

Abstract
Ca2+-stimulated adenylyl cyclases (ACs) have recently been shown to play important roles in pacemaking in the sino-atrial node. Here we present evidence that Ca2+-stimulated ACs are functionally active in guinea-pig atrial myocytes. Basal activity of an AC in isolated atrial myocytes was demonstrated by the observations that MDL 12,330A (10 μm), an AC inhibitor, reduced L-type Ca2+ current (ICaL) amplitude, while inhibition of phosphodiesterases with IBMX (100 μm) increased ICaL amplitude. Buffering of cytosolic Ca2+ by exposure of myocytes to BAPTA-AM (5 μm) reduced ICaL amplitude, as did inhibition of Ca2+ release from the sarcoplasmic reticulum with ryanodine (2 μm) and thapsigargin (1 μm). [Ca2+]i-activated calmodulin kinase II (CaMKII) inhibition with KN-93 (1 μm) reduced ICaL, but subsequent application of BAPTA-AM further reduced ICaL. This effect of BAPTA-AM, in the presence of CaMKII inhibition, demonstrates that there is an additional Ca2+-modulated pathway (not dependent on CaMKII) that regulates ICaL in atrial myocytes. The effects of BAPTA could be reversed by forskolin (10 μm), a direct stimulator of all AC isoforms, which would restore cAMP levels. In the presence of BAPTA-AM, the actions of IBMX were reduced. In addition, inclusion of cAMP in the patch electrode in the whole-cell configuration prevented the effects of BAPTA. These effects are all consistent with a role for Ca2+-stimulated AC in the regulation of atrial myocyte ICaL.
Key points
Adenylyl cyclases (ACs), the enzymes that synthesize cAMP, are essential for the inotropic response of cardiac muscle to adrenergic stimulation.
Ca2+-stimulated isoforms of AC have previously been identified in the sino-atrial node of the heart, where they contribute to pacemaking.
In this study, we demonstrate that Ca2+-stimulated isoforms of AC regulate the L-type Ca2+ current in atrial myocytes.
This regulation of the L-type Ca2+ current provides a feedback mechanism for the Ca2+ released from intracellular stores to control Ca2+ entry to atrial myocytes.
These Ca2+-stimulated ACs may be significant for the compartmentalization of cAMP signalling in atrial myocytes.
Introduction
The entry of Ca2+ ions through L-type Ca2+ channels is the signal that initiates contraction of atrial myocytes (Bers, 2002). Unlike ventricular myocytes, atrial cells contain no or few T-tubules (Huser et al. 1996; Smyrnias et al. 2010) and so the majority of Ca2+ entry occurs at the cell surface. The sub-cellular distribution of ryanodine receptors (RyRs) in atrial myocytes is similar to ventricular myocytes with the main difference being the expression of RyR clusters at the periphery of atrial myocytes (Carl et al. 1995; Mackenzie et al. 2001). As a result of the presence of these clusters, the frequency of spontaneous Ca2+ sparks is much higher at the periphery of atrial myocytes (Woo et al. 2003; Sheehan et al. 2006) and the amplitude of Ca2+ transients is also greater at the periphery than in the centre (Kockskamper et al. 2001). This higher likelihood of Ca2+ release at the periphery of atrial myocytes may be important for the regulation of the L-type Ca2+ current, particularly its inactivation (Sun et al. 1997).
The second messenger cAMP plays a key role in the regulation of cardiac function. The cytosolic level of cAMP is determined by the dynamic equilibrium between production by adenylyl cyclases (ACs) and degradation by phosphodiesterases (PDEs). cAMP signalling is thought to be highly compartmentalized within a single myocyte and there have been numerous studies that have highlighted the importance of various different PDE isoforms in accomplishing this (Jurevicius & Fischmeister, 1996; Mongillo et al. 2006; Rochais et al. 2006; Leroy et al. 2008). However, there has been relatively little focus on the possibility that the expression of different AC isoforms may also be important for cAMP signalling.
Since the first mammalian adenylyl cyclase was cloned (Krupinski et al. 1989), eight other species have been identified in various different tissues. Shortly after the discovery of calmodulin as a Ca2+-dependent regulator of PDE1 (Cheung, 1970), AC activity in membranes from brain tissue was found to be stimulated by Ca2+ (Brostrom et al. 1975). The expression of AC1 in Sf9 cells provided evidence that this isoform was potently stimulated by Ca2+–calmodulin (Tang et al. 1991) with an apparent Kd of 100 nm (Wu et al. 1993). AC8 was discovered a few years later (Krupinski et al. 1992) and was also shown to be stimulated by Ca2+–calmodulin, with an apparent Kd of 500 nm (Cali et al. 1994).
We have recently shown that Ca2+-stimulated isoforms of adenylyl cyclase (AC1 and AC8) are expressed in the atria and sino-atrial node (SAN), where they regulate If and cardiac pacemaking (Mattick et al. 2007). In ventricular myocytes, the predominant AC isoform expressed is AC5 (Pieroni et al. 1993), which is inhibited by Ca2+ with a Kd of 200 nm (Colvin et al. 1991). It appears from the following evidence that myocytes from different regions of the heart may differ in the resting activity of their adenylyl cyclases. Acetylcholine activates an inhibitory G protein that reduces AC activity. Acetylcholine inhibits If and ICaL in SAN myocytes in the absence of any β-adrenoceptor stimulation (DiFrancesco & Tromba, 1988; Petit-Jacques et al. 1993), whereas in ventricular myocytes acetylcholine only reduces ICaL following β-adrenoceptor stimulation (Hescheler et al. 1986). These observations suggest that SAN myocytes have a higher resting level of cAMP production than ventricular myocytes. Atrial myocytes have the same pattern of AC expression as SAN myocytes and therefore may also have a significant resting production of cAMP (Mattick et al. 2007). The purpose of the experiments presented here was to identify whether the Ca2+-stimulated AC isoforms contribute to the regulation of atrial myocyte ICaL.
Methods
Myocyte isolation
Forty-eight male guinea–pigs (weighing 350–500 g) were killed by cervical dislocation following stunning in accordance with the University of Oxford's Policy on the Use of Animals in Scientific Research and the Home Office Guidance on the operation of The Animals (Scientific Procedures) Act 1986 (H.M.S.O.). The heart was then rapidly excised and washed in a modified Tyrode medium containing EGTA and heparin to prevent blood from clotting in the small coronary vessels. Following this, the heart was mounted on a Langendorff apparatus for retrograde perfusion via the aorta. Perfusion was initially carried out in a modified Tyrode solution containing (mm): NaCl 136, KCl 5.4, NaHCO3 12, sodium pyruvate 1, NaH2PO4 1, MgCl2 1, EGTA 0.04, glucose 5; gassed with 95% O2–5% CO2 to maintain a pH of 7.4. This was then replaced with a modified Tyrode solution after 2 min that contained 50 μl of 0.1 m CaCl2 and 30 mg of collagenase (type II, Worthington Biochemical Corp., Lakewood, NJ, USA), but no EGTA in addition to the other salts.
After this enzymatic digestion, the heart was removed from the cannula and the atria were separated from the ventricles. For the isolation of atrial myocytes, slices of the atria were triturated (mechanical disruption using a flame-smoothed glass pipette) and stored at 4°C in high potassium medium containing (mm): KCl 70, MgCl2 5, potassium glutamine 5, taurine 20, EGTA 0.04, succinic acid 5, KH2PO4 20, Hepes 5, glucose 10; pH to 7.2 with KOH.
Ca2+ epifluorescence measurements
Myocytes were mounted in a perfusion bath on the surface of a coverslip and superfused with a solution that contained (mm): NaCl 125, NaHCO3 25, KCl 5.4, NaH2PO4 1.2, MgCl2 1, glucose 5.5, CaCl2 1.8; pH to 7.4 with NaOH and oxygenated with 95% O2–5% CO2. Whole-cell Ca2+ transients were recorded using an epifluorescence recording system with the ratiometric Ca2+ indicator indo-5F. Excitation light was provided by a xenon arc lamp with a 340 ± 15 nm interference filter while emitted light was split by dichroic mirrors and detected by two photomultipliers with 405 ± 15 and 495 ± 15 nm bandpass filters. All Ca2+ epifluorescence experiments were conducted at 36°C. The Ca2+ transient data have been normalized to the pre-drug baseline in each individual myocyte, which allows us to detect any changes to Ca2+ transient amplitude and diastolic Ca2+. In order to accurately determine the cellular auto-fluorescence, myocytes were superfused with a solution containing 0 Ca2+, the Ca2+ ionophore ionomycin and 5 mm Mn2+ to quench the fluorescence signal from any indo-5F.
Electrophysiology
Isolated atrial myocytes were patched in the whole-cell configuration using glass microelectrodes. All electrophysiology experiments were conducted at 36°C. Electrodes were pulled in a two-stage process which produced electrodes of resistance 2.5–3.5 MΩ when filled with patch pipette solution. Patch solution contained (in mm): potassium aspartate 110, KCl 10, NaCl 5, MgCl2 5.2, Hepes 5, K2ATP 5; pH to 7.2 with KOH. This solution was kept as close as possible to the expected physiological conditions since the aim of the experiments was to investigate effects that are thought to depend on changes in cytosolic calcium. For the measurement of the L-type Ca2+ current, myocytes were clamped at a holding potential of −40 mV (to inactivate Na+ and T-type Ca2+ currents) and 200 ms step depolarisations were applied up to 0 mV every 5 s. For the construction of current–voltage (I–V) curves, myocytes were held at −40 mV and 200 ms step depolarisations were applied from −30 mV in 10 mV steps up to +40 mV. The amplitude of the L-type Ca2+ current was measured as the peak minus the end of pulse current. The decay times were calculated by fitting a standard double exponential decay curve from the peak to the end of pulse. Voltage-gated potassium channels are thought not to influence peak ICaL at 0 mV (in the case of IKr (rapid K+ channel) since rectification would keep these currents negligibly small, and in the case of IKs (slow K+ channel) since activation would be negligibly small at times less than 10 ms at this potential, while IKur (ultra-rapid K+ channel) was taken to be approximately constant throughout the pulse). Potassium currents might, however, be a small contaminating influence for decay times. Currents were digitized at 2 kHz, filtered at 1 kHz and analysed with pCLAMP 9. Data are expressed as mean ± SEM. Statistical significance (P < 0.05) was assessed using Student's paired or unpaired t test as appropriate.
Immunocytochemistry
Isolated cardiac cells were plated onto flamed coverslips and left to adhere for 15 min. Cells were first fixed in 4% paraformaldehyde–phosphate buffered saline (PBS) for 15 min. In order to dissolve the paraformaldehyde in PBS it was necessary to heat the mixture to 55°C and to add 10 m NaOH. The paraformaldehyde–PBS was then cooled to room temperature before the pH was adjusted to 7.4 with HCl. Once the cells were fixed they were washed in PBS (3 changes, 10 min each) and then permeabilised using the detergent Triton X-100 (0.1% in PBS, Sigma-Aldrich) for 15 min. After permeabilisation the cells were washed in PBS (3 changes, 10 min each) then blocked with PBS −10% normal donkey serum for 60 min at room temperature to reduce non-specific binding. After blocking, the cells were incubated with primary antibodies at 4°C overnight. The next day, cells were first washed with PBS (3 changes, 10 min each) before being incubated with secondary antibodies at RT for 60 min (either AlexaFluor 488 or AlexaFluor 555 conjugates) then washed with PBS (3 changes, 10 min each). Finally, the cells were mounted using Vectashield and permanently sealed. Cells were stored in the dark at 4°C and imaged within 2 days. For control experiments either the primary or secondary antibody stage was omitted. Primary antibodies against AC1, AC8 (SantaCruz Biotechnology, sc25743 and sc32128) and CaMKII were used at a 1:100 dilution and the RyR2 at a 1:400 dilution (Abcam, ab52476 and ab2827). Observations were carried out using a Zeiss LSM 510 laser scanning confocal microscope (×40 oil objective). For detection of AlexaFluor 488, fluorescence excitation was at 488 nm with emission collected >515 nm. An excitation filter of 543 nm and an emission filter at 600 ± 15 nm were used to detect AlexaFluor 555.
In order to quantify the relationship between the red and green signals that were created during double-labelling experiments, we carried out a pixel-by-pixel co-localization analysis. The analysis used was to plot the grey values in the different channels against one another on a scatter plot, whereby the intensity of a pixel in the green channel is plotted as the x-coordinate while the intensity of the corresponding pixel from the red channel is plotted as the y-coordinate. This method of plotting values should produce a line whose gradient can be calculated by linear regression. The slope of the linear approximation provides an indication of the association between the two signals. The spread of signals around the line can then be used to generate the correlation coefficient, also called the Pearson coefficient, which provides an estimate of the goodness of the approximation. The Pearson overlap coefficients that were generated by the ImageJ plugin Just Another Co-localization Plugin (Bolte & Cordelieres, 2006) were between −1 (total exclusion of the signals) and +1 (complete co-localization of the signals).
Results
Single guinea-pig atrial myocytes were field stimulated (2 ms pulses) to fire action potentials at a frequency of 1 Hz. MDL 12,330A (MDL, 10 μm, an inhibitor of AC) reduced Ca2+ transient amplitude by 48 ± 8% (P < 0.001, n= 7), increased the time to peak by 45 ± 5% (P < 0.001, n= 7) and increased the time to 50% decay by 37 ± 13% (P < 0.05, n= 7) (Fig. 1A and B). These results are consistent with MDL inhibiting adenylyl cyclases that are active in the absence of any membrane receptor agonists. As a follow-up to the effects of MDL, 1 μm H89 (a PKA inhibitor) reduced Ca2+ transient amplitude by 37 ± 5% (P < 0.01, n= 6), increased the time to peak by 19 ± 3% (P < 0.001, n= 6) and increased the time to 50% decay by 20 ± 6% (P < 0.05, n= 6) (Fig. 1C and D). As expected, the effects of H89 are similar to the effects of MDL as H89 inhibits PKA, which is the downstream effector of cAMP.
Figure 1. Production of cAMP occurs in the absence of any agonists.
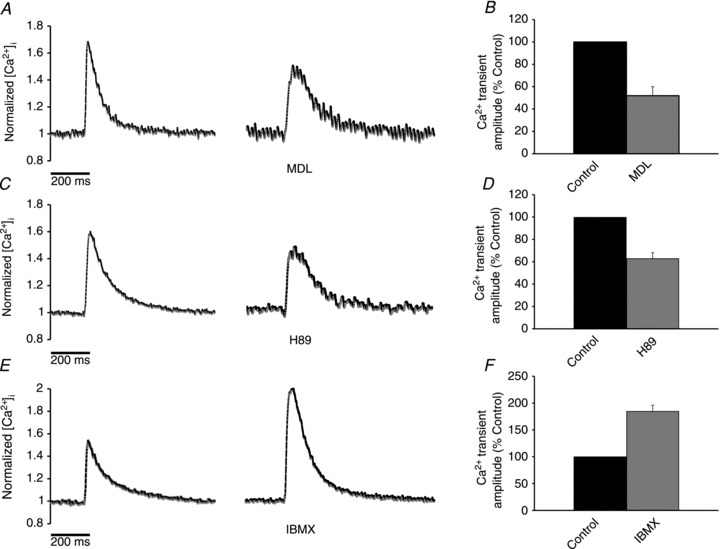
A, inhibition of adenylyl cyclases with MDL 12,330A (10 μm) reduced the Ca2+ transient amplitude and increased both the rise time and the decay time. B, bar graph showing the effect of MDL on Ca2+ transient amplitude. C, inhibition of PKA with H89 (1 μm) reduced the Ca2+ transient amplitude and increased both the rise time and the decay time. D, bar graph showing the effect of H89 on Ca2+ transient amplitude. E, inhibition of phosphodiesterases using IBMX (100 μm) increased Ca2+ transient amplitude. F, bar graph showing the effect of IBMX on Ca2+ transient amplitude.
The results with MDL and H89 support the hypothesis that there is a continual production of cAMP in atrial myocytes. It is reasonable to assume that this production is counterbalanced by the activity of phosphodiesterases to breakdown cAMP. This suggestion was tested using 100 μm IBMX (a non-selective PDE inhibitor), which is expected to inhibit over 95% of the activity of all phosphodiesterase sub-types that are present in the heart. IBMX (100 μm) increased Ca2+ transient amplitude by 85 ± 11% (P < 0.001, n= 6), but had no significant effect on the time to peak or the time to 50% decay (Fig. 1E and F).
Since the observations above are consistent with the suggestion that there is ongoing activity of adenylyl cyclase in the absence of activation of surface membrane receptors, the possibility was investigated that the cAMP produced by these pathways also has an effect on L-type Ca2+ currents (ICaL). Figure 2A shows the I–V curves for ICaL before and after superfusion with 10 μm MDL, an inhibitor of AC. Under these conditions MDL reduced ICaL amplitude by 39 ± 5% (P < 0.01, n= 7). As well as the reduction in current amplitude, MDL also increased the time to peak from 7.9 ± 2.0 to 9.8 ± 2.0 ms (32 ± 11%, P < 0.05, n= 7) and the τ (time constant of decay) from 22.0 ± 5.2 to 27.5 ± 5.0 ms (39 ± 12%, P < 0.05, n= 7). These effects of MDL are consistent with a reduction in cAMP levels that would lead to a decrease in the amount of active PKA and a reduction in the number of phosphorylated L-type Ca2+ channel subunits. This suggests that there is a basal production of cAMP (in the absence of any agonists).
Figure 2. Alterations in the synthesis or breakdown of cAMP regulate ICaL.
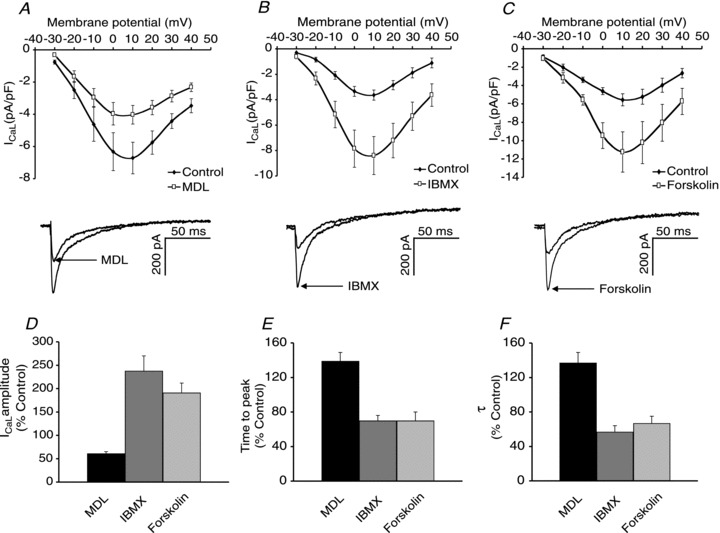
A, I–V curve and sample peak current showing the effect of MDL 12,330A (10 μm) to reduce ICaL. B, I–V curve and sample peak current showing the effect of IBMX (100 μm) to increase ICaL. C, I–V curve and sample peak current showing the effect of forskolin (10 μm) to increase ICaL. D, E and F, effect of MDL, IBMX and forskolin on ICaL amplitude (D), ICaL time to peak (E) and ICaLτ (F).
Figure 2B shows the I–V curves for ICaL before and after superfusion with 100 μm IBMX, a non-specific phosphodiesterase inhibitor, which increased peak ICaL by 107 ± 12% (P < 0.05, n= 6). As well as the increase in current amplitude, IBMX also reduced the time to peak from 8.1 ± 1.2 to 5.2 ± 0.8 ms (28 ± 7%, P < 0.01, n= 6) and the τ from 26.8 ± 4.6 to 14.7 ± 2.1 ms (43 ± 7%, P < 0.05, n= 6). Figure 2C shows the I–V curves for ICaL before and after superfusion with 10 μm forskolin, an activator of adenylyl cyclases. Forskolin increased peak ICaL by 91 ± 21% (P < 0.01, n= 8). As well as the increase in current amplitude, forskolin also reduced the time to peak from 7.8 ± 1.1 to 4.6 ± 0.5 ms (30 ± 10%, P < 0.01, n= 8) and the τ from 15.6 ± 2.1 to 10.4 ± 1.9 ms (33 ± 8%, P < 0.01, n= 8). This is consistent with the level of cAMP in atrial myocytes being regulated by the concurrent activity of AC and phosphodiesterases. Figure 2 also shows a summary of the effects of MDL and IBMX on ICaL peak amplitude (D), time to peak (E) and the τ (F).
Figure 3A shows that chelation of intracellular Ca2+ by superfusion with 5 μm BAPTA-AM reduced peak ICaL by 42 ± 4% (P < 0.01, n= 7). As well as the reduction in current amplitude, BAPTA-AM increased the time to peak from 6.9 ± 0.9 to 9.1 ± 1.1 ms (32 ± 8%, P < 0.05, n= 7) and the τ from 20.3 ± 3.4 to 24.8 ± 3.5 (28 ± 11%, P < 0.05, n= 7). Superfusion with BAPTA-AM results in the accumulation of a high concentration of free BAPTA in the cytosol (expected to be at least 150 μm on the basis of our experience comparing Ca2+ probes loaded via the patch pipette with application of the corresponding AM-ester; see Discussion). This concentration of cytosolic BAPTA would be expected to buffer free Ca2+ to levels (<10 nm) at which there would be no activity of Ca2+-stimulated ACs. The contention that free Ca2+ levels were lower than 10 nm was supported by a series of experiments in cells loaded with indo-5F and shown in Fig. 3B: BAPTA-AM reduced the fluorescence ratio to a level not statistically different (P > 0.05, n= 9) from the intrinsic fluorescence of the cell (taken to be the fluorescence left after Mn2+ is used to quench any indo-5F). These observations show that under the conditions of the experiments reported here exposure of myocytes to BAPTA-AM caused a fall in cytosolic Ca2+ to levels which are as low as can be detected with indo-5F. The observations are therefore consistent with regulation of peak ICaL by a Ca2+-dependent pathway.
Figure 3. Chelation of intracellular Ca2+ reduces ICaL.
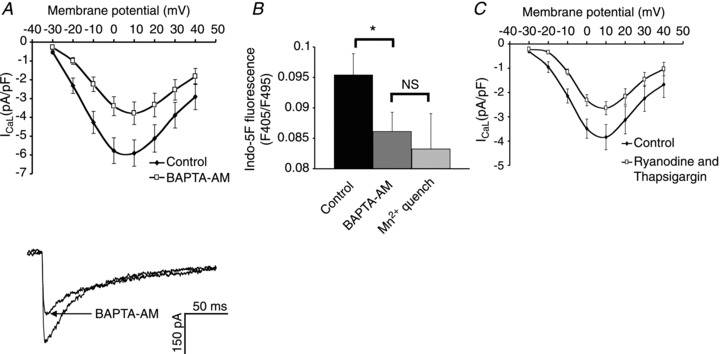
A, I–V curve and sample peak current showing the effect of superfusion with BAPTA-AM (5 μm) on ICaL. B, bar graph showing the reduction in indo-5F fluorescence that occurs with BAPTA-AM and following quenching of the dye with Mg2+. C, inhibition of sarcoplasmic reticulum Ca2+ release with ryanodine (2 μm) and thapsigargin (1 μm) reduces ICaL.
The application of ryanodine and thapsigargin to atrial myocytes depletes Ca2+ from the SR. Figure 3C shows the I–V curves for ICaL before and after superfusion with 2 μm ryanodine and 1 μm thapsigargin, and under these conditions the amplitude of ICaL was reduced by 33 ± 4% (P < 0.01, n= 7). As well as the reduction in current amplitude, ryanodine and thapsigargin increased the time to peak from 6.2 ± 1.1 to 7.8 ± 0.7 ms (38 ± 10%, P < 0.05, n= 7) and the τ from 23.8 ± 3.2 to 37.4 ± 3.0 ms (70 ± 17%, P < 0.01, n= 7). This indicates that Ca2+ released from the SR can modulate ICaL.
Figure 4A shows that superfusion with 1 μm KN-92 (an analogue of KN-93 that has little or no effect on CaMKII) had no effect on ICaL (P > 0.05, n= 6). In contrast, inhibition of CaMKII in Fig. 4B with 1 μm KN-93 reduced ICaL by 23 ± 5% (P < 0.01, n= 6; in other words a reduction to 77% of control). Application of 5 μm BAPTA-AM (in the presence of KN-93) further reduced peak ICaL to 53 ± 7% (P < 0.01, n= 6) of the control level (a further reduction of approximately 24%). This additional reduction (in the presence of CaMKII inhibition) demonstrates that there is an additional Ca2+-modulated pathway (not dependent on CaMKII) that regulates ICaL in atrial myocytes.
Figure 4. CaMKII inhibition in atrial and ventricular myocytes.
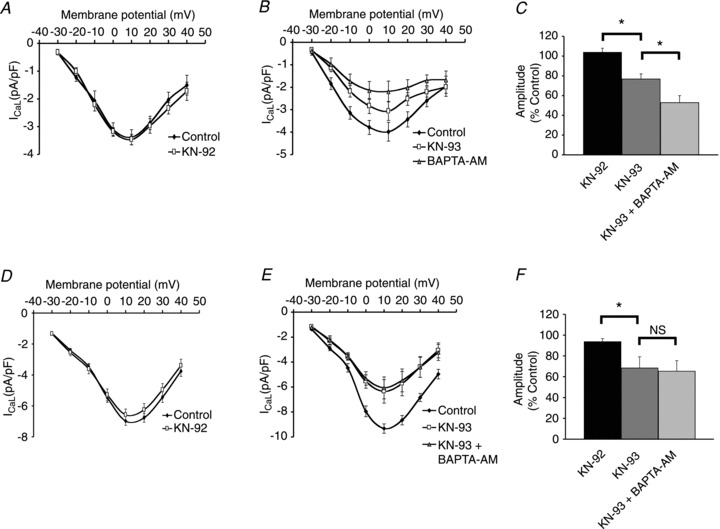
A, KN-92 (1 μm) had no effect on ICaL in atrial myocytes. B, KN-93 (1 μm) reduced peak ICaL amplitude. In the presence of KN-93, BAPTA-AM (5 μm) produced a further reduction in ICaL in atrial myocytes. C, comparison of the effects of KN-92, KN-93 and BAPTA-AM on peak ICaL amplitude in atrial myocytes. D, KN-92 (1 μm) produced a small but significant reduction in ICaL in ventricular myocytes. E, KN-93 (1 μm) reduced peak ICaL amplitude. In the presence of KN-93, BAPTA-AM (5 μm) had no further effect on ICaL in ventricular myocytes. F, comparison of the effects of KN-92, KN-93 and BAPTA-AM on peak ICaL amplitude in ventricular myocytes.
Similar experiments were carried out in ventricular myocytes. Superfusion with 1 μm KN-92 caused a small but significant reduction of ICaL by 6 ± 3% (P < 0.05, n= 7) in ventricular myocytes (Fig. 4D). Inhibition of CaMKII with 1 μm KN-93 (Fig. 4E) reduced ICaL by 31 ± 11% (P < 0.05, n= 6). In contrast to the observations in atrial myocytes, application of 5 μm BAPTA-AM (in the presence of KN-93) had no further effect on ICaL in ventricular myocytes (P > 0.05, n= 6). Figure 4C shows a bar graph summarizing the effects of KN-92, KN-93 and BAPTA-AM in atrial myocytes while Fig. 4F shows a bar graph summarizing the effects of KN-92, KN-93 and BAPTA-AM in ventricular myocytes.
The localization of CaMKII (Fig. 5A) was compared with the RyR2 (Fig. 5B) in atrial myocytes. CaMKII appears in a striated pattern throughout the cytosol of the cell and appears to co-localize with the RyRs (Fig. 5C and D). In order to quantify the relationship between the red and green signals, we carried out a pixel-by-pixel co-localization analysis (see Methods). The Pearson overlap coefficients was R= 0.63 ± 0.02 (n= 6) for CaMKII and RyR2 where the answer can be between −1 (total exclusion of the signals) and +1 (complete co-localization of the signals). The Pearson overlap coefficient value suggests that there is a reasonable degree of overlap of the two signals, which agrees with their visual appearance in which CaMKII and RyR2 appear to be closely associated throughout the majority of the cytosol but there is no CaMKII staining in the same place as the sub-sarcolemmal RyRs. Atrial myocytes lack T-tubules and so the L-type Ca2+ channels are located only at the sarcolemma. It therefore appears that CaMKII is separated from the L-type Ca2+ channels and so it is unable to exert a direct regulatory effect on them. Inhibition of CaMKII with KN-93 reduced the Ca2+ transient amplitude (indo-5F 405/495 fluorescence ratio reduced by 38 ± 2%, P < 0.001, n= 6, Supplemental Fig. S4) which, when combined with the observations about sub-cellular localization, is consistent with the suggestion that CaMKII is important for either maintaining the SR Ca2+ content (as phospholamban can be phosphorylated by CaMKII) or for maintaining the RyR release probability.
Figure 5. Immunolocalisation of RyRs and CaMKII.
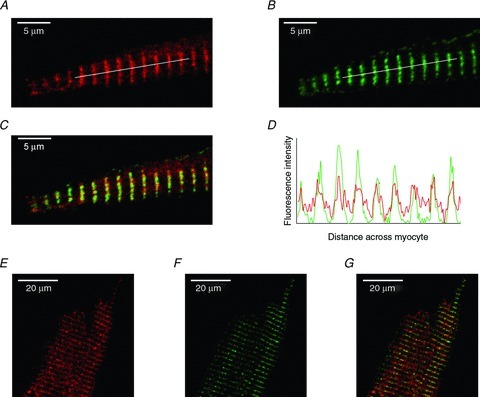
A, section of an atrial myocyte stained for CaMKII. B, section of an atrial myocyte stained for RyR2. C, overlay of CaMKII (A) and RyR2 (B). Any areas of yellow represent overlap of the red and green signals. D, fluorescence intensity profile for the lines shown in A and B. E, section of a ventricular myocyte stained for CaMKII. F, section of a ventricular myocyte stained for RyR2. G, overlay of CaMKII (E) and RyR2 (F). Any areas of yellow represent overlap of the red and green signals.
The localization of CaMKII (Fig. 5E) was compared with the RyR2 (Fig. 5F) in ventricular myocytes. CaMKII appears in a striated pattern throughout the cytosol of the cell and the Pearson overlap coefficient between CaMKII and the RyRs was R= 0.67 ± 0.03 (n= 6), which indicates a reasonable degree of co-localization between CaMKII and the RyRs (Fig. 5G). In ventricular myocytes, which contain an extensive network of T-tubules, the L-type Ca2+ channels are located very close to the RyRs throughout the cell and so CaMKII is in the correct location to directly influence L-type Ca2+ channels by phosphorylation. This observation, along with the finding that the effects of BAPTA-AM in ventricular myocytes can be solely attributed to the inhibition of CaMKII, suggests that Ca2+-stimulated ACs are not important in ventricular myocytes.
Standard immunocytochemical techniques were used to determine the localization of intracellular Ca2+ channels and AC isoforms. In order to compare the localization of various proteins, secondary antibodies conjugated to different fluorophores were used. Confocal images of atrial myocytes show staining in red and green, with any areas of yellow representing overlap of the red and green signals. As shown in Fig. 6A, labelling of the type 2 RyR (green) occurred in a striated pattern as well as sub-sarcolemmally. In contrast, AC1 (red) appeared to be localized in a ring just inside the RyR2 staining (Fig. 6B shows a close-up of the atrial myocyte in Fig. 6A). Figure 6C shows an atrial myocyte stained with primary antibodies against the RyR2 (green) and AC8 (red). AC8 appears to be co-localized with the RyRs that are located just beneath the sarcolemma (Fig. 6D shows a close-up of the atrial myocyte in Fig. 6C). This means that AC8, which has a Kd for Ca2+ of 500 nm is perfectly placed to be exposed to high levels of Ca2+ that occur in microdomains around the sub-sarcolemmal RyRs during cardiac EC coupling.
Figure 6. Immunolocalisation of RyRs and Ca2+-stimulated adenylyl cyclases.
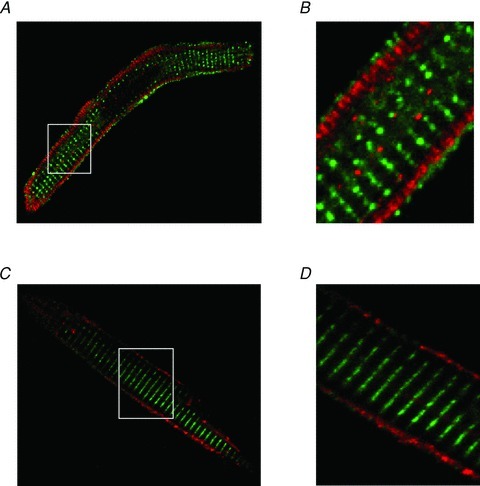
A, RyR2 (green) and AC1 (red). B, enlarged section of panel A identified by white box. C, RyR2 (green) and AC8 (red). D, enlarged section of panel C identified by white box.
In order to investigate the relative contribution of Ca2+-stimulated ACs (vs. Ca2+-insensitive AC) to the constitutive production of cAMP, a combination of PDE inhibition and Ca2+ chelation was used. Figure 7A shows the I–V curves for ICaL in atrial myocytes that were initially superfused with 100 μm IBMX, which increased peak ICaL by 109 ± 13% (P < 0.01, n= 7). The effects of IBMX were then reversed by returning to the control solution. Once the current returned to control levels, cells were superfused with 5 μm BAPTA-AM which reduced peak ICaL by 38 ± 4% (P < 0.01, n= 7). IBMX (100 μm) was then re-applied to the atrial myocytes (now loaded with BAPTA) which produced a 64 ± 20% (P < 0.05, n= 7) increase in peak ICaL (relative to the BAPTA-loaded value, Fig. 7B). The application of IBMX was able to return the current to values that were not statistically different from pre-BAPTA levels (Fig. 7C). Overall, the effects of IBMX were greatly reduced when atrial myocytes were loaded with BAPTA suggesting that a Ca2+-stimulated process contributes to the basal production of cAMP in these cells.
Figure 7. Chelation of intracellular Ca2+ reduces the production of cAMP.
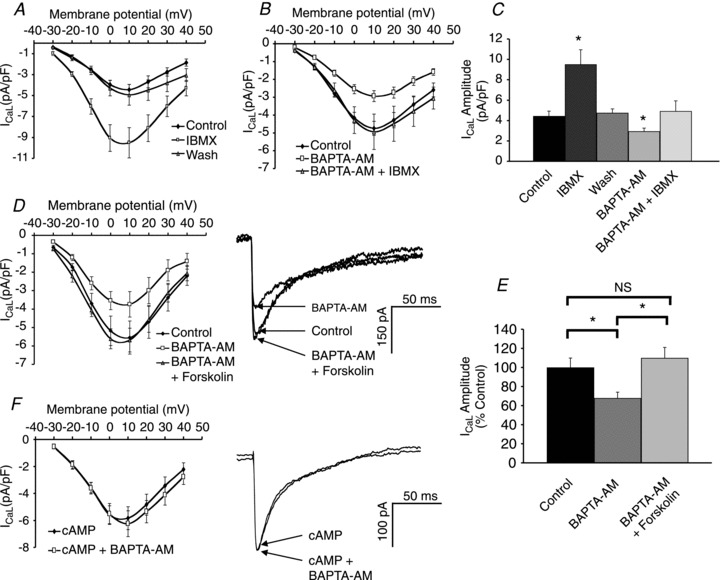
A, inhibition of phosphodiesterases with IBMX (100 μm) increased ICaL in a reversible manner. B, in the presence of BAPTA, inhibition of phosphodiesterases with IBMX had a greatly reduced effect on ICaL. C, comparison of the effects of IBMX in the presence and absence of BAPTA. D, I–V curve and sample peak currents showing that forskolin (10 μm) reversed the effects of BAPTA-AM on ICaL. E, in the presence of BAPTA-AM and forskolin, ICaL is not significantly different from control. F, I–V curve and sample peak currents showing that in the presence of 200 μm cAMP, BAPTA-AM has no effect on ICaL.
Figure 7D shows the I–V curves for ICaL in atrial myocytes that have been superfused with 5 μm BAPTA-AM and subsequently with 10 μm forskolin. BAPTA-AM reduced peak ICaL by 32 ± 6% (P < 0.01, n= 7). In the presence of BAPTA-AM, forskolin increased peak ICaL by 66 ± 15% (P < 0.01, n= 7). ICaL in the presence of BAPTA-AM and forskolin was not significantly different from control (P > 0.05, n= 7). The increase in ICaL with forskolin (shown in Fig. 2C) was significantly larger than the increase in ICaL with forskolin in the presence of BAPTA-AM (Fig. 7E). Forskolin directly activates ACs and the reduction of its effects in the presence of BAPTA-AM is consistent with the suggestion that the actions of BAPTA-AM on ICaL are mediated by an inhibition of ACs.
When 200 μm cAMP was included in the pipette filling solution there was a small increase in peak ICaL (16 ± 2%, P < 0.05, n= 6, Supplemental Fig. S5). Figure 7F shows the I–V curves before and after superfusion with 5 μm BAPTA-AM to myocytes that had their intracellular cAMP levels clamped in this way. Under these conditions, BAPTA-AM had no effect on ICaL (P > 0.05, n= 6). This provides additional evidence that the effects of BAPTA to decrease ICaL when cAMP is absent from the pipette, as reported above, are mediated by a reduction in cAMP synthesis since when cAMP is continuously present in the pipette solution (and therefore the cytosol) a reduction in the rate of production in cAMP by Ca2+-sensitive AC will have no effect on ICaL.
Discussion
The main findings reported here support the conclusion that Ca2+-stimulated ACs are active in the absence of substances that stimulate receptors in the surface membrane of guinea-pig atrial myocytes, and that these ACs regulate L-type Ca2+ channels.
Our conclusions depend on the ability of BAPTA-AM to enter the cells, and for the BAPTA subsequently released in the cytosol to chelate Ca2+. In a previous paper (Mattick et al. 2007) we have argued that exposure of atrial cells to 5 μm BAPTA-AM would be expected to result in accumulation of at least 150 μm cytosolic BAPTA. This estimate of at least a 30-fold higher concentration of free acid in the cytosol than the concentration of the AM-ester in the extracellular solution was based on a comparison of fluorescent Ca2+ probes loaded under similar conditions, where a similar level of fluorescence observed with a known concentration of free acid is achieved with a much lower concentration of AM-ester in the extracellular solution. BAPTA at 150 μm would be expected to reduce the cytosolic Ca2+ level in atrial cells from approximately 100 nm to an undetectable level (150 pm, calculated using Web Max C (http://www.stanford.edu/~cpatton/webmaxcS.htm) assuming cytosolic free Mg2+ of 0.7 mm). The reduction in free Ca2+ would be expected to inhibit Ca2+-stimulated adenylyl cyclase activity. In addition to the above calculations, it was observed that chelation of Ca2+ with BAPTA reduced Ca2+ to levels that are not detectable using indo-5F as a fluorescent probe for Ca2+.
BAPTA-AM reduced the L-type Ca2+ current amplitude and appeared to slow down the kinetics of channel opening and closing. Any reduction of inactivation of ICaL in the presence of BAPTA would be expected to increase rather than decrease the amplitude of ICaL. The observed reduction in amplitude of ICaL is therefore consistent with a reduction in PKA-dependent phosphorylation of the channel as a consequence of reduced activity of Ca2+-stimulated ACs. Reduced inactivation of ICaL due to changes in the Ca2+ signalling could contribute to the slowing of decay but pharmacological inhibition of ACs produced a similar magnitude reduction in ICaL and also slowed the kinetics of the current. Conversely, inhibition of PDEs increased ICaL amplitude and accelerated the kinetics of the current. These observations are consistent with the suggestion that there is a significant production of cAMP in the absence of membrane receptor stimulation that is mediated by AC1 or AC8. These isoforms appear to be activated by the resting levels of cytosolic Ca2+ in atrial myocytes. This activation of AC1 and AC8 in turn regulates ICaL. The observation that depletion of Ca2+ in the SR also reduces ICaL amplitude and slows the current's kinetics suggests that AC1 and AC8 are further stimulated by Ca2+ released from the SR. Additional evidence that the cytosolic level of Ca2+ influences the production of cAMP comes from the observation that PDE inhibition has less of a stimulatory effect on ICaL in the presence of BAPTA-AM (as shown by the contrast between Figs 2B and 7B). It should also be noted that the lack of effect of BAPTA-AM on ICaL when cAMP was present in the pipette (discussed in more detail below) shows that the effects of BAPTA-AM discussed above were not the result of non-specific toxic actions.
The acceleration of decay times observed with forskolin and IBMX, both of which will increase PKA-dependent phosphorylation of L-type Ca2+ channels, is most probably a secondary effect of increased SR Ca2+ release (which is also a result of the activation of PKA). Conversely, the slowing of ICaL decay observed with MDL, BAPTA-AM or ryanodine and thapsigargin is most probably due to a reduction in SR Ca2+ release or a reduction in the Ca2+ that can reach the channel mouth (due to Ca2+ buffering). These alterations in SR Ca2+ release will change the extent of Ca2+-dependent inactivation of ICaL in addition to any effects resulting directly from PKA-dependent phosphorylation of L-type Ca2+ channels.
Inhibition of CaMKII reduces Ca2+ transients and Ca2+ currents in atrial myocytes. In the presence of CaMKII inhibition, chelation of intracellular Ca2+ in atrial (but not ventricular) myocytes further reduced ICaL, demonstrating that in atrial myocytes there is an additional Ca2+-sensitive process that regulates ICaL under basal conditions. However, when intracellular cAMP levels were clamped by the inclusion of 200 μm cAMP in the patch pipette, chelation of Ca2+ had no effect on ICaL, even though this would be expected to inhibit CaMKII as well as any other Ca2+-dependent processes. This observation supports a model in which the effects of Ca2+ chelation on ICaL are mediated entirely by a reduction in cAMP levels, but this appears not to be consistent with the observed effects of CaMKII inhibition with KN-93. A possible explanation of this apparent contradiction is that the effect of CaMKII in atrial myocytes may be primarily on SR proteins and so inhibition of CaMKII will reduce the SR Ca2+ content and/or the release probability of RyRs. The resulting reduction in Ca2+ release from the SR will then produce a reduction in Ca2+-stimulated AC activity, leading to a reduction in ICaL. This hypothesis is supported by the observation that inhibition of CaMKII in the absence of a functional SR (i.e. in the presence of ryanodine and thapsigargin) has no further effect on ICaL (see Supplemental Fig. S4). Further support for this idea comes from the immunostaining experiments that looked at the intracellular localization of CaMKII. In atrial myocytes, CaMKII was co-localized with the RyRs. The lack of T-tubules in atrial myocytes means that all of the L-type Ca2+ channels are located at the cell surface, and so the majority of the CaMKII revealed by immunostaining close to the SR appears to be quite far removed from the L-type Ca2+ channels.
In this proposed mechanism, reducing cytosolic Ca2+ with BAPTA will have dual effects, both directly inhibiting Ca2+-stimulated ACs, and indirectly influencing Ca2+-stimulated ACs as a consequence of the reduced Ca2+ transient amplitude following CaMKII inhibition. Maintained cAMP concentrations from a patch pipette overcome both direct and indirect inhibition of Ca2+-stimulated ACs but should have no effect on the degree of CaMKII phosphorylation of L-type Ca2+ channels.
In ventricular myocytes, which do not express AC1 but appear to contain AC8 (Mattick et al. 2007), inhibition of CaMKII reduces ICaL and chelation of Ca2+ has no further effect, suggesting that Ca2+-stimulated ACs are not important for the regulation of ICaL in the ventricles. This observation also supports the suggestion that the concentration of KN-93 used here does cause maximal inhibition of CaMKII, since if this were not the case, BAPTA loading would be expected to further reduce ICaL in ventricular myocytes. The localization of CaMKII in atrial and ventricular myocytes may also help to explain the differences between the observations in the two cell types. In ventricular myocytes, CaMKII is co-localized with the RyRs. As ventricular myocytes have an extensive T-tubule network, L-type Ca2+ channels will be located in close apposition to the RyRs and so CaMKII is ideally placed to directly phosphorylate the L-type Ca2+ channels.
The close apposition between AC8 and the sub-sarcolemmal RyRs means that AC8 should be exposed to high levels of Ca2+ during a Ca2+ transient that will stimulate the production of cAMP. Small increases in the level of Ca2+-bound calmodulin during a Ca2+ transient have been detected in adult cardiac myocytes using a FRET-based probe (Maier et al. 2006). This finding supports the idea that AC8 activity will increase during Ca2+ transients, as AC8 stimulation requires Ca2+-bound calmodulin. The sub-cellular localization of signalling proteins is very important for determining their role in regulating different cellular processes. AC1 appears to be located just on the intracellular side of the sub-sarcolemmal RyRs and the Kd for Ca2+ in the case of AC1 is much lower than the Kd for Ca2+ for AC8. These differences may reflect different roles for AC1 and AC8. The lower Kd for Ca2+ means that AC1 should be active more of the time than AC8 and so it may be the isoform responsible for more of the basal production of cAMP than AC8. In the case of AC8, the higher Kd for Ca2+ may mean that it provides a mechanism to amplify increases in the Ca2+ transient by providing cAMP and subsequent PKA activation. Another possibility is that the cAMP produced by AC8 during an increase in Ca2+ transient amplitude may be important for maintaining the increased transient amplitude by increasing the entry of Ca2+ through L-type channels.
We have also provided direct measurements of changes in cAMP in the Supplemental material. Resting levels of cAMP were detected in the absence of agonists to stimulate surface receptors, and the changes with BAPTA-AM and IBMX were broadly in line with the functional evidence reported above. However, an important caveat relating to these observations is that we have measured global cellular cAMP while we would expect that microdomains of cAMP close to the surface membrane where AC1 and AC8 are located might be particularly important. It seems possible that proportionate effects of, for example, Ca2+ chelation with BAPTA, might be greater on cAMP levels close to the surface membrane than was detected in the concentrations averaged over the whole cell.
In summary, the observations reported here provide evidence that Ca2+-stimulated adenylyl cyclases are functionally active in guinea-pig atrial myocytes. The cAMP produced by Ca2+-stimulated adenylyl cyclases regulates the L-type Ca2+ current. The activity of the Ca2+-stimulated adenylyl cyclases is regulated by Ca2+ in the physiological range and can be influenced by Ca2+ release from the SR. These Ca2+-stimulated adenylyl cyclases may make an important contribution to the compartmentalization of cAMP signalling in atrial muscle. Abnormalities of intracellular Ca2+ signalling and reductions of ICaL density are part of the pathology of atrial fibrillation (Yue et al. 1997; Sun et al. 1998; Dobrev & Nattel, 2008) and it is possible that changes in the activity of Ca2+-stimulated ACs are part of the pathological progression of atrial fibrillation.
Acknowledgments
This work was funded by the British Heart Foundation (PhD Studentship FS/05/121 to T.P.C.) and the Wellcome Trust and Higher Education Funding Council for England (D.A.T.).
Glossary
Abbreviations
- AC
adenylyl cyclase
- CaMKII
[Ca2+]i-activated calmodulin kinase II
- ICaL
Ltype Ca2+ current
- PDE
phosphodiesterase
- RyR
ryanodine receptor
- SAN
sino-atrial node
- SR
sarcoplasmic reticulum
Author contributions
T.P.C. was involved in the design, collection, analysis and interpretation of the data as well as drafting, revising and approving the final version of the article for publication. D.A.T. was involved in the conception, design and interpretation of the data as well as drafting, revising and approving the final version of the article for publication.
References
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brostrom CO, Huang YC, Breckenridge BM, Wolff DJ. Identification of a calcium-binding protein as a calcium-dependent regulator of brain adenylate cyclase. Proc Natl Acad Sci U S A. 1975;72:64–68. doi: 10.1073/pnas.72.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali JJ, Zwaagstra JC, Mons N, Cooper DM, Krupinski J. Type VIII adenylyl cyclase. A Ca2+/calmodulin stimulated enzyme expressed in discrete regions of rat brain. J Biol Chem. 1994;269:12190–12195. [PubMed] [Google Scholar]
- Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ, Jr, Vaghy PL, Meissner G, Ferguson DG. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol. 1995;129:673–682. doi: 10.1083/jcb.129.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WY. Cyclic 3',5'-nucleotide phosphodiesterase. Demonstration of an activator. Biochem Biophys Res Commun. 1970;38:533–538. doi: 10.1016/0006-291x(70)90747-3. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Oibo JA, Allen RA. Calcium inhibition of cardiac adenylyl cyclase. Evidence for two distinct sites of inhibition. Cell Calcium. 1991;12:19–27. doi: 10.1016/0143-4160(91)90081-o. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tromba C. Inhibition of the hyperpolarization-activated current if induced by acetylcholine in rabbit sino-atrial node myocytes. J Physiol. 1988;405:477–491. doi: 10.1113/jphysiol.1988.sp017343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Nattel S. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J Cardiovasc Pharmacol. 2008;52:293–299. doi: 10.1097/FJC.0b013e318171924d. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Kameyama M, Trautwein W. On the mechanism of muscarinic inhibition of the cardiac Ca current. Pflugers Arch. 1986;407:182–189. doi: 10.1007/BF00580674. [DOI] [PubMed] [Google Scholar]
- Huser J, Lipsius SL, Blatter LA. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol. 1996;494:641–651. doi: 10.1113/jphysiol.1996.sp021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc Natl Acad Sci U S A. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA. Activation and propagation of Ca2+ release during excitation-contraction coupling in atrial myocytes. Biophys J. 2001;81:2590–2605. doi: 10.1016/S0006-3495(01)75903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupinski J, Coussen F, Bakalyar HA, Tang WJ, Feinstein PG, Orth K, Slaughter C, Reed RR, Gilman AG. Adenylyl cyclase amino acid sequence: possible channel- or transporter-like structure. Science. 1989;244:1558–1564. doi: 10.1126/science.2472670. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Lehman TC, Frankenfield CD, Zwaagstra JC, Watson PA. Molecular diversity in the adenylylcyclase family. Evidence for eight forms of the enzyme and cloning of type VI. J Biol Chem. 1992;267:24858–24862. [PubMed] [Google Scholar]
- Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol. 2001;530:417–429. doi: 10.1111/j.1469-7793.2001.0417k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LS, Ziolo MT, Bossuyt J, Persechini A, Mestril R, Bers DM. Dynamic changes in free Ca-calmodulin levels in adult cardiac myocytes. J Mol Cell Cardiol. 2006;41:451–458. doi: 10.1016/j.yjmcc.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the If pacemaker current. J Physiol. 2007;582:1195–1203. doi: 10.1113/jphysiol.2007.133439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- Petit-Jacques J, Bois P, Bescond J, Lenfant J. Mechanism of muscarinic control of the high-threshold calcium current in rabbit sino-atrial node myocytes. Pflugers Arch. 1993;423:21–27. doi: 10.1007/BF00374956. [DOI] [PubMed] [Google Scholar]
- Pieroni JP, Miller D, Premont RT, Iyengar R. Type 5 adenylyl cyclase distribution. Nature. 1993;363:679–680. doi: 10.1038/363679a0. [DOI] [PubMed] [Google Scholar]
- Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KA, Zima AV, Blatter LA. Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J Physiol. 2006;572:799–809. doi: 10.1113/jphysiol.2005.103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, Bootman MD. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium. 2010;47:210–223. doi: 10.1016/j.ceca.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Sun H, Gaspo R, Leblanc N, Nattel S. Cellular mechanisms of atrial contractile dysfunction caused by sustained atrial tachycardia. Circulation. 1998;98:719–727. doi: 10.1161/01.cir.98.7.719. [DOI] [PubMed] [Google Scholar]
- Sun H, Leblanc N, Nattel S. Mechanisms of inactivation of L-type calcium channels in human atrial myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H1625–H1635. doi: 10.1152/ajpheart.1997.272.4.H1625. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Krupinski J, Gilman AG. Expression and characterization of calmodulin-activated (type I) adenylylcyclase. J Biol Chem. 1991;266:8595–8603. [PubMed] [Google Scholar]
- Woo SH, Cleemann L, Morad M. Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res. 2003;92:e1-e11. doi: 10.1161/01.res.0000051887.97625.07. [DOI] [PubMed] [Google Scholar]
- Wu Z, Wong ST, Storms DR. Modification of the calcium and calmodulin sensitivity of the type I adenylyl cyclase by mutagenesis of its calmodulin binding domain. J Biol Chem. 1993;268:23766–23768. [PubMed] [Google Scholar]
- Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]