

Abstract
Acid–base transport in the vascular wall remains incompletely understood. Here, we investigated (a) implications of Na+/H+ exchanger NHE1 knockout for vascular smooth muscle (VSMC) and endothelial cell (EC) pHi regulation, mesenteric artery morphology, vasomotor function and blood pressure regulation, and (b) consequences of sustained EC and VSMC acidification for vasomotor function. Na+/H+ exchange activity was abolished in VSMCs and ECs from NHE1 knockout mice, but with CO2/HCO3− present, steady-state pHi was unaffected. Active tension was 30% smaller in arteries from NHE1 knockout than wild-type mice, and media thickness equally reduced. Number of VSMCs per unit artery length was unchanged whereas volume and cross-sectional area of individual VSMCs were reduced. Media stress, force production per VSMC cross-sectional area and VSMC Ca2+ responses were unaffected. Blood pressure was 25 mmHg lower in NHE1 knockout than wild-type mice. Omission of CO2/HCO3− caused VSMCs and ECs to acidify substantially more in NHE1 knockout (0.3–0.6 pH-units) than wild-type (0.02–0.1 pH units) mice. Removing CO2/HCO3− inhibited acetylcholine-induced NO-mediated relaxations in arteries from NHE1 knockout but not wild-type mice. Without CO2/HCO3−, effects of NO synthase and rho kinase inhibition on noradrenaline-induced contractions were smaller in arteries from NHE1 knockout than wild-type mice whereas the EC Ca2+ response to acetylcholine, VSMC Ca2+ response to noradrenaline and vasorelaxation to S-nitroso-N-acetylpenicillamine were unaffected. In conclusion, NHE1 mediates the Na+/H+ exchange in ECs and VSMCs. Under physiological conditions, CO2/HCO3−-dependent mechanisms mask the pHi-regulatory function of NHE1. NHE1 knockout causes hypotrophy of VSMCs, reduced artery tension and lower blood pressure. At acidic pHi, NO-mediated vasorelaxation and rho kinase-dependent VSMC Ca2+ sensitivity are reduced.
Key points
Small arteries are important for regulation of blood pressure and local blood flow.
Changes in intracellular pH alter artery tone although the mechanistic background has been unclear.
Using knockout mice for Na+/H+ exchanger NHE1 with reduced acid extrusion from cells in the arterial wall and low intracellular pH in the absence of CO2/HCO3-, we show that intracellular acidification alters enzymatic activity and consequently artery dilatation and contraction.
Although lack of NHE1 does not affect intracellular pH in the arterial wall when CO2/HCO3− is present, arteries from NHE1 knockout mice have thinner walls and produce less active force due to reduced volume and cross-sectional area of individual smooth muscle cells.
These results highlight the interplay between intracellular pH and artery function, provide new targets to consider for modulating artery structure, and underscore the need to evaluate acid–base transport in conditions of vascular disease and blood pressure disturbances.
Introduction
Na+/H+ exchange and Na+–HCO3− cotransport constitute the important net acid extrusion mechanisms in vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) of resistance arteries (Aalkjaer & Cragoe, 1988; Aalkjaer & Hughes, 1991; Boedtkjer et al. 2006, 2008, 2011; Boedtkjer & Aalkjaer, 2009). We recently showed that knockout (KO) of the electroneutral Na+–HCO3− cotransporter NBCn1 abolishes all Na+–HCO3− cotransport activity and causes a lower steady-state pHi in VSMCs and ECs of mouse mesenteric arteries (Boedtkjer et al. 2011). Furthermore, NBCn1 KO mice display inhibited endothelial NO production and a lower rho kinase-dependent VSMC Ca2+ sensitivity with implications for blood pressure regulation (Boedtkjer et al. 2011). The importance of Na+/H+ exchange for VSMC and EC pHi control, resistance artery function and morphology, and blood pressure regulation has not previously been conclusively determined.
The Na+/H+ exchangers (NHEs) are gathered in the slc9 family consisting of at least eight members with different expression profiles: NHE1–5 and NHE8 are expressed in plasma membranes while NHE6 and NHE7 are important for transport across organelle membranes (Orlowski & Grinstein, 2004; Nakamura et al. 2005). NHE1 (slc9a1) is ubiquitously expressed in mammalian cells and investigation of NHE1 KO mice has elucidated its importance for acid extrusion in neurons (Yao et al. 1999; Luo et al. 2005) and in epithelial cells from renal thick ascending limbs (Good et al. 2004), parotid glands (Evans et al. 1999), pancreatic acini (Brown et al. 2003) and choroid plexus (Damkier et al. 2009). We have previously shown that NHE1 is the only plasma membrane NHE-isoform expressed at mRNA level in mouse mesenteric arteries (Boedtkjer & Aalkjaer, 2009).
NHEs play a role for migration and proliferation of cultured cells (Orlowski & Grinstein, 2004). Recently, NHE1 KO mice were shown to be unsusceptible to hypoxia-induced pulmonary hypertension suggesting a role for NHE1 in artery remodelling (Yu et al. 2008). Whether NHE1 is important for the morphology of arteries in the systemic circulation is unknown. Structural changes in the arterial wall develop under a number of pathological conditions and are prominent during disturbances of blood pressure. As such, it has been shown that arteries from hypertensive patients have a larger media thickness to lumen diameter ratio than arteries from normotensive controls (Heagerty et al. 1993) and that the structural abnormalities improve during antihypertensive treatment (Heagerty et al. 1988). Importantly, recent studies have shown that abnormal artery structure is an independent predictor of cardiovascular events (Rizzoni et al. 2003; Mathiassen et al. 2007) underscoring the need for a more thorough understanding of the basic cellular functions controlling artery morphology and how these targets might be manipulated.
The role of sustained changes in EC and VSMC pHi for resistance artery function has until recently been difficult to investigate experimentally. The commonly used application of weak acids or bases to induce acute pHi changes is promptly followed by recovery towards physiological levels, and these substances may furthermore have direct, pHi-independent vascular effects (McKinnon et al. 1996; Aaronson et al. 1996; Aalkjaer et al. 1998). A number of targets and mechanistic explanations for pHi-induced changes in resistance artery function have, however, been proposed. Effects on endothelial enzymes such as endothelial nitric oxide synthase (Fleming et al. 1994; Boedtkjer et al. 2011) and endothelin converting enzyme (Ahn et al. 1992), vascular ion channels such as Ca2+-activated K+ channels (Peers & Green, 1991; Schubert et al. 2001) and L-type Ca2+ channels (Klockner & Isenberg, 1994), and the Ca2+ sensitivity of the contractile machinery (Mrwa et al. 1974; Gardner & Diecke, 1988; Boedtkjer et al. 2006, 2011) have been suggested.
We designed the present study to address two important questions. Firstly, we investigated the vascular effects of a sustained low EC and VSMC pHi. Previous conclusions rely largely on the vascular effects of NBCn1 knockout, and by targeting a different membrane transporter with equivalent transport function, we investigated whether altered pHi regulation or other changes secondary to the genetic manipulation are responsible. Secondly, we investigated to what extent knockout of NHE1 has vascular consequences under physiological conditions by studying the function and morphology of mesenteric small arteries and effects on blood pressure.
Methods
Ethical approval
All animal procedures were approved by the Danish Animal Care and Use Committee under the Danish Ministry of Justice.
NHE1 KO mice
Mice on an FVB/N genetic background and deficient in NHE1 were generously provided by Dr Gary E. Shull (University of Cincinnati, USA). Heterozygous mice were crossed to produce NHE1 KO and wild-type (WT) mice. As previously reported, a large proportion of the NHE1 KO mice die at an early age (Bell et al. 1999). Consequently, the mice were investigated at 3–4 weeks of age. Mice were killed by cervical dislocation and the mesenteric bed excised. First order mesenteric arteries were dissected free from surrounding connective tissue and mounted for isometric force recordings in wire myographs (DMT, Aarhus, Denmark).
Measurement of pHi and intracellular Ca2+ responses in ECs and VSMCs
We monitored EC and VSMC pHi in isolated mesenteric arteries from NHE1 KO and WT mice using the pH-sensitive fluorophores 2′,7′-bis-(2-carboxypropyl)-5-(and-6)-carboxyfluorescein (BCPCF) and 2′-7′-bis-(2-carboxyethyl)-5(and-6)-car boxyfluorescein (BCECF) as previously described (Boedtkjer et al. 2006; Boedtkjer & Aalkjaer, 2009). Intracellular buffering capacity of VSMCs was calculated from the change in pHi upon addition and washout of 20 mm NH4Cl and no apparent difference in intracellular buffering capacity was found between VSMCs from NHE1 KO and WT mice in the presence or absence of CO2/HCO3− (online Supplemental Material, Supplementary Fig. 1). Consistent with previous findings from smooth muscle cells (Aalkjaer & Cragoe, 1988; Aalkjaer & Hughes, 1991; Eiesland et al. 1991; Aickin, 1994; Boedtkjer et al. 2008, 2011), the intracellular buffering capacity did not appear to be affected by CO2/HCO3− in particular at low pHi values (Supplementary Fig. 1) where transport activities were quantified. VSMCs in arteries from both NHE1 KO and WT mice (with and without CO2/HCO3−) acidified to pHi values between 6.4 and 6.5 following removal of NH4Cl and Na+ from the bath. The rate of net Na+-dependent acid extrusion was calculated by multiplication of the intracellular buffering capacity with the initial rate of recovery from this pHi value upon re-addition of bath Na+ as previously described (Boedtkjer et al. 2006).
The intracellular Ca2+ response of ECs to acetylcholine was studied in isolated mesenteric arteries dually loaded with Calcium Green-1 and Fura Red using confocal microscopy as described elsewhere (Boedtkjer et al. 2011). The use of Fura-2 to evaluate intracellular Ca2+ responses in VSMCs has previously been described (Boedtkjer et al. 2006).
Measurement of lumen diameter, media thickness and artery tone
Wall dimensions were measured on arteries mounted in wire myographs (DMT, Denmark) as previously described (Mulvany et al. 1978). Arteries were gently stretched until a low tension was recorded. The adventitial, media and intimal thicknesses were measured at six individual points.
The lumen diameter at a transmural pressure of 100 mmHg was calculated using Laplace's law and arteries normalized to 90% of the internal diameter at this pressure (Mulvany & Halpern, 1977). Vaso-constriction to noradrenaline and elevated extracellular [K+] and vasorelaxation to acetylcholine and S-nitroso-N-acetylpenicillamine (SNAP) were investigated. NO synthase and rho kinase activity were inhibited using 100 μmNG-nitro-l-arginine methyl ester (l-NAME) and 10 μm Y-27632, respectively.
For histology, arteries were fixed in 2% glutaraldehyde for 1 h, plastic embedded, cut to 3 μm thick sections and Giemsa stained (Supplementary Fig. 2). The density and volume of cells in the media were evaluated by stereological analyses (disector) by light microscopy of two consecutive sections as previously described in detail (Baandrup et al. 1985; Mulvany et al. 1985).
Blood pressure measurements
Systemic blood pressure was measured non-invasively from 3- to 4-week-old NHE1 KO and WT mice by determining the tail blood volume with a volume pressure recording sensor and an occlusion tail-cuff (CODA System, Kent Scientific, Torrington, CT, USA). We have shown elsewhere that blood pressure effects of NBCn1 KO mice determined by tail-cuff measurements are in agreement with intra-arterial telemetry-based measurements (Boedtkjer et al. 2011).
Solutions
The HCO3− containing physiological salt solution (PSS) consisted of (in mm): 114 NaCl, 10 Hepes, 25 NaHCO3, 1.20 MgSO4, 4.70 KCl, 5.50 glucose, 0.026 EDTA, 1.18 KH2PO4, and 1.60 CaCl2. In bicarbonate-free solutions, NaHCO3 was replaced with an equimolar amount of NaCl. In Na+-free solutions, NaCl was replaced with an equimolar amount of N-methyl-D-glucamine titrated with HCl. The K-PSS was obtained by substituting KCl for NaCl. HCO3− containing solutions were bubbled with 5% CO2, balance air, whereas HCO3− free solutions were bubbled with air (nominally CO2 free); pH was adjusted to 7.40 at 37°C.
Statistics
Data are expressed as means ± SEM. Student's unpaired, two-tailed t test was used for comparison of two groups. To evaluate the effects of two variables on the measured variable, we used two-way ANOVA followed by Bonferroni's post hoc test. When the variable was measured multiple times for each mouse, a repeated measures two-way ANOVA was employed. Concentration–response relationships were analysed with sigmoidal curve fits and the derived log(EC50) and maximum values compared with extra sum-of-squares F tests. P < 0.05 was considered statistically significant; n equals number of mice. Statistical analyses were performed using GraphPad Prism 5.02 software (GraphPad Software Inc., La Jolla, CA, USA).
Results
NHE1 is the only functionally important Na+/H+ exchanger in VSMCs
The recovery of pHi from an intracellular acidification was investigated in VSMCs of isolated mesenteric arteries following an NH4+ prepulse. Original traces are shown in Supplementary Fig. 3. Addition of NH4Cl causes abrupt alkalinisation as NH3 enters the VSMCs. NH4+ influx contributes to the subsequent gradual fall in pHi. Upon washout of NH4Cl, NH3 leaves the cells with consequent intracellular accumulation of protons. NH4Cl was washed out into a Na+-free solution and in the absence of extracellular Na+ almost no recovery of pHi was seen (Fig. 1A). VSMCs of arteries from WT mice displayed a strong pHi recovery following addition of Na+ in the absence of CO2/HCO3− (Fig. 1A and C). This recovery was almost completely abolished in VSMCs of arteries from NHE1 KO mice (Fig. 1A and C). No efficient recovery of pHi was seen in VSMCs of arteries from NHE1 KO mice until CO2/HCO3− was introduced into the bath solution (Fig. 1A) with consequent activation of Na+–HCO3− cotransport (Boedtkjer et al. 2011).
Figure 1. NHE1 is the only functionally important Na+/H+ exchanger in VSMCs and becomes important for steady-state pHi regulation only in the absence of CO2/HCO3−.
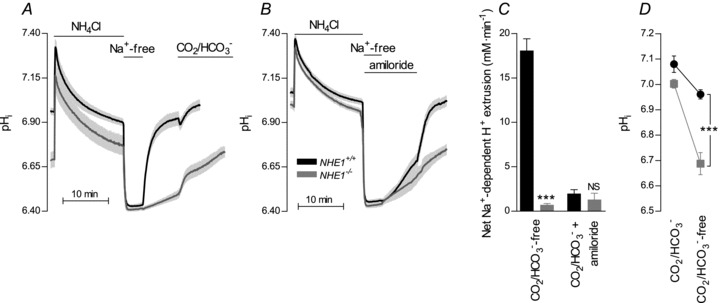
A, in the absence of CO2/HCO3−, the Na+-dependent pHi recovery was abolished in VSMCs of arteries from NHE1 KO mice (n= 5–7). Experiments were performed in the absence of CO2/HCO3− except for the final part (as indicated). B, the Na+- and HCO3−-dependent, amiloride-insensitive pHi recovery was unaltered in VSMCs of arteries from NHE1 KO mice (n= 5–6). Experiments were performed in the presence of CO2/HCO3−. C, average net H+ extrusion from VSMCs measured at average pHi values between 6.43 and 6.65. Na+/H+ exchange was abolished while Na+–HCO3− cotransport was unaffected in arteries from NHE1 KO mice. Comparisons were made with Student's two-tailed unpaired t test. D, in the absence of CO2/HCO3−, steady-state pHi was lower in VSMCs of arteries from NHE1 KO than WT mice. In the presence of CO2/HCO3−, no significant difference in steady-state pHi was seen between VSMCs of arteries from NHE1 KO and WT mice (n= 5–8). Comparisons were made with a two-way ANOVA followed by Bonferroni's post hoc test. ***P < 0.001; NS: not significantly different vs. WT.
In the absence of CO2/HCO3−, steady-state pHi was lower in VSMCs of arteries from NHE1 KO compared to WT mice (Fig. 1D). No significant difference in steady-state pHi was seen in the presence of CO2/HCO3− (Fig. 1D). In both NHE1 KO and WT mice, VSMC steady-state pHi was significantly higher in the presence of CO2/HCO3− than in its nominal absence (Fig. 1D) consistent with previous findings from mouse mesenteric artery VSMCs (Boedtkjer et al. 2006, 2011; Boedtkjer & Aalkjaer, 2009).
These results show that NHE1 is responsible for the Na+/H+ exchange in mouse mesenteric artery VSMCs and no other Na+/H+ exchanger compensates for the KO of NHE1. Under physiological conditions, CO2/HCO3−-dependent pHi regulatory mechanisms are sufficient to maintain normal VSMC steady-state pHi in arteries from NHE1 KO mice.
Na+–HCO3− cotransport in VSMCs is unaffected by NHE1 KO
We have previously shown that Na+–HCO3− cotransport mediated by NBCn1 plays an important role for pHi regulation in VSMCs from mouse mesenteric arteries (Boedtkjer et al. 2006, 2008, 2011). When the CO2/HCO3− system is in equilibrium, NBCn1 and NHE1 supposedly perform equivalent transport functions (i.e. net extrusion of the equivalent of one H+ in exchange for one Na+). Following KO of NBCn1, Na+/H+ exchange activity is increased (Boedtkjer et al. 2011). In contrast, we found no difference in Na+–HCO3− cotransport activity between VSMCs from NHE1 KO and WT mice (Fig. 1B and C).
NHE1 is the only functionally important Na+/H+ exchanger in ECs
In the absence of CO2/HCO3−, removal of bath Na+ reversibly acidified mesenteric artery ECs from WT mice (ΔpHi=−0.74 ± 0.16; Fig. 2A). ECs from NHE1 KO mice were not significantly acidified following Na+ removal (ΔpHi=−0.02 ± 0.03; P < 0.01 vs. WT; Fig. 2A). Furthermore, addition of 600 μm amiloride produced intracellular acidification of ECs from WT mice (ΔpHi=−0.29 ± 0.09; n= 5; P < 0.05) and this response was completely abolished in arteries from NHE1 KO mice (ΔpHi=−0.01 ± 0.05; n= 4; P < 0.05 vs. WT). Taken together, these results demonstrate that NHE1 is the only functionally relevant Na+/H+ exchanger in mouse mesenteric artery ECs.
Figure 2. NHE1 is the only functionally important Na+/H+ exchanger in ECs and becomes important for steady-state pHi regulation only in the absence of CO2/HCO3−.
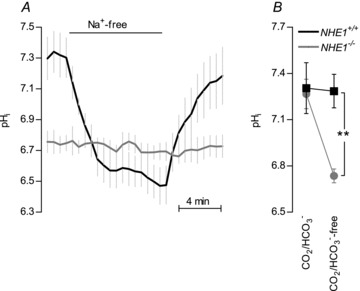
A, in the absence of CO2/HCO3−, the acidification upon Na+ removal was abolished in ECs of arteries from NHE1 KO mice (n= 5). B, in the absence of CO2/HCO3−, steady-state pHi was reduced in ECs of arteries from NHE1 KO mice compared to WT mice (n= 5). In the presence of CO2/HCO3−, no difference in steady-state pHi was seen between ECs from NHE1 KO and WT mice (n= 4). Comparisons were made with a two-way ANOVA followed by Bonferroni's post hoc test. **P < 0.01 vs. WT.
In the absence of CO2/HCO3−, ECs of arteries from NHE1 KO mice had a lower steady-state pHi than ECs of arteries from WT mice (Fig. 2B). In contrast, no significant difference was seen in steady-state pHi between ECs from NHE1 KO and WT mice when arteries were investigated in the presence of CO2/HCO3− (Fig. 2B). These data suggest that the effect of NHE1 KO on EC steady-state pHi is masked by CO2/HCO3−-dependent pHi regulatory mechanisms under physiological conditions.
NHE1 KO reduces vascular tension development at unaffected intracellular Ca2+ levels
Maximum tension development to noradrenaline was approximately 30% reduced in arteries from NHE1 KO compared to WT mice even in the presence of CO2/HCO3− (Fig. 3A) where pHi is unaffected (Fig. 1D). An original force trace is shown in Supplementary Fig. 4. No difference in the sensitivity (EC50) to noradrenaline with respect to tension development was found in the presence of CO2/HCO3− (Fig. 3A). The intracellular Ca2+ response of VSMCs to noradrenaline was not significantly different between arteries from NHE1 KO and WT mice in the presence of CO2/HCO3− (Fig. 3B).
Figure 3. Arteries from NHE1 KO mice produce lower tension to noradrenaline than arteries from WT mice while the VSMC [Ca2+]i-response is unaffected.
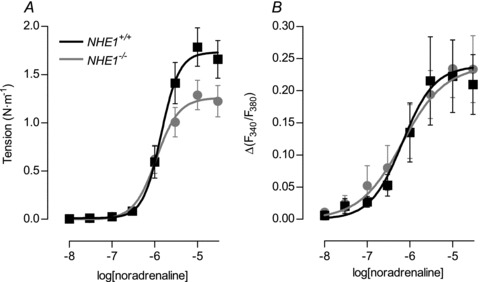
A, tension development to noradrenaline was reduced in arteries from NHE1 KO compared to WT mice (P < 0.05; n= 8–10). Experiments were performed in the presence of CO2/HCO3−. B, the VSMC Ca2+ response to noradrenaline was unaffected in arteries from NHE1 KO compared to WT mice (P= 0.72; n= 4–5). For comparisons, sigmoidal curve fits were performed and the derived log(EC50) and maximum values compared by extra sum-of-squares F tests.
To induce maximum constriction, we applied 10 μm noradrenaline in the presence of 123.6 mm extracellular K+ (K-PSS). In the presence of CO2/HCO3−, tension development was significantly reduced in arteries from NHE1 KO (1.83 ± 0.15 N m−1; n= 8) compared to WT mice (2.40 ± 0.16 N m−1; n= 10; P < 0.05). No significant difference in the intracellular Ca2+ response of VSMCs to 10 μm noradrenaline in CO2/HCO3− containing K-PSS was seen between arteries from NHE1 KO (Δ(F340/F380) = 0.31 ± 0.05; n= 5) and WT mice (Δ(F340/F380) = 0.28 ± 0.07; n= 4; P= 0.72).
KO of NHE1 results in smooth muscle hypotrophy
Differences in artery structure, and consequently in the amount of contractile elements in the artery wall, represent a plausible explanation for lower tension development in arteries from NHE1 KO compared to WT mice. No difference in the internal diameter of first order mesenteric arteries from NHE1 KO and WT mice was found (Table 1). In contrast, the media thickness was approximately 30% smaller in arteries from NHE1 KO compared to WT mice (Table 1). The media thickness to lumen diameter ratio and the media cross-sectional area were also reduced in arteries from NHE1 KO mice (Table 1).
Table 1.
Morphological characteristics of mesenteric arteries from NHE1 KO and WT mice
| NHE1 WT (n= 8–29) | NHE1 KO (n= 5–27) | P | |
|---|---|---|---|
| Myograph measurements | |||
| Media thickness (μm) | 9.2 ± 0.7 | 6.6 ± 0.4 | 0.005 |
| Lumen diameter (μm) | 180 ± 7 | 174 ± 5 | 0.49 |
| Media thickness/lumen diameter ratio | 0.053 ± 0.003 | 0.038 ± 0.003 | 0.006 |
| Media cross-sectional area (μm2) | 5086 ± 428 | 3712 ± 334 | 0.02 |
| Media stress (tension/media thickness) | |||
| 10 μm NA in PSS (×105 N m−2) | 1.90 ± 0.27 | 1.94 ± 0.21 | 0.91 |
| 10 μm NA in K-PSS (×105 N m−2) | 2.65 ± 0.22 | 2.65 ± 0.19 | 1.00 |
| Histological measurements | |||
| Volume fraction of VSMCs in media | 0.77 ± 0.01 | 0.77 ± 0.01 | 0.81 |
| Cell dimensions | |||
| Volume (μm3) | 1684 ± 177 | 1022 ± 221 | 0.04 |
| Length (μm) | 83 ± 14 | 77 ± 12 | 0.74 |
| Cross-sectional area (μm2) | 22 ± 3 | 13 ± 2 | 0.02 |
| Combined measurements | |||
| Cells per unit length (μm−1) | 2.5 ± 0.1 | 3.4 ± 0.7 | 0.12 |
| Cell layers | 1.7 ± 0.1 | 1.5 ± 0.1 | 0.16 |
| Active force | |||
| Per cell cross-section (kPa) | 335 ± 33 | 359 ± 44 | 0.66 |
| Per cell (μN) | 7.1 ± 0.8 | 4.5 ± 0.7 | 0.04 |
Values are expressed as means ± SEM. The probability values are derived from Student's unpaired two-tailed t test. NA: noradrenaline; PSS: physiological salt solution with control [K+]; K-PSS: PSS with KCl substituted for NaCl ([K+]= 123.6).
Stereological analyses showed that the reduced media area was caused by a reduced volume of the individual VSMCs in arteries from NHE1 KO compared to WT mice while the number of VSMCs per unit artery length was unchanged (Table 1). The reduced VSMC volume was caused by a smaller VSMC cross-sectional area while cell length was unaffected (Table 1).
NHE1 KO does not affect media stress or active force per cell cross-sectional area
Media stress (the ratio between tension and media thickness) after exposure to 10 μm noradrenaline in PSS or K-PSS was unaffected by NHE1 KO in the presence of CO2/HCO3− (Table 1). The maintained media stress and intracellular Ca2+ response of VSMCs suggest that reduced tension development by arteries from NHE1 KO mice is explained by the reduced media thickness. In support of this, we find that while active force production per VSMC was strongly reduced (Table 1), this was solely due to the difference in VSMC size. As such, active force production per VSMC cross-sectional area was similar in arteries from NHE1 KO and WT mice (Table 1).
NHE1 KO reduces systemic blood pressure without altering heart rate
Consistent with the reduced media thickness and lower artery tension development, mean arterial blood pressure (MAP; Fig. 4A) was significantly reduced (P < 0.05) from 109 ± 8 mmHg in WT mice (n= 5) to 84 ± 4 in NHE1 KO mice (n= 4). No significant difference (P= 0.98) in heart rate (HR; Fig. 4B) was seen between NHE1 KO (HR = 699 ± 22; n= 4) and WT mice (HR = 697 ± 53; n= 5).
Figure 4. NHE1 KO mice are hypotensive.
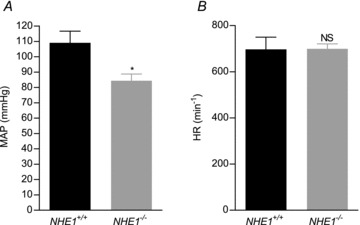
A, mean arterial blood pressure (MAP) was lower in NHE1 KO compared to WT mice. B, no difference in heart rate (HR) was seen between NHE1 KO and WT mice. Comparisons were made with Student's unpaired, two-tailed t test. *P < 0.05; NS: not significantly different vs. WT.
NHE1 KO results in reduced NO-mediated relaxations in the absence of CO2/HCO3−
We investigated the effect of NHE1 KO on vasorelaxation to acetylcholine. Representative force traces are shown in Supplementary Fig. 5. In the presence of CO2/HCO3−, pHi was similar in ECs from NHE1 KO and WT mice (Fig. 2B), and there was no difference in the relative relaxation to acetylcholine (Fig. 5A and B). Removing CO2/HCO3− from the bath solution did not affect EC pHi in WT mice (Fig. 2B) and vasorelaxation to acetylcholine was also unaffected (Fig. 5A). In arteries from NHE1 KO mice, ECs acidified upon removal of CO2/HCO3− (Fig. 2B) and the sensitivity to acetylcholine with respect to vasorelaxation was dramatically reduced (Fig. 5B). After application of 100 μm of the NO-synthase inhibitor l-NAME, no effect of removing CO2/HCO3− was seen on the relaxation to acetylcholine in arteries from neither NHE1 KO nor WT mice (Fig. 5A and B).
Figure 5. NO-mediated relaxations induced by acetylcholine are inhibited by omission of CO2/HCO3− in arteries from NHE1 KO mice while arteries from WT mice are unaffected by removal of CO2/HCO3−.
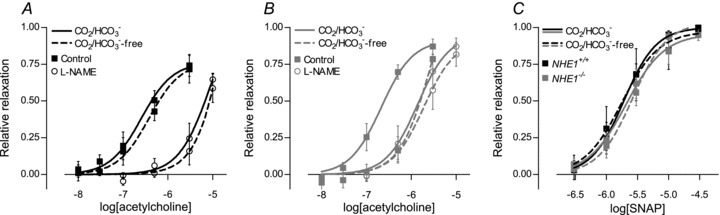
A, relaxation of arteries from WT mice (n= 5) to acetylcholine was unaffected by removal of CO2/HCO3− in the presence (P= 0.72) and in the absence (P= 0.73) of 100 μm l-NAME. B, relaxation of arteries from NHE1 KO mice (n= 5) to acetylcholine was attenuated upon removal of CO2/HCO3− (P < 0.001). After pretreatment with 100 μm l-NAME, no effect of removing CO2/HCO3− was seen (P= 0.65). C, no significant difference (P= 0.96) in the relaxation to the NO donor SNAP was found between arteries from NHE1 KO and WT mice in the presence or absence of CO2/HCO3−. Sigmoidal curve fits were performed and the derived log(EC50) and maximum values compared by extra sum-of-squares F tests.
Reduced l-NAME-sensitive relaxations to acetylcholine in arteries from NHE1 KO mice in the absence of CO2/HCO3− could be explained by either reduced NO delivery to the VSMCs or reduced effect of NO on the VSMCs. We investigated the response to exogenous NO using the NO donor SNAP. As shown in Fig. 5C, vasorelaxation to SNAP was unaffected by KO of NHE1 as well as removal of CO2/HCO3− (P= 0.90) suggesting that the reduced vasorelaxation to acetylcholine is explained by a lower NO concentration.
Since no difference in acetylcholine-induced relaxation was seen between NHE1 KO and WT mice in the presence of CO2/HCO3−, our findings are unlikely to be explained by a change in endothelial nitric oxide synthase (eNOS) expression. Since the NO synthase is known to be regulated by the intracellular concentration of Ca2+ in ECs, we investigated the EC Ca2+ response to acetylcholine but found no effect of removing CO2/HCO3− in arteries from NHE1 KO or WT mice (Fig. 6A and B).
Figure 6. The EC Ca2+ response to acetylcholine is not affected by omission of CO2/HCO3− in NHE1 KO or WT mice.
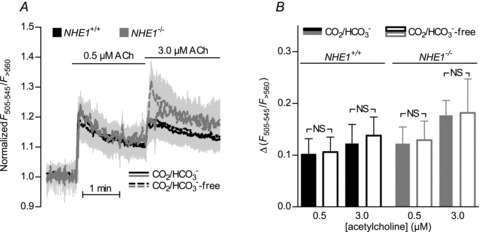
A, no difference was found in the EC Ca2+ response to acetylcholine between arteries from NHE1 KO and WT mice (n= 5) investigated in the presence or absence of CO2/HCO3−. B, average increases in Ca2+-dependent fluorescence calculated from panel A. Comparisons were made with repeated measures two-way ANOVA followed by Bonferroni's post hoc test. NS: not significantly different.
Taken together, our data support the hypothesis (Boedtkjer et al. 2011) that endothelial pHi modulates NO-mediated relaxations due to the intrinsic pHi sensitivity of the NO synthase (Fleming et al. 1994).
NHE1 KO inhibits rho kinase-dependent Ca2+ sensitivity with CO2/HCO3− absent
Similar to our findings in the presence of CO2/HCO3− (Fig. 3A), suppression of maximum tension development to noradrenaline was observed in arteries from NHE1 KO mice in the absence of CO2/HCO3− (Fig. 7A and B). Without CO2/HCO3−, noradrenaline sensitivity with respect to tension development was similar in arteries from NHE1 KO and WT mice (Fig. 7A and B). Application of l-NAME resulted in an increased noradrenaline sensitivity with respect to tension development in arteries from WT mice (Fig. 7A and B). In arteries from NHE1 KO mice, this change in noradrenaline sensitivity following l-NAME treatment was abolished (Fig. 7B) consistent with lower basal and/or noradrenaline-stimulated NO production in arteries from NHE1 KO mice. Treatment with l-NAME also unmasked a difference in vascular contractility between arteries from NHE1 KO and WT mice (Fig. 7A and B). The difference in contractile function with l-NAME present was not caused by a difference in the intracellular VSMC Ca2+ response (Fig. 7C) suggesting an effect on VSMC Ca2+ sensitivity. Treatment with the rho kinase inhibitor Y-27632 (10 μm) completely abolished the difference in noradrenaline sensitivity (Fig. 7A). Y-27632 reduced the intracellular VSMC Ca2+ response to noradrenaline similarly in arteries from WT and NHE1 KO mice (Fig. 7C). In accordance with our previous results from NBCn1 KO mice (Boedtkjer et al. 2011), our current findings suggest that intracellular acidification of ECs and VSMCs inhibits NO production and rho kinase-dependent VSMC Ca2+ sensitivity.
Figure 7. VSMC rho kinase-dependent Ca2+ sensitivity is reduced in arteries from NHE1 KO mice in the absence of CO2/HCO3−.
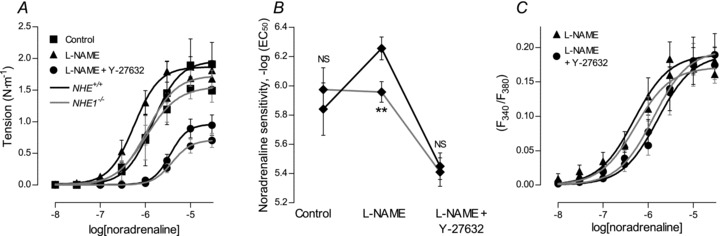
A, concentration–response curves showing average tension development to noradrenaline with or without 100 μm l-NAME and 10 μm Y-27632 in arteries from WT and NHE1 KO mice (n= 4–9) in the absence of CO2/HCO3−. B, summary of the changes in noradrenaline sensitivity with respect to tension development in arteries from NHE1 KO and WT mice (n= 4–9) investigated in the absence of CO2/HCO3−. C, concentration–response curves showing the average VSMC Ca2+ response to noradrenaline in the presence of 100 μm l-NAME or the combination of l-NAME and 10 μm Y-27632 in arteries from NHE1 KO and WT mice (n= 5) in the absence of CO2/HCO3−. Sigmoidal curve fits were performed and the derived log(EC50) and maximum values compared by extra sum-of-squares F tests. **P < 0.01; NS: not significantly different vs. WT.
Discussion
Here, we investigated the importance of Na+/H+ exchanger NHE1 and the effects of sustained intracellular acidification in VSMCs and ECs of mouse mesenteric small arteries, which are known to contribute to peripheral vascular resistance (Fenger-Gron et al. 1995) and consequently to blood pressure regulation.
We show that NHE1 is the only Na+/H+ exchanger relevant for pHi regulation in mouse mesenteric artery ECs and VSMCs. Although NHE1 acts as a major acid extruder at low pHi, it is functionally inactive at normal resting pHi and not important for control of bulk steady-state pHi in the presence of CO2/HCO3−. We recently showed (Boedtkjer et al. 2011) that KO of Na+–HCO3− cotransporter NBCn1 results in reduced EC and VSMC steady-state pHi in the presence of CO2/HCO3− while no difference in pHi between arteries from NBCn1 KO and WT mice was seen in the absence of CO2/HCO3−. In combination, these findings suggest that EC and VSMC steady-state pHi under resting, physiological conditions (i.e. at an extracellular pH of 7.40 with CO2/HCO3− present) depends primarily on the activity of NBCn1.
The results from the present study combined with our previous results (Boedtkjer et al. 2006, 2011; Boedtkjer & Aalkjaer, 2009) show that acid extrusion from acidified VSMCs relies exclusively on Na+–HCO3− cotransport mediated by NBCn1 and Na+/H+ exchange mediated by NHE1. While there are considerable quantitative differences between the different genetic backgrounds that we have investigated (C57BL/6J, FVB/N, NMRI), NBCn1 and NHE1 seem to be of overall similar quantitative importance for acid extrusion from VSMCs during intracellular acidification (Boedtkjer et al. 2006, 2011; Boedtkjer & Aalkjaer, 2009).
With respect to ECs, our present and previous results show that NBCn1 and NHE1 are the only Na+-dependent acid extruders (Boedtkjer & Aalkjaer, 2009; Boedtkjer et al. 2011). As previously reported, the NH4+-prepulse technique cannot be reliably employed to produce intracellular acidification of ECs in situ loaded with BCPCF without causing cellular damage with extensive endothelial blebbing (Boedtkjer & Aalkjaer, 2009). We cannot, therefore, rule out that Na+-independent mechanisms contribute to acid extrusion from these cells. Since steady-state pHi, however, is exquisitely sensitive to KO of NBCn1 (in the presence of CO2/HCO3−) (Boedtkjer et al. 2011) and NHE1 (in the absence of CO2/HCO3−), it is unlikely that other acid extruders play a significant functional role in the physiological pHi range.
In the present study, we demonstrate that NHE1 KO affects mesenteric artery structure and maximum contractile responses under physiological conditions (i.e. with CO2/HCO3− present) and is associated with a lower systemic MAP. Since no effect of NHE1 KO is seen on bulk steady-state pHi of VSMCs and ECs in the presence of CO2/HCO3−, the vascular effects of NHE1 KO under these conditions may be independent of pHi in the vascular wall or depend on regulation of pHi in subcellular domains without affecting bulk pHi. It is of interest that NHE1 has been suggested to have a number of pHi-independent effects including volume control through functional coupling with Cl−/HCO3−exchange (Mason et al. 1989), modulation of cell signalling pathways and interactions with the cytoskeleton (Denker et al. 2000; Denker & Barber, 2002). Although many aspects of artery morphology associated with hypertension have been investigated in detail, the causal relationship between changes in blood pressure and artery structure remains unexplained (Mulvany, 2002). Blood pressure disturbances and changes in artery structure, however, clearly go hand in hand. While it is plausible that the reduced media thickness is directly caused by the absence of NHE1 in VSMCs or ECs and responsible for the change in blood pressure, it is also possible that the change in media thickness is secondary to the low blood pressure, which in turn could be caused by disruption of NHE1 function in other tissues (e.g. brain or kidney). In support of a primary role for NHE1 expressed in the vascular wall for determining artery structure, inhibitory effects of NHE1 knockdown on proliferation, hypertrophy and migration of cultured VSMCs have previously been reported (Yu & Hales, 2011). It should be noted that in the present study, cell hypertrophy but not proliferation appears to be attenuated by the disruption of NHE1 based on the normal VSMC density but smaller VSMC volume observed in arteries from the NHE1 KO mice. The cellular responses to changes in blood pressure have been extensively studied using the disector method (Mulvany et al. 1985). While there is general consensus that arteries from patients with essential hypertension display eutrophic inward remodelling (i.e. rearrangement of otherwise normal cells around a smaller diameter) (Mulvany, 2002), arteries from patients with renovascular hypertension are characterized by hypertrophic inward remodelling (i.e. increased amounts of media material around a smaller diameter) mainly due to cell hypertrophy (Rizzoni et al. 2000). In spontaneously hypertensive rats and rats chronically infused with adrenergic receptor agonists, VSMC hyperplasia has been reported (Mulvany et al. 1985; Dao et al. 2001). These diverse findings indicate the involvement of multiple signalling pathways influenced by local (e.g. transmural pressure and flow) and systemic (e.g. neurohumoral) factors. Based on the above considerations, it is apparent that the temporal and mechanistic relationship between changes in blood pressure and changes in resistance artery structure are complex and not understood in any detail, and consequently, defining direct targets of NHE1 KO and indeed most other effectors on these complex processes is extremely challenging.
By use of the NHE1 KO mice, we provide important new information regarding the significance of a normal EC and VSMC pHi level for artery function. We find that intracellular acidification in the artery wall inhibits NO-mediated vasorelaxation and rho kinase-dependent VSMC Ca2+ sensitivity. The reduced acetylcholine-induced NO-mediated relaxation of arteries from NHE1 KO mice in the absence but not in the presence of CO2/HCO3− strongly supports the hypothesis that NO synthase activity is modulated by EC pHi (Boedtkjer et al. 2011). The isolated NO synthase displays a strong intrinsic pH dependency and deduced from in vitro activity measurements (Fleming et al. 1994), the changes in steady-state pHi observed in ECs of arteries from NHE1 KO mice upon removal of CO2/HCO3− would greatly reduce NO synthase activity. In light of the reduced NO-mediated vasorelaxation, lower intraluminal NO concentrations and inhibited NO synthase activity observed in NBCn1 KO mice (Boedtkjer et al. 2011), we propose that membrane acid–base transport and pHi in ECs play an important role for maintaining NO synthesis in resistance arteries. It is important to note that the magnitude of NO synthase inhibition, which is expected even with relatively small, physiologically relevant changes in pHi, is sufficient to have physiological effects (Fleming et al. 1994; Boedtkjer et al. 2011). We report a reduced rho kinase-dependent VSMC Ca2+ sensitivity in arteries from NHE1 KO mice when investigated in the absence of CO2/HCO3−. This is strongly supported by our recent findings that intracellular acidification of VSMCs caused by KO of NBCn1 inhibits rho kinase-dependent Ca2+ sensitivity and phosphorylation of the myosin light chain phosphatase targeting subunit at Thr-850 (Boedtkjer et al. 2011). In NBCn1 KO mice, the lower NO synthase activity and reduced rho kinase-dependent signalling were also evident in vivo accounting for prominent changes in blood pressure regulation (Boedtkjer et al. 2011). Since ECs and VSMCs from NHE1 KO mice are acidified only in the absence of CO2/HCO3−, the NHE1 KO mice cannot be used to study the importance of pHi-mediated inhibition of NO- and rho kinase-dependent signalling in vivo.
While the functional effects of NHE1 KO in the absence of CO2/HCO3− contribute greatly towards a comprehensive understanding of the intracellular signalling pathways linking changes in pHi to changes in artery tone, it should be emphasized that vasodilatory and contractile functions of arteries from NHE1KO mice were normal under physiological conditions with CO2/HCO3− present. The reduced maximum tension development observed under these conditions was fully accounted for by the structural changes of the artery wall since media stress and active force production per VSMC cross-sectional area were similar in arteries from NHE1 KO and WT mice.
In conclusion, we find that NHE1 is the only functionally important Na+/H+ exchanger in mouse mesenteric arteries where it is a major acid extruder at low pHi but has no effect on steady-state pHi in the presence of CO2/HCO3−. Under physiological conditions, KO of NHE1 reduces blood pressure and the media to lumen ratio of resistance arteries. Experiments performed in the absence of CO2/HCO3− strongly corroborate that EC pHi modulates NO synthase activity while VSMC pHi affects rho kinase-dependent Ca2+ sensitivity. Based on these findings, acid–base transport and pHi regulation in the artery wall need to be taken into consideration when evaluating conditions of disturbed artery function and blood pressure regulation.
Acknowledgments
The authors would like to thank Dr Jeppe Praetorius for fruitful discussions. Jørgen Andresen, Helle Zibrandtsen, Jane Rønn and Christian Westberg are thanked for expert technical assistance. The Water and Salt Research Centre at Aarhus University was established and supported by the Danish National Research Foundation. This work was supported by the Danish Council for Independent Research (grant no. 10-094816 to E.B. and 271-06-0472 to C.A.) and the Danish Heart Foundation (grant no. 08–10-R68-A2179-B719–22494 to E.B.). The authors have no conflicts of interest.
Glossary
Abbreviations
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- HR
heart rate
- KO
knockout
- K-PSS
physiological salt solution with high extracellular [K+]
- MAP
mean arterial pressure
- NA
noradrenaline
- NHE
Na+/H+ exchanger
- PSS
physiological salt solution
- SNAP
S-nitroso-N-acetylpenicillamine
- VSMCs
vascular smooth muscle cells
- WT
wild-type
Author contributions
Experiments were performed at the Department of Biomedicine, Aarhus University, Denmark. E.B. and C.A. conceived the project, designed experiments and interpreted data. E.B. and H.H.D. collected data. E.B. analysed the data and wrote the manuscript. All authors revised the manuscript for important intellectual content and approved the final version.
References
- Aalkjaer C, Cragoe EJ., Jr Intracellular pH regulation in resting and contracting segments of rat mesenteric resistance vessels. J Physiol. 1988;402:391–410. doi: 10.1113/jphysiol.1988.sp017211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalkjaer C, Hughes A. Chloride and bicarbonate transport in rat resistance arteries. J Physiol. 1991;436:57–73. doi: 10.1113/jphysiol.1991.sp018539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalkjaer C, Mortensen FV, Jensen PE, Nielsen H. The role of [Ca2+]i, membrane potential and pHi in the relaxation of rat mesenteric arteries to hyperosmolar acetate. Pflugers Arch. 1998;436:705–711. doi: 10.1007/s004240050692. [DOI] [PubMed] [Google Scholar]
- Aaronson PI, McKinnon W, Poston L. Mechanism of butyrate-induced vasorelaxation of rat mesenteric resistance artery. Br J Pharmacol. 1996;117:365–371. doi: 10.1111/j.1476-5381.1996.tb15200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Beningo K, Olds G, Hupe D. The endothelin-converting enzyme from human umbilical vein is a membrane-bound metalloprotease similar to that from bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1992;89:8606–8610. doi: 10.1073/pnas.89.18.8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aickin CC. Regulation of intracellular pH in the smooth muscle of guinea-pig ureter: Na+ dependence. J Physiol. 1994;479:301–316. doi: 10.1113/jphysiol.1994.sp020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baandrup U, Gundersen HJG, Mulvany MJ. Is it possible to solve the problem: hypertrophy/hyperplasia of smooth muscle cells in the vessel wall of hypertensive subjects? Advan Appl Microcirc. 1985;8:122–128. [Google Scholar]
- Bell SM, Schreiner CM, Schultheis PJ, Miller ML, Evans RL, Vorhees CV, Shull GE, Scott WJ. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am J Physiol Cell Physiol. 1999;276:C788–C795. doi: 10.1152/ajpcell.1999.276.4.C788. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Aalkjaer C. Insulin inhibits Na+/H+ exchange in vascular smooth muscle and endothelial cells in situ: involvement of H2O2 and tyrosine phosphatase SHP-2. Am J Physiol Heart Circ Physiol. 2009;296:H247–H255. doi: 10.1152/ajpheart.00725.2008. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Aalkjaer C. NBCn1 (slc4a7) mediates the Na+-dependent bicarbonate transport important for regulation of intracellular pH in mouse vascular smooth muscle cells. Circ Res. 2006;98:515–523. doi: 10.1161/01.RES.0000204750.04971.76. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Fuchtbauer EM, Aalkjaer C. Antibody-independent localization of the electroneutral Na+-HCO3− cotransporter NBCn1 (slc4a7) in mice. Am J Physiol Cell Physiol. 2008;294:C591–C603. doi: 10.1152/ajpcell.00281.2007. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Fuchtbauer EM, Aalkjaer C. Disruption of Na+, HCO3− cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca2+-sensitivity and hypertension development in mice. Circulation. 2011;124:1819–1829. doi: 10.1161/CIRCULATIONAHA.110.015974. [DOI] [PubMed] [Google Scholar]
- Brown DA, Melvin JE, Yule DI. Critical role for NHE1 in intracellular pH regulation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G804–G812. doi: 10.1152/ajpgi.00150.2003. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Prasad V, Hubner CA, Praetorius J. Nhe1 is a luminal Na+/H+ exchanger in mouse choroid plexus and is targeted to the basolateral membrane in Ncbe/Nbcn2-null mice. Am J Physiol Cell Physiol. 2009;296:C1291–C1300. doi: 10.1152/ajpcell.00062.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao HH, Lemay J, de CJ, deBlois D, Moreau P. Norepinephrine-induced aortic hyperplasia and extracellular matrix deposition are endothelin-dependent. J Hypertens. 2001;19:1965–1973. doi: 10.1097/00004872-200111000-00006. [DOI] [PubMed] [Google Scholar]
- Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol. 2002;159:1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker SP, Huang DC, Orlowski J, Furthmayr H, Barber DL. Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol Cell. 2000;6:1425–1436. doi: 10.1016/s1097-2765(00)00139-8. [DOI] [PubMed] [Google Scholar]
- Eiesland J, Baro I, Raimbach S, Eisner DA, Wray S. Intracellular pH and buffering power measured in isolated single cells from pregnant rat uterus. Exp Physiol. 1991;76:815–818. doi: 10.1113/expphysiol.1991.sp003548. [DOI] [PubMed] [Google Scholar]
- Evans RL, Bell SM, Schultheis PJ, Shull GE, Melvin JE. Targeted disruption of the Nhe1 gene prevents muscarinic agonist-induced up-regulation of Na+/H+ exchange in mouse parotid acinar cells. J Biol Chem. 1999;274:29025–29030. doi: 10.1074/jbc.274.41.29025. [DOI] [PubMed] [Google Scholar]
- Fenger-Gron J, Mulvany MJ, Christensen KL. Mesenteric blood pressure profile of conscious, freely moving rats. J Physiol. 1995;488:753–760. doi: 10.1113/jphysiol.1995.sp021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Hecker M, Busse R. Intracellular alkalinization induced by bradykinin sustains activation of the constitutive nitric oxide synthase in endothelial cells. Circ Res. 1994;74:1220–1226. doi: 10.1161/01.res.74.6.1220. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Diecke FP. Influence of pH on isometric force development and relaxation in skinned vascular smooth muscle. Pflugers Arch. 1988;412:231–239. doi: 10.1007/BF00582502. [DOI] [PubMed] [Google Scholar]
- Good DW, Watts BA, III, George T, Meyer JW, Shull GE. Transepithelial HCO3− absorption is defective in renal thick ascending limbs from Na+/H+-exchanger NHE1 null mutant mice. Am J Physiol Renal Physiol. 2004;287:F1244–F1249. doi: 10.1152/ajprenal.00176.2004. [DOI] [PubMed] [Google Scholar]
- Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension. 1993;21:391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- Heagerty AM, Bund SJ, Aalkjaer C. Effects of drug treatment on human resistance arteriole morphology in essential hypertension: direct evidence for structural remodelling of resistance vessels. Lancet. 1988;2:1209–1212. doi: 10.1016/s0140-6736(88)90808-2. [DOI] [PubMed] [Google Scholar]
- Klockner U, Isenberg G. Intracellular pH modulates the availability of vascular L-type Ca2+ channels. J Gen Physiol. 1994;103:647–663. doi: 10.1085/jgp.103.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MJ, Smith JD, Garcia-Soto JJ, Grinstein S. Internal pH-sensitive site couples Cl−-HCO3− exchange to Na+-H+ antiport in lymphocytes. Am J Physiol Cell Physiol. 1989;256:C428–C433. doi: 10.1152/ajpcell.1989.256.2.C428. [DOI] [PubMed] [Google Scholar]
- Mathiassen ON, Buus NH, Sihm I, Thybo NK, Morn B, Schroeder AP, Thygesen K, Aalkjaer C, Lederballe O, Mulvany MJ, Christensen KL. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens. 2007;25:1021–1026. doi: 10.1097/HJH.0b013e32805bf8ed. [DOI] [PubMed] [Google Scholar]
- McKinnon W, Aaronson PI, Knock G, Graves J, Poston L. Mechanism of lactate-induced relaxation of isolated rat mesenteric resistance arteries. J Physiol. 1996;490:783–792. doi: 10.1113/jphysiol.1996.sp021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrwa U, Achtig I, Ruegg JC. Influences of calcium concentration and pH on the tension development and ATPase activity of the arterial actomyosin contractile system. Blood Vessels. 1974;11:277–286. doi: 10.1159/000158021. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ. Small artery remodeling and significance in the development of hypertension. News Physiol Sci. 2002;17:105–109. doi: 10.1152/nips.01366.2001. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Baandrup U, Gundersen HJ. Evidence for hyperplasia in mesenteric resistance vessels of spontaneously hypertensive rats using a three-dimensional disector. Circ Res. 1985;57:794–800. doi: 10.1161/01.res.57.5.794. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Hansen OK, Aalkjaer C. Direct evidence that the greater contractility of resistance vessels in spontaneously hypertensive rats is associated with a narrowed lumen, a thickened media, and an increased number of smooth muscle cell layers. Circ Res. 1978;43:854–864. doi: 10.1161/01.res.43.6.854. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005;280:1561–1572. doi: 10.1074/jbc.M410041200. [DOI] [PubMed] [Google Scholar]
- Orlowski J, Grinstein S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004;447:549–565. doi: 10.1007/s00424-003-1110-3. [DOI] [PubMed] [Google Scholar]
- Peers C, Green FK. Inhibition of Ca2+-activated K+ currents by intracellular acidosis in isolated type I cells of the neonatal rat carotid body. J Physiol. 1991;437:589–602. doi: 10.1113/jphysiol.1991.sp018613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation. 2003;108:2230–2235. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- Rizzoni D, Porteri E, Guefi D, Piccoli A, Castellano M, Pasini G, Muiesan ML, Mulvany MJ, Rosei EA. Cellular hypertrophy in subcutaneous small arteries of patients with renovascular hypertension. Hypertension. 2000;35:931–935. doi: 10.1161/01.hyp.35.4.931. [DOI] [PubMed] [Google Scholar]
- Schubert R, Krien U, Gagov H. Protons inhibit the BKCa channel of rat small artery smooth muscle cells. J Vasc Res. 2001;38:30–38. doi: 10.1159/000051027. [DOI] [PubMed] [Google Scholar]
- Yao H, Ma E, Gu XQ, Haddad GG. Intracellular pH regulation of CA1 neurons in Na+/H+ isoform 1 mutant mice. J Clin Invest. 1999;104:637–645. doi: 10.1172/JCI6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hales CA. Silencing of sodium-hydrogen exchanger 1 attenuates the proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells via E2F1. Am J Respir Cell Mol Biol. 2011;45:923–930. doi: 10.1165/rcmb.2011-0032OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Quinn DA, Garg HG, Hales CA. Deficiency of the NHE1 gene prevents hypoxia-induced pulmonary hypertension and vascular remodeling. Am J Respir Crit Care Med. 2008;177:1276–1284. doi: 10.1164/rccm.200710-1522OC. [DOI] [PMC free article] [PubMed] [Google Scholar]