides (MCT) and killing was decreased from 30 to 20 min. On the first day, mice were weighed and anaesthetized using a vaporizer with 5% isoflurane in O2. For analgesia, a subcutaneous injection of flunixin (1 mg kg−1) was administered pre-operatively, post-operatively and again on the following morning. Laparotomy was performed aseptically with a 1 cm midline incision of the upper abdomen. A Polyethylene-10 catheter with 0.28 mm i.d. and 0.61 mm o.d., sterilized by ethylene oxide, was inserted into the duodenum 5 mm distal to the pylorus. The catheter, with its tip pointing away from the stomach, was secured with 6-0 polypropylene sutures. The other end of the catheter was externalized through the peritoneum, heat-sealed and implanted under the skin. The abdominal incision was closed in two layers with polypropylene sutures. Mice were allowed to recover for 48 h in individual cages, receiving a paste of ground diet mixed with water and water ad libitum. Mice were then fasted for 20 h with access to water only.
On the fourth day, mice were re-anaesthetized, and the implanted end of the duodenal catheter was re-exposed and cut open. 3H-sitostanol (2 μCi), dissolved in 100 μl MCT, was injected. After being transferred to individual cages, mice regained consciousness within 1–3 min. Exactly 20 min after the 3H-sitostanol injection, the animals were killed with CO2, the abdomen was opened, and ligatures were placed circumferentially around the intestine distal to the tip of the duodenal catheter and at the ileocecal junction. The stomach and small and large intestines were excised and removed. The small intestine was flash-frozen in liquid N2 and cut into 20 equal segments that were placed in vials containing 10 ml solvent consisting of chloroform:methanol (2:1, v/v) and stored at 4°C for 48–72 h. The stomach, including the short duodenal segment containing the catheter, and the large intestine were placed in individual vials. The vials were then vortex-mixed to achieve a uniform solution of 3H-sitostanol and centrifuged at 1000 g for 30 min. Aliquots (1 ml) were transferred to counting vials, the solvent was evaporated under reduced pressure at 25°C overnight, and 8 ml of scintillation fluid (Ecolite(+), MP Biomedicals, Solon, OH, USA) was added. Radioactivity was assayed using a scintillation counter. The stomach and large intestine, analysed in the same manner, did not show any appreciable radioactivity above background.
Results for small intestinal transit were quantified by two methods. The percentage of total radioactivity in each segment was determined and plotted, producing a distribution of radioactivity across the full length of the small intestine. In addition, the geometric centre of radioactivity, a dimensionless number representing intestinal transit function, was calculated as the sum of the fraction of 3H-sitostanol per segment times the segment number (Miller et al. 1981).
Preparation of proximal small intestine smooth muscle sarcolemmae and lipid analysis
A total of 36 male and 8 female C57L mice, 4- and 5-weeks old and free of Helicobacter spp. infection, were obtained from The Jackson Laboratory and fed a standard chow diet. The mice were allowed to reach 8 weeks of age, at which time they were either maintained on chow or fed one of two experimental diets for 14 days, the standard LD or LD + EZ, containing the maximum dose of EZ (approx. 60 mg kg−1 diet). On day 14, each mouse was fasted for 8 h to stimulate overnight feeding and then supplied with the same diet. The following morning, the mouse was weighed and then killed in a bell jar with an overdose of isoflurane in O2. A laparotomy was performed, and the entire small intestine from the pylorus to the ileocecal junction was excised, and all connective and adipose tissues were removed. The small intestine was cut in half and the distal part (ileum to ileocecal junction) discarded. The proximal small intestine was placed in a Petri dish of cold PBS, prepared according to Sambrook et al. (1989). A lateral incision was made along the length of the intestine to expose the inner mucosal layer. After emptying the intestine of its contents by gentle agitation in PBS, the tissue was weighed in a dry Petri dish. Tissues of two mice were pooled and placed in a 10 ml solution composed of 3 mm Na2EDTA dihydrate and 0.05 mm dithiothreitol in PBS for 45 min at 4°C, according to Whitehead et al. (1999). The wash solution was decanted, 10 ml of PBS was added, and the sample was vortex-mixed for 30 s to dislodge the mucosa. The supernatant containing the mucosal layer was discarded and the remainder rinsed in another 10 ml of PBS. This tissue, in 5 ml 0.25 m sucrose buffer (Medium A), prepared according to Rybal'chenko et al. (1984), was shredded with an Ultra Turrax T25 disperser employing two 15 s bursts at moderate-high speed (setting 5). The tissue was further homogenized in a Potter-Elvehjem homogenizer with a polytetrafluoroethylene pestle (Chen et al. 1999) and the homogenate placed in a thick-walled polycarbonate centrifuge tube and balanced. Preliminary centrifugation steps (Chen et al. 1999) were performed in a Beckman L8-80M Ultracentrifuge using a Beckman 70.1 Ti rotor and Medium A as the buffered solvent for pellet reconstitution (Rybal'chenko et al. 1984). Samples were separated over a stepwise sucrose density gradient (20%, 3 ml; 30%, 4 ml; 35%, 3 ml; 40%, 2 ml) at 100,000 g (SW 40 Ti rotor) for 3 h without braking during deceleration. Bands were separated, diluted in Medium A, and centrifuged at 150,000 g (Type 70.1 Ti rotor) for 30 min. The pellets were harvested, placed on ice and, depending on the pellet yield, reconstituted in 40–100 μl of 0.25 m sucrose in 5 mm Tris-HCl. The reconstituted pellets were stored separately in four 1 ml Eppendorf tubes at −80°C until thawed for analysis.
Total protein content and Western blotting
Total protein content was determined by the standard procedure for the Pierce bicinchoninic acid (BCA) total protein assay (Thermo Fisher Scientific, Rockford, IL, USA). Following the standard protocol for Western blotting, we employed ‘Caveolin-3 (C-2)’ (Santa Cruz Biotechnology, Santa Cruz, CA, USA) as a primary antibody against Caveolin-3 (Cav3), a protein intrinsic to sarcolemmae, to verify the membrane's presence and relative yield in the reconstituted pellets. The blots were visualized using HRP chemiluminescence, and bands were quantified as optical densities and normalized to total protein content using ImageJ (National Institutes of Health, Bethesda, MD, USA). The highest yield of sarcolemmae was found at the 20–30% sucrose interface of the gradient; therefore, both electron microscopy and the subsequent lipid assays were performed on pellets from this interface.
Electron microscopy
Isolated sarcolemmae from three samples were fixed briefly in 2.5% glutaraldehyde and 2% formaldehyde in 0.1 m Tris-HCl buffer. They were then gently pelletted and embedded in 2% SeaPrep Agarose (Lonza, Rockland, ME, USA) prepared in water, allowing the membranes to be equally distributed in the agarose, and then hardened. The solid agarose block was additionally fixed for 2 days in the same buffer, washed in water, fixed in 2% aqueous uranyl acetate, and then dehydrated through graded alcohols and propylene oxide. The block containing membranes was embedded in LX112 resin, sectioned with an UltracutE ultramicrotome, and imaged with a Jeol 1400 electron microscope.
Extraction of lipids and analyses
Lipids from the aqueous membrane samples were extracted into 2:1 chloroform:methanol following the method of Folch et al. (1957). This procedure removed all traces of inorganic phosphate left by the PBS washes following surgery. Using methods identical to those employed for gallbladder bile (see below), cholesterol and phospholipid contents were assayed and absolute concentrations were calculated.
Analysis of gallbladder bile
Immediately after killing, mouse bile was obtained by puncture and aspiration from the gallbladder. Total bile salts were assayed by the 3α-hydroxysteroid dehydrogenase method (Turley & Dietschy, 1978). Biliary phospholipids were measured as inorganic phosphorus (Bartlett, 1959) and, after extraction into hexane, biliary cholesterol was quantified by HPLC (Duncan et al. 1979; Vercaemst et al. 1989). CSIs were calculated according to critical tables (Carey, 1978).
Statistical analyses
Prism 4.0 software (GraphPad, San Diego, CA, USA) was utilized for statistical tests, including two-way ANOVA, one-way ANOVA with Tukey honestly significant difference (HSD) or Bonferroni post hoc tests, and two-tailed t tests. Data are presented as mean ± SEM, and P < 0.05 is considered a significant difference.
Results
Influence of lithogenic diet and Helicobacter hepaticus infection on small intestinal transit times
Figure 1A displays the per cent intestinal distribution of 3H-sitostanol in uninfected mice on a chow diet, with radioactivity peaking in segment 13. These results are essentially similar to those of mice on the chow diet infected with H. hepaticus (Fig. 1B). Figure 1C and D, respectively, show that the distribution of radioactivity in uninfected or infected mice fed LD is shifted toward proximal intestinal segments (i.e. slower transit) compared with uninfected or infected mice on the chow diet (Fig. 1A and B). Peak radioactivity occurs in segment 10 in uninfected mice, and in segment 8 in H. hepaticus-infected mice on LD. There were no differences in mouse weights and small intestinal lengths among all four groups (data not displayed).
Figure 1. Influence of lithogenic diet (LD) and helicobacter infection on small intestinal transit times in male C57L mice.
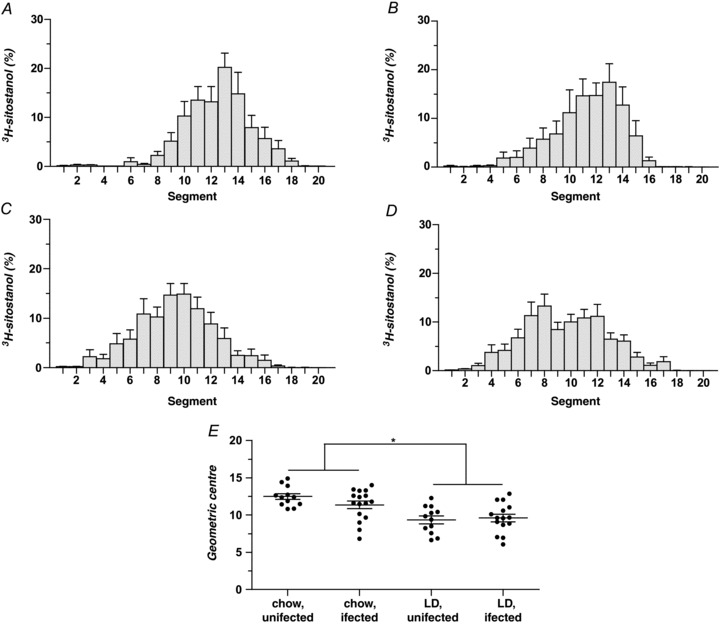
A–D, small intestinal transit is plotted as per cent distribution of the non-absorbed marker 3H-sitostanol along the length of the small intestine 20 min after intra-duodenal injection (Time 0). Peak radioactivity on a chow diet occurs in segment 13 of uninfected mice (A) and H. hepaticus-infected mice (B). On the LD, peak radioactivity is found in segment 10 of uninfected mice (C) and in segment 8 of H. hepaticus-infected mice (D), indicating that LD feeding slows intestinal transit irrespective of infection status. E, small intestinal transit in the four groups is plotted as dimensionless geometric centres (Σ (fraction of 3H per segment × segment number)) of the radioactive marker for individual mice, with means ± SEM plotted for each group. The geometric centres of uninfected and infected mice on the chow diet (12.5 ± 0.4 and 11.4 ± 0.5, respectively) are significantly (P < 0.05) larger (i.e. intestinal transit is faster) than the geometric centres of the uninfected and infected groups fed the LD (9.4 ± 0.5 and 9.6 ± 0.5, respectively, as determined by two-way ANOVA). No statistically significant differences exist between the geometric centres of the two chow-fed or two LD-fed groups. *P < 0.05.
Figure 1E records the geometric centre of radioactivity in each group. A two-way ANOVA revealed a significant main effect of diet (F= 23.40, P < 0.0001) with the geometric centres of radioactivity being lower (i.e. slower) in uninfected (9.4 ± 0.5) and infected (9.6 ± 0.5) mice on LD compared with uninfected (12.5 ± 0.4) and infected (11.4 ± 0.5) mice on the chow diet. In contrast, H. hepaticus infection shows no significant effect (F= 0.77, P= 0.39) on motility, nor is there any interaction of diet and infection status (F= 1.89, P= 0.18). These data confirm that the LD prolongs small intestinal transit, but infection with H. hepaticus does not alter transit times significantly, nor does the combination of LD and H. hepaticus infection induce an additive effect compared with LD alone.
Induction and reversal of prolonged small intestinal transit times
Figure 2A illustrates the speed of onset of slowed intestinal transit for uninfected mice fed LD and its reversal by replacing LD with chow. Relative to mice on the chow diet (12.5 ± 0.4), a one-way ANOVA revealed a significant effect (F= 8.33, P= 0.0001) at 8 days on LD (10.3 ± 0.2), and post hoc analysis with Tukey HSD tests indicated that small intestinal transit is prolonged significantly (P < 0.01). This effect is even more pronounced at 14 days of LD feeding relative to the chow-fed mice (9.7 ± 0.2, P < 0.001), although the 8- and 14-day LD groups are statistically equivalent (P > 0.05). Figure 2A also reveals that, after replacement of LD with chow for 3 days, small intestinal transit (11.9 ± 1.0) does not differ significantly from the chow-fed group but is significantly faster than in the 14-day LD group. However, the difference in small intestinal transit between the mice fed LD for 14 days followed by 3 days of chow is not significantly different from the 8-day LD group. Likewise, after replacing LD with chow for 6 days, transit (10.8 ± 0.8) is statistically equivalent to the chow-fed group, and the two chow replacement groups (i.e. at 3 and 6 days) are also statistically equivalent. Taken together, these observations indicate that prolonged small intestinal transit induced by the LD is reversed rapidly by discontinuing the diet.
Figure 2. Effect of LD duration and reversal on small intestinal transit times and cholesterol saturation indices (CSIs) of gallbladder bile.
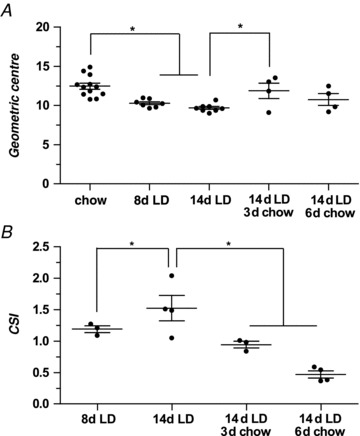
A, geometric centres with means ± SEM of 3H-sitostanol in uninfected male C57L mice fed chow, LD for 8 or 14 days, or LD for 14 days followed by 3 or 6 days of chow diet, respectively. Geometric centres are significantly reduced after 8 and 14 days on LD compared with the chow group. In 14 day LD-fed mice subsequently returned to chow diet for 3 or 6 days, the geometric centres increase, becoming statistically equivalent to the chow-fed group. The values for mice returned to chow for 3 days are also significantly different from the 14-day LD-fed group. B, Means ± SEM of CSIs for the same C57L mice as in A. At 8 days of LD feeding, the CSI is 1.2 ± 0.05, and in mice fed the LD for 14 days, the CSI is further increased to 1.5 ± 0.2. By returning the mice to chow diet for 3 days, gallbladder bile becomes unsaturated with cholesterol (CSI = 0.9 ± 0.05), and after 6 days of chow feeding, bile becomes even more unsaturated (CSI = 0.5 ± 0.06). *P < 0.05.
Time course of cholesterol supersaturation of gallbladder bile
Figure 2B displays CSI values calculated from results of lipid analyses of gallbladder bile in the same animals. This panel shows that bile is supersaturated with cholesterol (i.e. CSI > 1) at 8 days on LD (CSI = 1.2 ± 0.05) and more supersaturated at 14 days (CSI = 1.5 ± 0.2). When chow was reinstated for 3 or 6 days following 14 days of LD feeding, bile becomes unsaturated (CSI = 0.9 ± 0.05 and 0.5 ± 0.06, respectively), corresponding to literature values for chow-fed mice (Wang et al. 1997). These data indicate that cholesterol supersaturation of bile is induced by 8 days of LD feeding and that supersaturation is reversed within 3 days on the chow diet, corresponding to the time required for induction and reversal of slowed intestinal transit (Fig. 2A).
Comparison of small intestinal transit times in male and female mice
In contrast to humans, most strains of female mice on LD display a lower cholesterol gallstone prevalence rate than males (Paigen & Carey, 2002). We therefore tested whether the LD would slow small intestinal transit in uninfected females as in uninfected males. Female mice (n= 4 per group) fed the LD for 8 weeks revealed significantly (P= 0.002) prolonged intestinal transit compared with those fed the chow diet (9.7 ± 0.6 and 13.1 ± 0.6, respectively). Mouse weights and small intestinal lengths did not differ between the two groups (data not displayed). Furthermore, small intestinal transit times, as derived from the geometric centres, are statistically equivalent in females (13.1 ± 0.6) and males (12.5 ± 0.4, viz. Fig. 1E) on the chow diet (P= 0.38), as well as in females (9.7 ± 0.6, data not displayed) and males (9.4 ± 0.5, viz. Fig. 1E) on the LD (P= 0.65).
Small intestinal transit times on diets containing LD components: high cholesterol, high cholic acid or a combination of both
Employing three additional chow-based diets, we studied the influence of specific components of the LD on small intestinal motility in H. hepaticus-free male mice (Fig. 3A).
Figure 3. Influence of cholesterol and/or cholic acid and added ezetimibe (EZ) on geometric centres with respect to intestinal transit times.
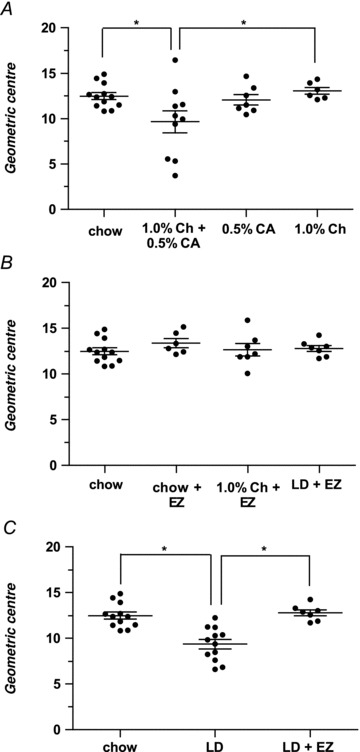
A, mice fed the 1.0% cholesterol + 0.5% cholic acid diet display significantly prolonged small intestinal transit (9.7 ± 1.2) compared with the chow diet group (12.5 ± 0.4) and the 1.0% cholesterol diet group (13.1 ± 0.4). Small intestinal transit of mice on either the 1.0% cholesterol or 0.5% cholic acid (12.1 ± 0.6) diet is equivalent to that of mice on chow diet. B, the addition of EZ (60 mg kg−1) to chow diet does not alter small intestinal transit (13.4 ± 0.5). In the EZ + 1.0% cholesterol diet group (12.7 ± 0.7) and in the LD + EZ group (12.8 ± 0.3), small intestinal motility is also statistically equivalent to the chow diet group. C, small intestinal transit in the LD + EZ group is significantly faster than in mice fed LD (geometric centres are 12.8 ± 0.3 and 9.4 ± 0.5, respectively; P= 0.0002), and the LD + EZ values are essentially the same as for mice fed chow (12.5 ± 0.4). Therefore, slowing of small intestinal transit can be reversed completely by blocking intestinal cholesterol absorption with the drug. *P < 0.05; Ch, cholesterol; CA, cholic acid.
A one-way ANOVA revealed a significant effect (F= 3.66, P= 0.02), and post hoc analysis with Tukey HSD tests showed that mice fed the 0.5% cholic acid diet (12.1 ± 0.6) or the 1.0% cholesterol diet (13.1 ± 0.4) for 8 weeks exhibit statistically equivalent small intestinal transit times to mice maintained on the chow diet (12.5 ± 0.4). In addition, Fig. 3A shows that mice fed the 1.0% cholesterol + 0.5% cholic acid diet for 8 weeks demonstrate (P < 0.05) slowed small intestinal transit with a value (9.7 ± 1.2) that differs significantly from chow-fed mice, suggesting also that the high triglyceride content (not studied individually) of the LD is not critically important in influencing gut motility. Moreover, the combination of dietary cholesterol and cholic acid markedly promotes cholesterol absorption in mice (Wang et al. 1999a).
After 8 weeks on the 1.0% cholesterol diet, mice display equivalent body weights (34.4 ± 0.9 g) to mice on chow (35.7 ± 1.1 g). However, mice in the 0.5% cholic acid group become significantly lighter at the end of the experiment (29.7 ± 0.7 g; P < 0.01) than either the chow- or 1.0% cholesterol-fed groups (P < 0.001 and P < 0.01, respectively). At the end of the feeding period, mice in the 1.0% cholesterol + 0.5% cholic acid group weigh even less (25.0 ± 0.7 g) than the chow-fed (P < 0.001), 1.0% cholesterol-fed (P < 0.001) and 0.5% cholic acid-fed (P < 0.01) groups. Small intestinal lengths did not differ between these cohorts (data not displayed).
Small intestinal transit times on diets supplemented with EZ
Figure 3B shows small intestinal transit times in uninfected male mice after cholesterol absorption was completely blocked using EZ at 60 mg kg−1 of diet. A one-way ANOVA revealed no significant differences in the geometric centres (F= 3.66, P= 0.61) between mice fed for 8 weeks with chow diet (12.5 ± 0.4), chow + EZ (13.4 ± 0.5), 1.0% cholesterol + EZ (12.7 ± 0.7) and LD + EZ (12.8 ± 0.3). Therefore, in contrast to mice on LD alone, mice on LD + EZ display completely normal small intestinal motility compared with the chow-fed group. Furthermore, Fig. 3C demonstrates that the LD + EZ-fed group exhibits significantly faster small intestinal transit compared with mice fed LD alone (12.8 ± 0.3 and 9.4 ± 0.5, respectively; P < 0.001). This indicates that the slowing of small intestinal transit by the LD is reversed completely by blocking intestinal cholesterol absorption with EZ. Small intestinal length was equivalent between EZ-treated, LD and chow diet groups.
Proximal small intestinal weights, imaging of isolated sarcolemmae and lipid analyses
Figure 4A shows a typical electron photomicrograph depicting that reconstituted pellets reveal vesicular plasma membranes at high yield following sucrose density centrifugation, similar to those in images of smooth muscle cell sarcolemmae from rats (Kidwai, 1974). Figure 4B shows Western blots of the Cav3 distribution across three ultracentrifuged sucrose gradients (see Methods) and confirms the success of sarcolemmal isolation in the expected interface. These representative samples from chow-, LD- and LD + EZ-fed mice displayed bands with maximum relative Cav3 densities of 35%, 41% and 42%, respectively, in the 20–30% (No. 2) interface.
Figure 4. Electron micrograph and Caveolin-3 (Cav3) distribution of isolated sarcolemmae from upper small intestinal smooth muscle cells of male C57L mice.

A, an electron photomicrograph (original magnification, ×2000) of the plasma membranes, as multiple vesicular structures, isolated by sucrose density gradient centrifugation from the small intestinal smooth muscle cells of mice. B, the relative distributions of sarcolemmal-specific Cav3 protein at the sucrose interfaces (numbered consecutively) of three representative samples: (1) 0.25 m (∼8%)–20%, (2) 20–30%, (3) 30–35% and (4) 35–40% sucrose. The per cent total Cav3 at each interface, calculated using optical density and normalized to total protein content, gives the highest yield of Cav3 at the 20–30% sucrose interface. These samples from chow-, LD- and LD + EZ-fed mice, with per cent Cav3 totals of 35%, 41% and 42%, respectively, were assayed for cholesterol and phospholipids.
Figure 5A plots proximal small intestinal weights for mice in the chow, LD and LD + EZ groups. For identical lengths of intestine, the weights of the latter (0.87 ± 0.02 g) are significantly higher (P < 0.001) than for chow-fed controls (0.71 ± 0.03 g) and LD mice (0.65 ± 0.04 g). Phospholipid concentrations in the sarcolemmae are not significantly different between groups (data not displayed). However, as Fig. 5B shows, cholesterol concentrations in the LD group (0.25 ± 0.04 μmol (mg protein)−1) are significantly higher than in the chow-fed control group (0.16 ± 0.01 μmol (mg protein)−1, P < 0.01). Furthermore, the cholesterol concentration in the LD + EZ group (0.14 ± 0.03 μmol (mg protein)−1) reveals a significant decrease (P < 0.01) compared with the LD group, but no difference when compared with chow-fed controls. Figure 5C plots the cholesterol to phospholipid (Ch/PL) ratios in the sarcolemmae, which are significantly greater in the LD group (0.57 ± 0.03, P < 0.01) compared with chow-fed controls (0.45 ± 0.02) but decreased by the addition of EZ in the LD + EZ group (0.38 ± 0.03; P < 0.001 for comparison of LD and LD + EZ groups). The cholesterol to phospholipid ratios of the chow- and LD + EZ-fed groups are statistically equivalent.
Figure 5. Proximal small intestinal weights, cholesterol concentrations and cholesterol to phospholipid ratios (Ch/PL) in sarcolemmae of the proximal small intestine in male C57L mice.
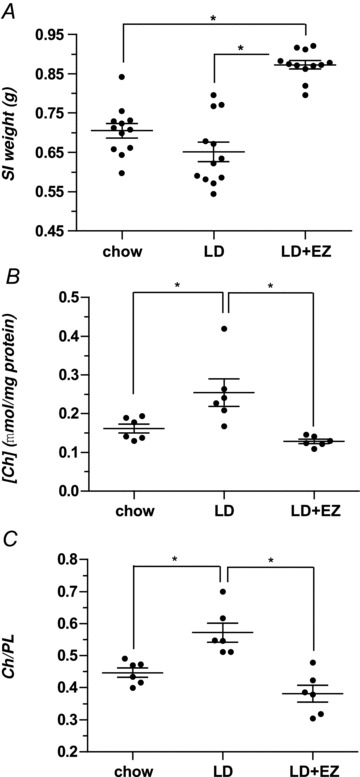
A, the proximal small intestinal (SI, mucosa and smooth muscle) weight (g) of the LD + EZ group (0.87 ± 0.02 g) is significantly (P < 0.001) higher compared with identical lengths of small intestine in both the chow- (0.71 ± 0.03 g) and LD-fed (0.65 ± 0.04 g) groups. B, cholesterol concentrations (normalized for total protein content) in the three groups reveal that cholesterol concentrations in the LD group (0.25 ± 0.04 μmol (mg protein)−1) are significantly (P < 0.01) greater than in either LD + EZ (0.14 ± 0.03 μmol (mg protein)−1) or chow (0.16 ± 0.01 μmol (mg protein)−1, P < 0.01) groups. C, in the LD group, Ch/PL ratios (0.57 ± 0.03) are significantly larger than in the LD + EZ (0.38 ± 0.03, P < 0.001) and in the chow (0.45 ± 0.02, P < 0.01) groups. *P < 0.01 and <0.001, as noted above.
Discussion
In this study we demonstrated that feeding a standard LD containing both cholesterol and cholic acid to C57L inbred mice with multiple Lith genes and a marked proclivity towards cholesterol gallstone formation (Paigen & Carey, 2002) significantly slows small intestinal transit (Fig. 1). Intestinal hypomotility, a well-documented accompaniment to gallstones and the lithogenic state in humans (Heaton et al. 1993, 2000; Azzaroli et al. 1999), is believed to promote cholesterol cholelithogenesis by at least two mechanisms. First, LD feeding permits greater cholesterol absorption from the upper small intestine and, in mice, increases biliary cholesterol secretion (Wang et al. 1999a, 2004). In the present work and in other lithogenic animals (Xu et al. 1996; Fan et al. 2007), a LD results in slower small intestinal transit, engendering a vicious cycle that further augments cholesterol absorption. Second, prolonged small intestinal transit also increases deoxycholic acid (DCA) formation in the distal small and large intestines (Thomas et al. 2005). DCA promotes cholesterol gallstone formation by augmenting cholesterol secretion into bile (Carulli et al. 1984; Shoda et al. 1995) and by destabilizing cholesterol-rich biliary vesicles, leading to accelerated cholesterol crystallization in gallbladder bile (Stolk et al. 1994; Hussaini et al. 1995), as also exemplified in parallel studies on model systems (van Erpecum & Carey, 1997).
Prior to this study there was a dearth of information on whether enterohepatic Helicobacter spp. infection influences intestinal transit (Fox, 2002); moreover, studies of gastrointestinal motility in patients infected with the more common gastrotropic H. pylori have yielded inconclusive results (Pieramico et al. 1993; Minocha et al. 1994; Leontiadis et al. 2004). From the present results (Fig. 1B and D), we can rule out altered small intestinal motility as a mechanism by which enterohepatic helicobacter infection influences cholelithogenesis. Gallstone promotion by these bacteria is more probably related to the host gallbladder's immune responses that promote cholesterol monohydrate crystal separation from liquid crystals, an irreversible phase transition during cholelithogenesis (Maurer et al. 2007, 2009).
We then explored cholesterol hyperabsorption as a possible explanation for small intestinal hypomotility (Traber & Ostwald, 1978; Ponz de Leon et al. 1982). Cholesterol molecules absorbed from bile and their intercalation into sarcolemmae of the gallbladder have a profound effect on smooth muscle function (Yu et al. 1996). In the current scenario, intestinal cholesterol absorption takes place primarily in the duodenum and proximal jejunum. We therefore questioned whether similar studies on sarcolemmae from upper small intestinal smooth muscle would provide an explanation for our observations. As in lithogenic human gallbladders (Yu et al. 1996), we noted augmented cholesterol incorporation (Fig. 5B and C) into isolated proximal small intestinal sarcolemmae without concomitant change in phospholipid content. However, in contrast to the gallbladder, the small intestine esterifies and then incorporates absorbed cholesterol molecules into lipoproteins (i.e. chylomicrons and nascent HDL); our observations of sarcolemmal enrichment with free (i.e. unesterified) cholesterol molecules therefore suggest that some of the absorbed cholesterol from the upper small intestine is not immediately esterified for lipoprotein assembly but is free to enter the lamina propria and, subsequently, smooth muscle plasma membranes.
In the gallbladder, cholesterol-induced hypomotility is believed to occur through at least two intracellular mechanisms: (1) the inhibitory effect of cholesterol on action potentials and Ca2+ currents in smooth muscle (Jennings et al. 1999), and (2) CCK-1R (previously CCK-A)-mediated activation of phospholipase C leading to signal-transduction decoupling of CCK when gallbladder sarcolemmae become enriched in cholesterol (Yu et al. 1998; Xiao et al. 1999). It appears that an increase in caveolar cholesterol inhibits Cav3 phosphorylation in gallbladder sarcolemmae, a vital step in the recycling and reintegration of CCK-1R–G protein complexes into non-caveolar portions of the smooth muscle plasma membranes (Cong et al. 2010). This inhibition of Cav3 phosphorylation, along with sequestration of CCK-1R–G protein complexes in caveolae, most probably explains why CCK ‘finds’ fewer CCK-1Rs to bind to in cholesterol-enriched, as compared with normal sarcolemmae (Cong et al. 2010). As the gene for CCK-1R is expressed not only on smooth muscle cells of the gallbladder and stomach, but also on those of the small intestine (Lacourse et al. 1997), we propose that the hyperabsorbed cholesterol similarly inhibits the contraction of proximal intestinal smooth muscle cells, resulting in marked slowing of small intestinal transit. The function of the CCK-1R on the small intestine is known to be physiologically relevant since its motor action on small intestinal motility occurs with post-prandial levels of CCK (Wang et al. 2004). Furthermore, the identical LD exhibited no effect on intestinal transit times in CCK-1R-knockout mice (Wang et al. 2004), supporting our hypothesis that cholesterol's inhibitory effect on intestinal motility occurs through a CCK-1R-dependent pathway (Yu et al. 1998; Xiao et al. 1999). For this reason, we chose not to test other motility agonists, such as acetylcholine (Price et al. 1969), since our results could be fully explained by CCK antagonism; moreover, a different ex vivo experimental set-up would have been required. Nonetheless, as highlighted earlier, pro-kinetic agents, including cisapride in humans (Veysey et al. 2001) and erythromycin in ground squirrels (Xu et al. 1998), reverse presumed cholesterol-induced small intestinal hypomotility under these lithogenic conditions, indicating that other pro-motility agents clearly function by CCK-1R-independent pathways.
A number of parallelisms and differences between our work and studies of prolonged small intestinal transit in hamsters (Fan et al. 2007) and ground squirrels (Xu et al. 1996) fed a high cholesterol diet need emphasis. In our attempt to mimic such experiments in mice, we did not observe any effect on small intestinal motility in animals fed a chow diet supplemented with a high cholesterol concentration alone (Fig. 3A). This can be explained by the poor absorption efficiency of cholesterol from proximal small intestinal mixed bile salt micelles in mice, including the C57L strain, due to their high levels (40–60%) of hydrophilic muricholate and ursodeoxycholate conjugates in the bile salt pool (Wang et al. 1997). In contrast, ground squirrels and hamsters absorb dietary cholesterol much more efficiently without cholic acid dosing because of their more hydrophobic bile salt pools (Heuman, 1989; Xu et al. 1996). Nonetheless, both the LD and the cholesterol + cholic acid combination markedly increased intestinal cholesterol absorption in mice because cholic acid becomes taurocholate after uptake and hepatic conjugation, displacing most of the very hydrophilic bile salts (Wang et al. 2004, 2009). Accordingly, we observed prolonged small intestinal transit in both of these groups (Figs 2A and 3A).
Despite cholate conjugates being the better promoters of intestinal cholesterol absorption (Wang et al. 2003; Woollett et al. 2004), DCA conjugates augment micellar cholesterol solubilisation in the small intestine of animals with hydrophilic bile salt pools (Wang et al. 2009). Because endogenous bile salts would otherwise incorporate cholesterol molecules into micelles poorly (Wang et al. 2003), this solubilising effect is critically important for efficient intestinal cholesterol absorption in mice (Xu et al. 1996; Wang et al. 1997, 1999b, 2003).
Potentially, the altered molecular species composition of the bile salt pool from feeding 0.5% cholic acid in the LD could itself influence small intestinal motility. Even though the murine liver efficiently rehydroxylates DCA to cholic acid, levels of DCA conjugates in bile increase markedly in mice fed LD (Wang et al. 2003). While taurocholate, a bile salt of intermediate hydrophobic–hydrophilic balance (Heuman, 1989), accelerates intestinal transit in rats, DCA prolongs transit, possibly via an ‘ileal-brake’ effect (Brown et al. 1990). Nonetheless, in the present study we did not observe any alterations whatsoever in intestinal motility from feeding cholic acid alone (Fig. 3A), nor from the obligatory accumulation of its secondary catabolic product DCA, as also demonstrated earlier (Wang et al. 2003).
Finally, we tested the effects of reversing the accumulation of hyperabsorbed cholesterol molecules on small intestinal motility by desorbing cholesterol from intestinal sarcolemmae. In vitro, cholesterol-induced alterations in gallbladder smooth muscle contractility are reversed rapidly when the sterol is extracted into cholesterol-free phospholipid liposomes (Yu et al. 1996). This demonstration is consistent with our in vivo observation that slowed intestinal transit was reversed within 3 days when the LD was discontinued (Fig. 2A) or when EZ and LD were co-administered (Fig. 3B). The observed 23% and 34% increases in the weights of identical lengths of proximal small intestine (containing both smooth muscle and mucosa) from the LD + EZ-fed mice when compared with those of mice fed chow or LD, respectively, are most probably explained by EZ's high density of 1.334 g cm−3, as reported on the basis of its crystal structure (Patel et al. 2011). With the maximal doses of EZ employed in our murine diets (see Methods), the number of molecules ingested was probably sufficient to saturate all Niemann-Pick C1 Like-1 (NPC1L1) receptors expressed on the apical plasma membranes of proximal small intestinal enterocytes of mice (Garcia-Calvo et al. 2005). In this respect, if passive sterol (i.e. cholesterol) enrichment of the brush border membranes had occurred in the EZ-treated mice, the densities of the monohydrate (1.09 g cm−3; Craven, 1976) and anhydrous (1.052 g cm−3; Shieh et al. 1977) forms of cholesterol would not account for the weight differential we observed with EZ. Moreover, we found that small intestinal transit times did not differ significantly between the high-cholesterol and high-cholesterol + EZ diet groups (Fig. 3B), indicating that very little passive absorption of cholesterol took place. Predictably, NPC1L1-knockout mice are insensitive to EZ and display dramatically lower cholesterol absorption rates that are not altered by dietary supplementation with bile acids (Altmann et al. 2004). In addition, our evidence that the chow + EZ diet did not produce a change in transit times compared with mice on the chow diet alone (Fig. 3B) is fully consistent with the low baseline level of dietary cholesterol absorption in mice (Wang et al. 2003). From the extensive earlier work on the effect of absorbed cholesterol on gallbladder hypomotility (Yu et al. 1996; Chen et al. 1999; Cong et al. 2010) and the present study, it appears that both the proximal small intestine and the gallbladder lose their contractile function and become hypomotile during cholelithogenesis because of increased luminal cholesterol absorption in both organs.
How do our dietary observations in mice relate pathophysiologically to the slowed intestinal transit so well documented in cholesterol gallstone patients (Heaton et al. 1993; Azzaroli et al. 1999)? It has been established that cholesterol gallstone patients do not customarily ingest diets rich in cholesterol (Cuevas et al. 2004; Méndez-Sánchez et al. 2007). Despite this, the proximal small intestine of lithogenic humans is flooded continuously with excess cholesterol molecules, but these are derived from hepatic hypersecretion into bile, by as much as 2- to 4-fold compared with controls. Cholesterol hypersecretion occurs during all stages of gallstone disease, including the early pre-stone stage, after gallstones have formed, and even in the post-cholecystectomy state (Shaffer & Small, 1977; Leiss & von Bergmann, 1985; Reuben et al. 1985). This is further supported by the recent discovery of a gain-in-function from a p.D19H coding variant of ABCG8, the heterodimeric canalicular cholesterol transporter of the liver, in genome-wide association, case-control and linkage studies of human gallstone populations from Germany, Chile, Romania, China, Sweden, India and Denmark, where the odds ratio varied from 1.9 to 3.5 (Krawczyk et al. 2011).
In earlier clinical studies, both high biliary DCA levels and CSIs in gallstone patients were decreased clinically with drug- or diet-induced acceleration of intestinal transit times, such as by employing the laxative senna (Marcus & Heaton, 1986) or unrefined carbohydrate- (Thornton et al. 1983) and wheat bran-supplemented diets (Pomare et al. 1976). Since EZ has been shown to diminish cholesterol saturation of bile and prevent gallstones in mice at the very high doses employed in the present study (Wang et al. 2008; Zúñiga et al. 2008), there is ample reason to believe that its influence in normalizing intestinal motility could be an ancillary factor in preventing a hypomotile gut's contribution to cholesterol gallstone formation in humans.
Acknowledgments
We thank Ms Bian B. Yu for technical assistance and Dr Kirk Maurer for advice on helicobacter infection experiments and other aspects of the study. The authors wish to express their gratitude for editorial assistance to Ms Monika R. Leonard and Ms Julie Szymaniak. This work was supported in part by NIH (USPHS) grant R37DK036588 (M.C.C., PI), training grant T32DK007533 (R. S. Blumberg, PI) and R01CA067529 (J.G.F., PI). No conflicts of interest, financial or otherwise, are declared by the authors.
Glossary
Abbreviations
- BW
body weight
- C57L/J (C57L)
an inbred cholesterol gallstone-susceptible mouse strain
- Cav3
Caveolin-3
- CCK
cholecystokinin
- CCK-1R
CCK-1 receptor
- CSI
cholesterol saturation index
- DCA
deoxycholic acid
- EZ
ezetimibe (Zetia)
- LD
lithogenic diet
- Lith
murine lithogenic gene
- MCT
medium-chain triglycerides
- NPC1L1
Niemann-Pick C1 Like-1 transporter
Author contributions
M.X., then a post-doctoral research fellow, played a central role in the conception, design and execution of most of the small intestinal transit time studies. She drafted the earlier versions of the paper, incorporating analysis and interpretation of motility data. V.R.K. took over where M.X. left off and designed, performed, collected, interpreted and wrote up the remaining motility studies, including most studies involving the cholesterol absorption blocking drug ezetimibe's effects on small intestinal motility. J.D.P.A. played a key role in working out the molecular mechanism for the intestinal hypomotility observed with the lithogenic diet. This required setting up ab initio a protocol for isolating the sarcolemmae of intestinal smooth muscle cells, proving their identity and purity, and carrying out all the studies in the paper involving alterations in cholesterol levels in these plasma membranes. He also wrote up and interpreted these results and extensively reworked the Discussion. J.G.F. and M.C.C. are Principal Investigators at MIT (Cambridge, MA, USA) and Brigham & Women's Hospital/Harvard Medical School (Boston, MA) in whose laboratories these studies were performed. J.G.F. conceived, designed and interpreted all intellectual aspects of the Helicobacter hepaticus infection experiments, which were carried out in his laboratory. M.C.C. conceived, designed, interpreted and edited all other aspects of this study. All authors approved the final version.
References
- Altmann SW, Davis HR, Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- Azzaroli F, Mazzella G, Mazzeo C, Simoni P, Festi D, Colecchia A, Montagnani M, Martino C, Villanova N, Roda A, Roda E. Sluggish small bowel motility is involved in determining increased biliary deoxycholic acid in cholesterol gallstone patients. Am J Gastroenterol. 1999;94:2453–2459. doi: 10.1111/j.1572-0241.1999.01375.x. [DOI] [PubMed] [Google Scholar]
- Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- Brown NJ, Read NW, Richardson A, Rumsey RD, Bogentoft C. Characteristics of lipid substances activating the ileal brake in the rat. Gut. 1990;31:1126–1129. doi: 10.1136/gut.31.10.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MC. Critical tables for calculating the cholesterol saturation of native bile. J Lipid Res. 1978;19:945–955. [PubMed] [Google Scholar]
- Carulli N, Loria P, Bertolotti M, de Leon PonzM, Menozzi D, Medici G, Piccagli I. Effects of acute changes of bile acid pool composition on biliary lipid secretion. J Clin Invest. 1984;74:614–624. doi: 10.1172/JCI111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Amaral J, Biancani P, Behar J. Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology. 1999;116:678–685. doi: 10.1016/s0016-5085(99)70190-3. [DOI] [PubMed] [Google Scholar]
- Cong P, Pricolo V, Biancani P, Behar J. Effects of cholesterol on CCK-1 receptors and caveolin-3 proteins recycling in human gallbladder muscle. Am J Physiol Gastrointest Liver Physiol. 2010;299:G742–G750. doi: 10.1152/ajpgi.00064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven BM. Crystal structure of cholesterol monohydrate. Nature. 1976;260:727–729. doi: 10.1038/260727a0. [DOI] [PubMed] [Google Scholar]
- Cuevas A, Miquel JF, Reyes MS, Zanlungo S, Nervi F. Diet as a risk factor for cholesterol gallstone disease. J Am Coll Nutr. 2004;23:187–196. doi: 10.1080/07315724.2004.10719360. [DOI] [PubMed] [Google Scholar]
- Dowling RH. Review: pathogenesis of gallstones. Aliment Pharmacol Ther. 2000;14:39–47. doi: 10.1046/j.1365-2036.2000.014s2039.x. [DOI] [PubMed] [Google Scholar]
- Dowling RH, Hussaini SH, Murphy GM, Besser GM, Wass JA. Gallstones during octreotide therapy. Metabolism. 1992;41:22–33. doi: 10.1016/0026-0495(92)90027-8. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan IW, Culbreth PH, Burtis CA. Determination of free, total, and esterified cholesterol by high-performance liquid chromatography. J Chromatogr. 1979;162:281–292. doi: 10.1016/s0378-4347(00)81515-7. [DOI] [PubMed] [Google Scholar]
- Fan Y, Wu SD, Fu BB. Effect of intestinal transit on the formation of cholesterol gallstones in hamsters. Hepatobiliary Pancreat Dis Int. 2007;6:513–515. [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane StanleyGH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Fox JG. The non-H pylori helicobacters: their expanding role in gastrointestinal and systemic diseases. Gut. 2002;50:273–283. doi: 10.1136/gut.50.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1) Proc Natl Acad Sci U S A. 2005;102:8132–8137. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton KW. Review article: epidemiology of gall-bladder disease – role of intestinal transit. Aliment Pharmacol Ther. 2000;14:9–13. doi: 10.1046/j.1365-2036.2000.014s2009.x. [DOI] [PubMed] [Google Scholar]
- Heaton KW, Emmett PM, Symes CL, Braddon FE. An explanation for gallstones in normal-weight women: slow intestinal transit. Lancet. 1993;341:8–10. doi: 10.1016/0140-6736(93)92479-d. [DOI] [PubMed] [Google Scholar]
- Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30:719–730. [PubMed] [Google Scholar]
- Hussaini SH, Pereira SP, Murphy GM, Dowling RH. Deoxycholic acid influences cholesterol solubilization and microcrystal nucleation time in gallbladder bile. Hepatology. 1995;22:1735–1744. [PubMed] [Google Scholar]
- Jennings LJ, Xu QW, Firth TA, Nelson MT, Mawe GM. Cholesterol inhibits spontaneous action potentials and calcium currents in guinea pig gallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol. 1999;277:G1017–G1026. doi: 10.1152/ajpgi.1999.277.5.G1017. [DOI] [PubMed] [Google Scholar]
- Khanuja B, Cheah YC, Hunt M, Nishina PM, Wang DQ, Chen HW, Billheimer JT, Carey MC, Paigen B. Lith1, a major gene affecting cholesterol gallstone formation among inbred strains of mice. Proc Natl Acad Sci U S A. 1995;92:7729–7733. doi: 10.1073/pnas.92.17.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwai AM. Isolation of plasma membrane from smooth, skeletal, and heart muscle. Methods Enzymol. 1974;31:134–144. doi: 10.1016/0076-6879(74)31013-0. [DOI] [PubMed] [Google Scholar]
- Krawczyk M, Wang DQ, Portincasa P, Lammert F. Dissecting the genetic heterogeneity of gallbladder stone formation. Semin Liver Dis. 2011;31:157–172. doi: 10.1055/s-0031-1276645. [DOI] [PubMed] [Google Scholar]
- Lacourse KA, Lay JM, Swanberg LJ, Jenkins C, Samuelson LC. Molecular structure of the mouse CCK-A receptor gene. Biochem Biophys Res Commun. 1997;236:630–635. doi: 10.1006/bbrc.1997.7030. [DOI] [PubMed] [Google Scholar]
- Lammert F, Carey MC, Paigen B. Chromosomal organization of candidate genes involved in cholesterol gallstone formation: a murine gallstone map. Gastroenterology. 2001;120:221–238. doi: 10.1053/gast.2001.20878. [DOI] [PubMed] [Google Scholar]
- Leiss O, von Bergmann K. Comparison of biliary lipid secretion in non-obese cholesterol gallstone patients with normal, young, male volunteers. Klin Wochenschr. 1985;63:1163–1169. doi: 10.1007/BF01740592. [DOI] [PubMed] [Google Scholar]
- Leontiadis GI, Minopoulos GI, Maltezos E, Kotsiou S, Manolas KI, Simopoulos K, Hatseras D. Effects of Helicobacter pylori infection on gastric emptying rate in patients with non-ulcer dyspepsia. World J Gastroenterol. 2004;10:1750–1754. doi: 10.3748/wjg.v10.i12.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MA, Wittenburg H. Cholesterol gallstone susceptibility loci: a mouse map, candidate gene evaluation, and guide to human LITH genes. Gastroenterology. 2006;131:1943–1970. doi: 10.1053/j.gastro.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Marcus SN, Heaton KW. Intestinal transit, deoxycholic acid and the cholesterol saturation of bile – three inter-related factors. Gut. 1986;27:550–558. doi: 10.1136/gut.27.5.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer KJ, Carey MC, Fox JG. Roles of infection, inflammation, and the immune system in cholesterol gallstone formation. Gastroenterology. 2009;136:425–440. doi: 10.1053/j.gastro.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer KJ, Ihrig MM, Rogers AB, Ng V, Bouchard G, Leonard MR, Carey MC, Fox JG. Identification of cholelithogenic enterohepatic Helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023–1033. doi: 10.1053/j.gastro.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Maurer KJ, Rao VP, Ge Z, Rogers AB, Oura TJ, Carey MC, Fox JG. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology. 2007;133:1304–1315. doi: 10.1053/j.gastro.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer KJ, Rogers AB, Ge Z, Wiese AJ, Carey MC, Fox JG. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: a prospective study. Am J Physiol Gastrointest Liver Physiol. 2006;290:G175–G182. doi: 10.1152/ajpgi.00272.2005. [DOI] [PubMed] [Google Scholar]
- Méndez-Sánchez N, Zamora-Valdés D, Chávez-Tapia NC, Uribe M. Role of diet in cholesterol gallstone formation. Clin Chim Acta. 2007;376:1–8. doi: 10.1016/j.cca.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Miller MS, Galligan JJ, Burks TF. Accurate measurement of intestinal transit in the rat. J Pharmacol Methods. 1981;6:211–217. doi: 10.1016/0160-5402(81)90110-8. [DOI] [PubMed] [Google Scholar]
- Minocha A, Mokshagundam S, Gallo SH, Rahal PS. Alterations in upper gastrointestinal motility in Helicobacter pylori-positive nonulcer dyspepsia. Am J Gastroenterol. 1994;89:1797–1800. [PubMed] [Google Scholar]
- Paigen B, Carey MC. Gallstones. In: King R, Rotter J, Motulsky A, editors. The Genetic Basis of Common Diseases. 2nd edn. New York: Oxford University Press; 2002. pp. 298–335. [Google Scholar]
- Patel KM, Shah DH, Patel JB, Patel JS, Garg CS, Sen DJ. Chemistry of four membered heterocyclic ezetimibe as lipid lowering agent. Int J Drug Dev Res. 2011;3:104–110. [Google Scholar]
- Pieramico O, Ditschuneit H, Malfertheiner P. Gastrointestinal motility in patients with non-ulcer dyspepsia: a role for Helicobacter pylori infection? Am J Gastroenterol. 1993;88:364–368. [PubMed] [Google Scholar]
- Pomare EW, Heaton KW, Low-Beer TS, Espiner HJ. The effect of wheat bran upon bile salt metabolism and upon the lipid composition of bile in gallstone patients. Am J Dig Dis. 1976;21:521–526. doi: 10.1007/BF01464757. [DOI] [PubMed] [Google Scholar]
- de Leon PonzM, Iori R, Barbolini G, Pompei G, Zaniol P, Carulli N. Influence of small-bowel transit time on dietary cholesterol absorption in human beings. N Engl J Med. 1982;307:102–103. doi: 10.1056/NEJM198207083070207. [DOI] [PubMed] [Google Scholar]
- Price WE, Shehadeh Z, Thompson GH, Underwood LD, Jacobson ED. Effects of acetylcholine on intestinal blood flow and motility. Am J Physiol. 1969;216:343–347. doi: 10.1152/ajplegacy.1969.216.2.343. [DOI] [PubMed] [Google Scholar]
- Reuben A, Maton PN, Murphy GM, Dowling RH. Bile lipid secretion in obese and non-obese individuals with and without gallstones. Clin Sci (Lond) 1985;69:71–79. doi: 10.1042/cs0690071. [DOI] [PubMed] [Google Scholar]
- Rybal'chenko VK, Pogrebnoi PV, Gruzina TG, Karamushka VI. Extraction of plasma membranes from smooth muscle cells of the rabbit small intestine (in Russian) Biull Eksp Biol Med. 1984;97:106–108. [PubMed] [Google Scholar]
- Sambrook J, Fritsch E, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn, Appendix B12. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shaffer EA, Small DM. Biliary lipid secretion in cholesterol gallstone disease. The effect of cholecystectomy and obesity. J Clin Invest. 1977;59:828–840. doi: 10.1172/JCI108705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh HS, Hoard LG, Nordman CE. Crystal structure of anhydrous cholesterol. Nature. 1977;267:287–289. doi: 10.1038/267287a0. [DOI] [PubMed] [Google Scholar]
- Shoda J, He BF, Tanaka N, Matsuzaki Y, Osuga T, Yamamori S, Miyazaki H, Sjövall J. Increase of deoxycholate in supersaturated bile of patients with cholesterol gallstone disease and its correlation with de novo syntheses of cholesterol and bile acids in liver, gallbladder emptying, and small intestinal transit. Hepatology. 1995;21:1291–1302. [PubMed] [Google Scholar]
- Stolk MF, van de Heijning BJ, van Erpecum KJ, van den Broek AM, Renooij W, van Berge-Henegouwen GP. The effect of bile acid hydrophobicity on nucleation of several types of cholesterol crystals from model bile vesicles. J Hepatol. 1994;20:802–810. doi: 10.1016/s0168-8278(05)80153-9. [DOI] [PubMed] [Google Scholar]
- Thomas LA, Veysey MJ, Murphy GM, Russell-Jones D, French GL, Wass JA, Dowling RH. Octreotide induced prolongation of colonic transit increases faecal anaerobic bacteria, bile acid metabolising enzymes, and serum deoxycholic acid in patients with acromegaly. Gut. 2005;54:630–635. doi: 10.1136/gut.2003.028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JR, Emmett PM, Heaton KW. Diet and gall stones: effects of refined and unrefined carbohydrate diets on bile cholesterol saturation and bile acid metabolism. Gut. 1983;24:2–6. doi: 10.1136/gut.24.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber MG, Ostwald R. Cholesterol absorption and steroid excretion in cholesterol-fed guinea pigs. J Lipid Res. 1978;19:448–456. [PubMed] [Google Scholar]
- Turley SD, Dietschy JM. Re-evaluation of the 3 alpha-hydroxysteroid dehydrogenase assay for total bile acids in bile. J Lipid Res. 1978;19:924–928. [PubMed] [Google Scholar]
- van Erpecum KJ, Carey MC. Influence of bile salts on molecular interactions between sphingomyelin and cholesterol: relevance to bile formation and stability. Biochim Biophys Acta. 1997;1345:269–282. doi: 10.1016/s0005-2760(97)00002-7. [DOI] [PubMed] [Google Scholar]
- van Erpecum KJ, van Berge-Henegouwen GP. Gallstones: an intestinal disease? Gut. 1999;44:435–438. doi: 10.1136/gut.44.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercaemst R, Union A, Rosseneu M, De Craene I, De Backer G, Kornitzer M. Quantitation of plasma free cholesterol and cholesteryl esters by high performance liquid chromatography. Study of a normal population. Atherosclerosis. 1989;78:245–250. doi: 10.1016/0021-9150(89)90230-x. [DOI] [PubMed] [Google Scholar]
- Veysey MJ, Malcolm P, Mallet AI, Jenkins PJ, Besser GM, Murphy GM, Dowling RH. Effects of cisapride on gall bladder emptying, intestinal transit, and serum deoxycholate: a prospective, randomised, double blind, placebo controlled trial. Gut. 2001;49:828–834. doi: 10.1136/gut.49.6.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DQ, Cohen DE, Carey MC. Biliary lipids and cholesterol gallstone disease. J Lipid Res. 2009;50(Suppl):S406–S411. doi: 10.1194/jlr.R800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DQ, Lammert F, Cohen DE, Paigen B, Carey MC. Cholic acid aids absorption, biliary secretion, and phase transitions of cholesterol in murine cholelithogenesis. Am J Physiol Gastrointest Liver Physiol. 1999a;276:G751–G760. doi: 10.1152/ajpgi.1999.276.3.G751. [DOI] [PubMed] [Google Scholar]
- Wang DQ, Lammert F, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: pathophysiology of biliary lipid secretion. J Lipid Res. 1999b;40:2066–2079. [PubMed] [Google Scholar]
- Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395–1411. [PubMed] [Google Scholar]
- Wang DQ, Paigen B, Carey MC. Genetic factors at the enterocyte level account for variations in intestinal cholesterol absorption efficiency among inbred strains of mice. J Lipid Res. 2001;42:1820–1830. [PubMed] [Google Scholar]
- Wang DQ, Schmitz F, Kopin AS, Carey MC. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J Clin Invest. 2004;114:521–528. doi: 10.1172/JCI16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DQ, Tazuma S, Cohen DE, Carey MC. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am J Physiol Gastrointest Liver Physiol. 2003;285:G494–G502. doi: 10.1152/ajpgi.00156.2003. [DOI] [PubMed] [Google Scholar]
- Wang HH, Portincasa P, Méndez-Sánchez N, Uribe M, Wang DQ. Effect of ezetimibe on the prevention and dissolution of cholesterol gallstones. Gastroenterology. 2008;134:2101–2110. doi: 10.1053/j.gastro.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead RH, Demmler K, Rockman SP, Watson NK. Clonogenic growth of epithelial cells from normal colonic mucosa from both mice and humans. Gastroenterology. 1999;117:858–865. doi: 10.1016/s0016-5085(99)70344-6. [DOI] [PubMed] [Google Scholar]
- Woollett LA, Buckley DD, Yao L, Jones PJ, Granholm NA, Tolley EA, Tso P, Heubi JE. Cholic acid supplementation enhances cholesterol absorption in humans. Gastroenterology. 2004;126:724–731. doi: 10.1053/j.gastro.2003.11.058. [DOI] [PubMed] [Google Scholar]
- Xiao ZL, Chen Q, Amaral J, Biancani P, Jensen RT, Behar J. CCK receptor dysfunction in muscle membranes from human gallbladders with cholesterol stones. Am J Physiol Gastrointest Liver Physiol. 1999;276:G1401–G1407. doi: 10.1152/ajpgi.1999.276.6.G1401. [DOI] [PubMed] [Google Scholar]
- Xu QW, Scott RB, Tan DT, Shaffer EA. Slow intestinal transit: a motor disorder contributing to cholesterol gallstone formation in the ground squirrel. Hepatology. 1996;23:1664–1672. doi: 10.1002/hep.510230650. [DOI] [PubMed] [Google Scholar]
- Xu QW, Scott RB, Tan DT, Shaffer EA. Effect of the prokinetic agent, erythromycin, in the Richardson ground squirrel model of cholesterol gallstone disease. Hepatology. 1998;28:613–619. doi: 10.1002/hep.510280302. [DOI] [PubMed] [Google Scholar]
- Yu P, Chen Q, Biancani P, Behar J. Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am J Physiol Gastrointest Liver Physiol. 1996;271:G56–G61. doi: 10.1152/ajpgi.1996.271.1.G56. [DOI] [PubMed] [Google Scholar]
- Yu P, Chen Q, Xiao Z, Harnett K, Biancani P, Behar J. Signal transduction pathways mediating CCK-induced gallbladder muscle contraction. Am J Physiol Gastrointest Liver Physiol. 1998;275:G203–G211. doi: 10.1152/ajpgi.1998.275.2.G203. [DOI] [PubMed] [Google Scholar]
- Zúñiga S, Molina H, Azocar L, Amigo L, Nervi F, Pimentel F, Jarufe N, Arrese M, Lammert F, Miquel JF. Ezetimibe prevents cholesterol gallstone formation in mice. Liver Int. 2008;28:935–947. doi: 10.1111/j.1478-3231.2008.01808.x. [DOI] [PubMed] [Google Scholar]