

Abstract
Adenosine 5′-triphosphate (ATP) has long been considered to be the purine inhibitory neurotransmitter in gastrointestinal (GI) muscles, but recent studies indicate that another purine nucleotide, β-nicotinamide adenine dinucleotide (β-NAD+), meets pre- and postsynaptic criteria for a neurotransmitter better than ATP in primate and murine colons. Using a small-volume superfusion assay and HPLC with fluorescence detection and intracellular microelectrode techniques we compared β-NAD+ and ATP metabolism and postjunctional effects of the primary extracellular metabolites of β-NAD+ and ATP, namely ADP-ribose (ADPR) and ADP in colonic muscles from cynomolgus monkeys and wild-type (CD38+/+) and CD38−/− mice. ADPR and ADP caused membrane hyperpolarization that, like nerve-evoked inhibitory junctional potentials (IJPs), were inhibited by apamin. IJPs and hyperpolarization responses to ADPR, but not ADP, were inhibited by the P2Y1 receptor antagonist (1R,2S,4S,5S)-4-[2-iodo-6-(methylamino)-9H-purin-9-yl]-2-(phosphonooxy)bicyclo[3.1.0]hexane-1-methanol dihydrogen phosphate ester tetraammonium salt (MRS2500). Degradation of β-NAD+ and ADPR was greater per unit mass in muscles containing only nerve processes than in muscles also containing myenteric ganglia. Thus, mechanisms for generation of ADPR from β-NAD+ and for termination of the action of ADPR are likely to be present near sites of neurotransmitter release. Degradation of β-NAD+ to ADPR and other metabolites appears to be mediated by pathways besides CD38, the main NAD-glycohydrolase in mammals. Degradation of β-NAD+ and ATP were equal in colon. ADPR like its precursor, β-NAD+, mimicked the effects of the endogenous purine neurotransmitter in primate and murine colons. Taken together, our observations support a novel hypothesis in which multiple purines contribute to enteric inhibitory regulation of gastrointestinal motility.
Key points
Normal gastrointestinal activity depends upon orderly movement of nutrients and wastes through the alimentary canal. These movements require coordinated contractions of the muscular wall and regulation by excitatory and inhibitory motor neurons of the enteric nervous system.
We examined the nature of candidate purine neurotransmitters (ATP and β-NAD) and their metabolites (ADP and ADP-ribose) and the effects of these compounds on electrical and mechanical responses of colonic muscles.
After release, ATP and β-NAD were rapidly degraded to ADP and ADP-ribose, suggesting that inhibitory neural responses may include actions of primary transmitters and metabolites.
Metabolites of both neurotransmitter candidates elicited responses similar to responses to inhibitory nerve stimulation. However, only ADP-ribose had pharmacology that mimicked the effects of the endogenous inhibitory neurotransmitter.
These results help us better understand neural regulation of colonic motility and provide new insights about how defects in neural responses might lead to motility disorders such as constipation.
Introduction
Enteric inhibitory motor neurotransmission in the gastrointestinal (GI) tract is mediated largely by nitric oxide (Bult et al. 1990; Sanders & Ward, 1992) and a purine nucleotide, thought for many years to be ATP (Burnstock et al. 1970; Burnstock, 2008). Recent studies of murine colon (Mutafova-Yambolieva et al. 2007) and human and non-human primate (Macaca fascicularis) colons (Hwang et al. 2011) have shown that β-nicotinamide adenine dinucleotide (β-NAD+) better meets presynaptic and postsynaptic criteria for the inhibitory purine neurotransmitter than ATP. Frequency-dependent, neurotoxin-sensitive release of β-NAD+, but not of ATP, was demonstrated in colonic muscles, and exogenous β-NAD+ caused postsynaptic hyperpolarization that mimicked the pharmacology of inhibitory junction potentials (IJPs), generated by the endogenous purine neurotransmitter, better than ATP. In fact, most ATP appeared to originate from non-neuronal sources in colonic muscles, because a large part of the ATP release was insensitive to neurotoxins. Nevertheless, some portion of ATP may have been released from neurons, and the failure of exogenous ATP to mimic neural responses might have been due to activation of multiple postjunctional receptors, compartmentalization of postjunctional purine receptors, and/or rapid enzymatic degradation of ATP.
β-NAD+ released into the interstitium is degraded to nicotinamide and ADPR and to cADPR by NAD glycohydrolase and ADP-ribosyl cyclase, respectively, which in mammals are mainly associated with the CD38 protein (Munshi et al. 2000; Lee, 2001; Graeff et al. 2009). NAD glycohydrolase and ADP-ribosyl cyclase account for about 98% and 2% of the CD38 enzymatic activities (Lee, 2001). ADPR is further degraded to AMP by ectonucleotide pyrophosphatases (E-NPPs, CD203 family) and AMP is degraded to adenosine (ADO) by 5′-nucleotidase, CD73 (Di Girolamo et al. 1997; Zimmermann, 2000). ATP is degraded to ADP, AMP and ADO by ectonucleoside 5′-triphosphate diphosphohydrolases (E-NTPDases, CD39 family), nucleotide pyrophosphatases (NPPs), and 5′-nucleotidase, respectively (Zimmermann, 2000). These enzymes not only control the lifetime of purine nucleotides, they also generate agonists for additional purine receptors expressed by various cells within GI muscles (Abbracchio et al. 2009). We have demonstrated previously that tissue superfusates contain β-NAD+ and ATP, as well as their metabolites, ADP, AMP, ADPR, cADPR and ADO (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). However, it is currently unknown whether the direct metabolites of β-NAD+ and ATP, ADPR and ADP, also contribute to the postjunctional responses elicited by purinergic inhibitory neurotransmission.
The effects of purinergic neurotransmitter(s) released from enteric inhibitory neurons are known to be mediated by postjunctional P2Y1 receptors (Gallego et al. 2006, 2011; Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). P2Y1 receptor-mediated effects can be activated by ADP and ATP (von Kugelgen, 2006; Mutafova-Yambolieva et al. 2007) and by β-NAD+ (Mutafova-Yambolieva et al. 2007; Klein et al. 2009; Hwang et al. 2011) and ADPR (Gustafsson et al. 2011). The effects of exogenous β-NAD+, but not ATP, are blocked by selective and specific P2Y1 receptor inhibitors (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). It is currently unknown whether ADPR and ADP mimic the pharmacology of purinergic neurotransmission. In the current study we compared the degradation of β-NAD+ and ATP in the tunica muscularis and in isolated circular muscles of monkey colon at rest and during nerve stimulation and examined whether CD38, the major extracellular β-NAD+-metabolizing protein in mammals, is responsible for the degradation of β-NAD+ in the large intestine. We also tested whether metabolites of β-NAD+ and ATP (ADPR and ADP, respectively) display postjunctional effects that mimic the endogenous neurotransmitter(s). Our data suggest that ADPR, whether it is produced by metabolism of extracellular β-NAD+ or released as a primary neurotransmitter, may contribute to enteric inhibitory neurotransmission in GI muscles.
Methods
Ethical procedures
Cynomolgus monkeys (Macaca fascicularis), untreated with other drugs, were sedated with ketamine (10 mg kg−1) and 0.7 ml Beuthanasia-D (Schering-Plough AH, Kenilworth, NJ, USA) and were exsangunated at Charles River Laboratories Preclinical Services (Reno, NV, USA) for the purpose of clinical trials unrelated to this project. No cynomolgus monkeys were killed specifically for the experiments described in this paper. Proximal colon was retrieved from untreated, control animals by the Charles River Laboratories staff, placed in ice-cold Krebs–Ringer buffer (KRB) and transported to the University of Nevada site of experimental work. Use of muscles from these animals and transport of tissues to laboratories at the University of Nevada was approved by the Institutional Animal Care and Use Committee at University of Nevada (IACUC). C57/BL6 wild-type and CD38−/− mice were killed by sedation with isoflurane followed by cervical dislocation and exsanguination. The entire gastrointestinal tract, from oesophagus to the internal anal sphincter, was removed and placed in oxygenated ice-cold KRB for further dissection. All experimental procedures were approved by the IACUC.
Tissue preparation
Monkey circular muscles (CMs) were prepared by peeling away the longitudinal muscle with attached myenteric ganglia. Other experiments were carried out using whole tunica muscularis (whole muscle, WM). Proximal colons of C57BL/6 mice (wild-type, CD38+/+, Charles River Laboratories, Wilmington, MA, USA) and CD38 knockout mice (CD38−/−, The Jackson Laboratory, Bar Harbor, ME, USA) were prepared by peeling away the mucosa and submucosa.
Degradation of purine nucleotides in mouse and monkey colonic smooth muscle
To determine enzymatic degradation of β-NAD+, ATP, and ADPR colonic preparations were superfused with 1,N6-etheno-NAD (eNAD) and 1,N6-etheno-ATP (eATP) substrates. 1,N6-Etheno-nucleotides have been used previously as exogenous substrates for nucleotidases (Secrist et al. 1972; Jamal et al. 1988; Todorov et al. 1997; Mihaylova-Todorova et al. 2001; Bobalova & Mutafova-Yambolieva, 2003), since they are highly fluorescent and allow about 1,000,000-fold more sensitive detection of nucleotide metabolism compared to authentic nucleotides (Bobalova et al. 2002). Non-derivatized purine nucleotides cannot be used in studies in small tissue preparations, because the authentic purine nucleotides generally have very low fluorescence coefficients, not allowing detection of small changes in substrate or product concentrations.
Smooth muscle tissue segments of mouse or monkey tunica muscularis or monkey circular muscle were placed in 200 μl water-jacket Brandel superfusion chambers equipped with platinum electrodes as used in neurotransmitter release studies and described previously (Bobalova & Mutafova-Yambolieva, 2001; Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). Chambers were mounted vertically and tissues were superfused with oxygenated Krebs solution (at 37°C) with the following composition (mm): 118.5 NaCl, 4.2 KCl, 1.2 MgCl2, 23.8 NaHCO3, 1.2 KH2PO4, 11.0 dextrose, 1.8 CaCl2 (pH 7.4). After a 45 min equilibration, tissues were superfused with either eATP or eNAD, at maximum saturating concentrations of 0.05 and 0.2 μm, respectively. Maximal saturating concentrations of eATP or eNAD were determined in preliminary concentration-metabolism experiments (data not shown). Three hundred microlitres of the superfusion solution were collected from the beaker containing the substrate (S0, (–) tissue). Typically the tissue samples were treated with the substrates for 30 or 60 s and the contents of the chamber were then collected in ice-cold Eppendorf tubes (S1, (+) tissue); the enzymatic reactions stopped by immersing the tubes in liquid nitrogen. In some experiments tissues were treated with substrate for 1 s and 5 s (30 min apart) to determine whether the same degradation of substrate occurs at shorter contact with tissue. In some experiments tissues were subjected to electrical field stimulation (EFS; 480 pulses; 0.3 ms or 0.5 ms; 16 Hz) once the substrate had reached the chamber to evaluate potential ‘releasable’ nucleotidase activity (S2, (+) tissue, (+) EFS).
The degradation of eNAD was evaluated by (1) the formation of its end product eADO, and (2) the sum of its products eADPR, eAMP and eADO. For the latter analysis we had to determine first the composition of the 12.5 min peak and evaluate the quantity of eAMP and eNAD as described in HPLC fraction analysis. Note that the formation of cADPR from eNAD cannot be measured in these experiments, because the N6 of the adenine moiety of eNAD is unavailable for cyclization and, therefore, 1,N6-etheno-cADPR cannot be formed (Smyth et al. 2006). Therefore, to evaluate the ADP-ribosyl cyclase activity degrading β-NAD+ to cADPR we used nicotinamide guanine dinucleotide (NGD, 0.2 mm) as substrate (Graeff et al. 1998) and measured the formation of cyclic guanosine diphosphate ribose (cGDPR) as shown previously (Smyth et al. 2006). eATP metabolism was determined by the production of eADO as well as by the appearance of eADP + eAMP + eADO in superfusates collected during superfusion of tissues with eATP. eADPR degradation was determined by the production of eAMP and eADO. HPLC coupled with fluorescence detection was used to measure formation of 1,N6-etheno-nucleotides and nucleosides and cGDPR as described below. The amount of products formed in S1 was compared with S0 to measure the spontaneous enzyme activity. The amount of products in S2 was compared with S1 to determine whether additional ‘releasable’ activity (Todorov et al. 1997) is present in the large intestine. Endogenously released nucleotides were not derivatized in these experiments; thus, the formation of 1,N6-etheno-nucleotides and nucleosides corresponds to direct products of 1,N6-etheno-substrates without interference of endogenous nucleotides.
HPLC assay of etheno-nucleotides/nucleosides and cGDPR
A reverse-phase gradient Agilent Technologies 1200 liquid chromatography system (Agilent Technologies, Wilmington, DE, USA) was used to detect the 1,N6-etheno-products and cGDPR as described previously (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). The mobile phase consisted of 0.1 m KH2PO4 (pH 6.0) as eluent A. Eluent B consisted of 65% eluent A and 35% methanol. Gradient elution was employed according to the following linear programme: time 0, 0% eluent B; 18 min, 100% eluent B. The flow rate was 1 ml min−1 and run time was 20 min. Column and autosampler temperatures were maintained at 25°C and 4°C, respectively. The fluorescence detector was set to record 1,N6-etheno-derivatized nucleotide and nucleoside signals at an excitation wavelength of 230 nm and emission wavelength of 420 nm. cGDPR was detected at an excitation wavelength of 270 nm and emission wavelength of 400 nm. These are optimum conditions for detection of 1,N6-etheno-derivatized (Bobalova et al. 2002) and non-etheno-derivatized (Smyth et al. 2004) nucleotides/nucleosides, respectively. The amount of nucleotide/nucleoside in each sample was calculated from calibration curves of nucleotide standards run simultaneously with each set of unknown samples. Results were normalized for sample volume and tissue weight and product formation was expressed in fmol (mg tissue)-1.
HPLC fraction analysis
eNAD elutes in the HPLC column at ∼12.5 min which is also the retention time of the eNAD product eAMP. Therefore, HPLC fraction analysis was carried out to determine the compounds present in the 12.5 min peak and the proportions of eAMP and eNAD in this peak. An Agilent Technologies 1200 Analytical Fraction Collector was employed to collect the peak at 12.5 min. The sample was etheno-derivatized (see below) and re-injected in the HPLC system. Etheno-derivatization at high temperature caused eNAD to be converted to eADPR (Smyth et al. 2004) which elutes at 11.5 min in the HPLC column. The peak that remained at 12.5 min was eAMP.
Preparation of etheno-substrates
ATP, ADO and ADPR were purchased from Sigma-Aldrich (St Louis, MO, USA) and etheno-derivatized as follows: 0.2 mm ATP, ADO or ADPR (dissolved in double distilled water) was acidified to pH 4.0 with citrate phosphate buffer. Chloroacetaldehyde (1 m) was added and substrates were heated to 80°C for 40 min to form 1,N6-ethenoderivatives eATP, eADO and eADPR (Levitt et al. 1984; Bobalova et al. 2002). eNAD was purchased from BioLog Life Science Institute (Bremen, Germany). Substrates were further diluted in the superfusion solution to final concentration needed.
Preparation of cGDPR
cGDPR, used as a standard for the HPLC analysis of enzymatic degradation of NGD, was prepared as previously described (Smyth et al. 2006). Briefly, nicotinamide-guanine dinucleotide (NGD, 2 mm, Sigma-Aldrich, MO, USA) was incubated with 2.5 units of Aplysia californica cyclase (Sigma-Aldrich, MO, USA) in reaction solution containing Tris-HCl (20 mm) in double distilled water (total volume 1 ml) for 1 h at 37°C. Quantitative conversion of NGD to cGDPR was confirmed by HPLC analysis of 10 μl aliquots of the reaction mixture.
Intracellular electrical activity and force measurements
Circular muscles from monkey and mouse (approximately 10 mm × 10 mm) were pinned to the Sylgard elastomer-lined floor of a recording chamber with the myenteric plexus side of the circular muscle facing upward. The bath chamber was constantly perfused with oxygenated Krebs–Ringer buffer (KRB) of the following composition (mm): 118.5 NaCl, 4.5 KCl, 1.2 MgCl2, 23.8 NaHCO3, 1.2 KH2PO4, 11.0 dextrose, 2.4 CaCl2, pH 7.4, at 37 ± 0.5°C. After 1 h equilibration circular muscle cells were impaled with glass microelectrodes with 80–100 MΩ resistance filled with 3 m KCl as described previously (Burns et al. 1996; Mutafova-Yambolieva et al. 2007). Experiments were performed in the presence of nifedipine (1 μm) to reduce contractions and facilitate impalements of cells for extended periods except where stated. Parallel platinum electrodes were placed on either side of the muscle strips and neural responses were evoked by square wave pulses of electrical field stimulation (EFS; 0.5 ms pulse duration) using a Grass S48 stimulator (Quincy, MA, USA). In each series of experiments agonists (ATP, ADP or ADPR each at a concentration of 10 mm or 50 mmβ-NAD+) were applied (10 ms pulses at 10 psi) via picospritz micropipettes (Picospritzer; General Valve, East Hanover, NJ, USA) positioned close to the site of electrical recording before and after addition of purinergic antagonists onto tissues. Responses to picospritzed agonists were compared with responses evoked by electrical field stimulation of intrinsic nerves. In one series of experiments concentration–response effects of ADP and ADPR and the effects of the P2Y1 antagonist, MRS2500, on these responses were compared. These experiments were performed by loading spritz pipettes with the same concentrations of ADP and ADPR (10 mm) and then applying multiple spritzes, ranging in pulse duration from 10 to 100 ms, while maintaining impalements in the same cell. Spritz pipette tips were positioned slightly farther from tissues in this series of experiments so impalements were not lost when longer duration spritzes (50–100 ms) were applied. In experiments in which mechanical activity was recorded, circular muscle strips of mouse colon (6 mm wide) were fixed at one end and attached to a Fort 10 isometric force transducer (World Precision Instruments, Sarasota, FL, USA). Isometric force was measured as described previously (Burns et al. 1996).
RNA isolation and RT-PCR
Total RNA was isolated from colonic tunica muscularis of wild-type and CD38−/− mice using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA was treated with 1 U μl−1 DNase I (Promega Corp., Madison, WI, USA) and cDNA was prepared using Oligo dT(12–18) primer and SuperScript II reverse transcriptase (Invitrogen). Polymerase chain reaction (PCR) was performed with gene-specific primers for CD38 (Kato et al. 1999), see below, on 2 μl complementary DNA using Advantage 2 polymerase mix reagents (Clontech, Mountain View, CA, USA). A two-step PCR method (95°C for 10 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min) was used to amplify the primer pairs. After PCR, 2 μl RT-PCR products were analysed on a 2.0% agarose gel. The following primers were used in this assay: Cd38-F ACAGACCTGGCTGCCGCCTCTCTAG (Tm= 64°C) and Cd38-R, GGGGCGTAGTCTTCTCTTGTGATGT (Tm= 59°C); accession number NM-007646.
Western immunoblot analysis
Colons from monkey (WM and CM preparations) were frozen by immersion in liquid nitrogen. Frozen tissues were pulverized and total protein was extracted by glass–glass homogenization with a RIPA buffer composed of 20 mm Tris, 150 mm NaCl, 10% glycerol, 1% NP40, 2.5 mm NaF, 0.1 mm sodium orthovanadate, 1 mm benzamidine, 2.5 mmβ-glycerophosphate, 100 μm AEBSF and 1 μm leupeptin. Insoluble material was pelleted by centrifugation at 15,000 g for 20 min at 4°C. Total protein concentration of the supernatant was determined by the BCA assay using bovine serum albumin for standards. Tissue homogenates were reduced with Laemmli reagent and equal amounts of total protein (10 μg) were resolved by SDS-PAGE (12% acrylamide) and transferred onto PVDF membranes for 1.5 h at 100 V and 4°C (Bio-Rad, Hercules, CA, USA). HuT78 T lymphocyte lymphoma whole cell lysate (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a positive control to identify the CD38 band. Membranes were blocked for 1 h with 2% non-fat dry milk + 0.2% Tween and probed for 18 h at 4°C with an anti-CD38 primary rabbit polyclonal antibody (Santa Cruz Biotechnology, cat no. sc-15362), diluted 500-fold in the blocking solution. After removal of excess primary antibody, membranes were incubated for 1.5 h at room temperature with a secondary horseradish peroxidase-conjugated rabbit antibody. Immunostained protein bands were detected using ECL Advantage (GE HealthCare Biosciences, Piscataway, NJ, USA), and visualized with a CCD camera-based detection system (Epi Chem II, UVP Laboratory Products, Upland, CA, USA).
Statistics
Data are presented as means ± SEM. Means were compared by Student's two-tailed, unpaired t test or by one-way ANOVA for comparison of more than two groups followed by a post hoc Bonferroni multiple comparison test (GraphPad Prism, v. 3 (GraphPad Software, Inc., San Diego, CA) or SigmaPlot (Systat Software Inc., Richmond, CA, USA)). A probability value of less than 0.05 was considered significant.
Drugs and Chemicals
ATP, ADP, adenosine, ADP-ribose (ADPR), apamin, atropine, erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA), nicotinamide guanine dinucleotide (NGD), NG-nitro-l-arginine (l-NNA), nifedipine, pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonate (PPADS) and suramin were purchased from Sigma-Aldrich. 6-S-[(4-Nitrophenly)methyl]-6-thioinosine (NBMPR or NBTI) and (1R,2S,4S,5S)-4-[2-iodo-6-(methylamino)-9H-purin,-9-yl]-2-(phosphonooxy)bicyclo[3.1.0]hexane-1-methanol dihydrogen phosphate ester tetraammonium salt (MRS2500) were purchased from Tocris Bioscience (Ellisville, MO, USA). β-Nicotinamide-1,N6- ethenoadenine dinucleotide (eNAD) came from BioLog Life Science Institute (Bremen, Germany). All drugs were dissolved in deionized H2O, apart from nifedipine (dissolved in ethanol) and NBMPR (dissolved in DMSO), and further diluted in perfusion solutions; final concentration of ethanol or DMSO in superfusion solution was 0.001%.
Results
β-NAD+ forms ADPR in primate colonic smooth muscles
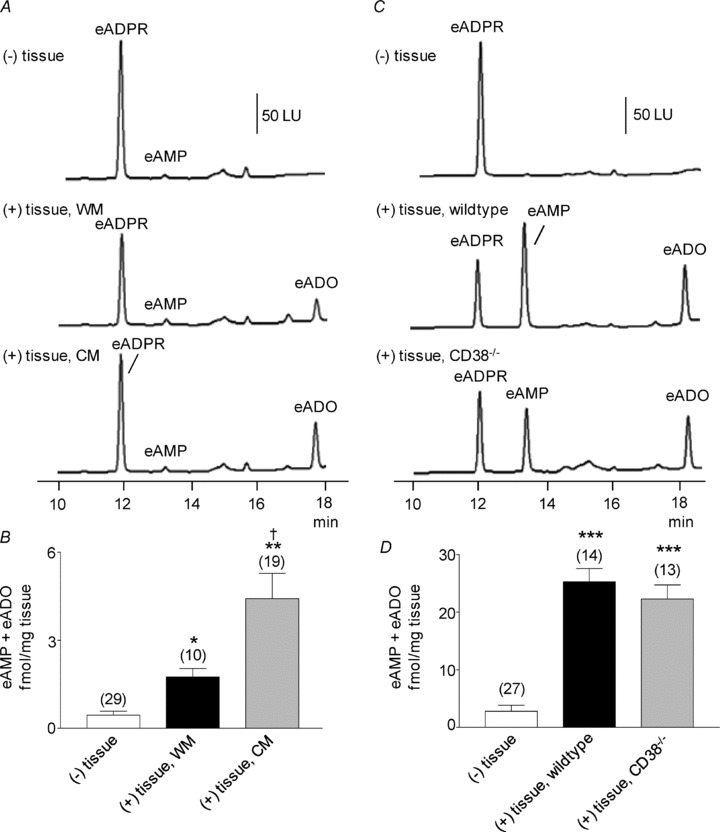
We have previously shown that tissue superfusates collected during EFS of primate and murine colonic muscles contain ADPR, β-NAD+, ATP and ADP (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). ADPR, in particular, comprised about 20–30% of the β-NAD++ ADPR + cADPR combination in primate colons (Hwang et al. 2011). However, it is not clear whether ADPR is formed as a result of metabolism of β-NAD+ released from nerves or whether it is a substance that is released upon activation of nerve terminals. Therefore, we first tested whether β-NAD+ is metabolized to ADPR in colonic muscles. Primate WM and CM colonic muscles were superfused with eNAD (0.2 μm), and the formation of eADPR and the end metabolite, eADO, was measured. To match neurotransmitter overflow studies (i.e. Mutafova-Yambolieva et al. 2007; Hwang et al. 2011), tissues were superfused with the substrate for 30–60 s, as this was the duration of EFS in previous transmitter overflow experiments. As shown in Fig. 1, eNAD substrate from BioLog Life Science Institute contains a small amount of eADPR (Fig. 1A, (–) tissue). Clearly, eADPR is formed from eNAD in both WM and CM of the colon (Fig. 1A, middle and bottom chromatograms). Whereas eADO was not detected in the absence of tissue (Fig. 1A, top panel), eADO became detectable in the tissue superfusates when WM or CM was superfused with eNAD (Fig. 1A, middle and bottom panels). When normalized against tissue mass, the amount of eADO produced from eNAD was significantly higher in CM than in WM, P < 0.05 (Fig. 1B), suggesting that degradation of eNAD was greater in preparations containing a higher proportion of motor nerve processes and terminals per unit weight. Western immunoblot analysis of CD38, however, showed no significant differences in the levels of CD38 in WM and CM (Fig. 1C). No further degradation of eADO was observed when tissues were superfused with eADO (0.05 μm) as a substrate (Table 1). Furthermore, the amounts of eADO remained unchanged in the presence of either the adenosine deaminase inhibitor (EHNA, 10 μm) or the adenosine uptake blocker NBMPR (10 μm) (Table 1). Similar results were obtained in murine colons (Table 1). Therefore, the formation of eADO appeared to be a relatively reliable measure for eNAD metabolism since no further degradation by adenosine deaminase (Agteresch et al. 1999) or adenosine transport (Zimmermann et al. 1998) was detected in these preparations. Formation of eADPR and eADO from eNAD was observed even when the tissues were in contact with eNAD for 1 s or 5 s only. As discussed above eADO was not observed in the absence of tissue. However, eADO was 0.94 ± 0.17 (n= 8) and 1.34 ± 0.23 fmol (mg tissue)−1 (n= 7) after contact of eNAD with WM for 1 s and 5 s, respectively. Likewise, in CM superfusates eADO was measured to be 1.45 ± 0.18 (n= 8) and 1.57 ± 0.21 fmol (mg tissue)-1 (n= 8) after contact with eNAD for 1 s and 5 s, respectively.
Figure 1. Degradation of eNAD in monkey whole muscle (WM) and circular muscle (CM) colon preparations.
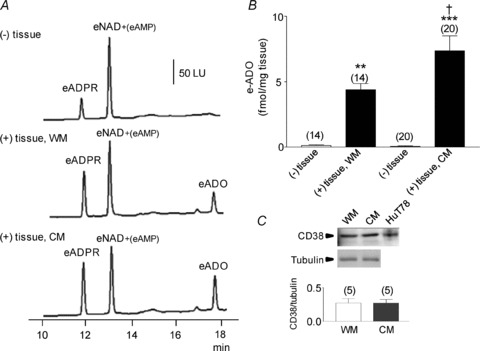
A, original chromatograms of eNAD (0.2 μm) in the absence, (–) tissue, and presence, (+) tissue, of either WM or CM (30 s contact of substrate with tissue). The formation of eADPR and eADO was increased in the (+) tissue samples. No noticeable changes were observed in the peak of eNAD at 12.5 min, because this peak also contains eAMP formed from eNAD; LU, luminescence units. B, graphic representation of eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Note the increased formation of eADO in both WM and CM; the formation of eADO was greater in CM than WM. Asterisks denote significant differences from the amounts of eADO in (–) tissue samples (**P < 0.01, ***P < 0.001). There is a significant difference a significant increase in CM as compared to WM (†P < 0.05); number of experiments in parentheses. C, Western immunoblot analysis of CD38 showed no significant differences between the protein levels of CD38 in WM and CM. Density of each band is normalized to tubulin, which was used to control equal protein loading.
Table 1.
Degradation of eADO (0.05 μm) by monkey WM, monkey CM and murine colon under control conditions or in the presence of either EHNA (10 μm), NBMPR (10 μm), or EHNA + NBMPR combined
| (–) tissue | (+) tissue, control | (+) tissue, + EHNA | (+) tissue, + NBMPR | (+) tissue, + EHNA + NBMPR | |
|---|---|---|---|---|---|
| Monkey WM | 60.0 ± 5.1 (10) | 59.01 ± 10.9 (4) | 63.51 ± 12.2 (2) | 41.93 ± 9.1 (2) | 57.15 ± 3.5 (2) |
| Monkey CM | 110.6 ± 4.9 (8) | 108.87 ± 4.9 (2) | 102.05 ± 7.4 (2) | 61.79 ± 1.8 (2) | 124.77 ± 5.5 (2) |
| Mouse CD38+/+ | 112.8 ± 13.9 (9) | 104.2 ± 16.5 (7) | — | — | 110.0 ± 34.7 (2) |
Values are fmol (mg tissue)−1. No significant differences in (–) tissue versus (+) tissue, control for all groups (P > 0.05). No significant differences between (+) tissue, control and (+) tissue, + EHNA, or (+) tissue, + NBTI, or (+) tissue, + EHNA + NBTI for all groups (P > 0.05). Number of experiments in parentheses.
We determined the relative content of eAMP and eNAD in the 12.5 min (eNAD + eAMP) peak and analysed the degradation of eNAD by the formation of the total e-product eADPR + eAMP + eADO after 30–60 s superfusion with eNAD. For this, we first carried out an HPLC fractional analysis to determine the portion of eAMP in the 12.5 min chromatography peak: eAMP comprised ∼2.2% of the 12.5 min peak of the eNAD provided by BioLog Life Science Institute when no muscle was present. When muscles were superfused with eNAD, the proportion of eAMP in the 12.5 min peak was increased to 6.1% of the peak in WM and 3.12% of the peak in CM. In WM the sum of eADPR + eAMP + eADO was 7.37 ± 1.24 and 17.11 ± 1.64 fmol (mg tissue)−1 (n= 14, P < 0.05) in the absence and presence of muscle, respectively. In experiments testing CM eADPR + eAMP + eADO was 11.18 ± 1.37 and 24.28 ± 2.72 fmol (mg tissue)−1 (n= 20, P < 0.001) in the absence and presence of muscle, respectively. Thus, the amount of eADPR + eAMP + eADO formed in CM was higher than in WM (P < 0.05), as also observed when eNAD degradation was evaluated by eADO formation.
We also tested whether degradation of eNAD increased in colonic muscles during nerve stimulation, as suggested for metabolism of purines in some smooth muscles innervated by autonomic neurons (Todorov et al. 1997). The degradation of eNAD in WM and CM was not significantly increased during EFS. For example, in WM eADO was 4.39 ± 0.47 fmol (mg tissue)−1 in the absence of EFS (S1, n= 14) and 4.98 ± 0.67 fmol (mg tissue)−1 in the presence of EFS (S2, n= 14, P > 0.05). Likewise, in CM eADO was 7.37 ± 1.14 fmol (mg tissue)−1 in the absence of EFS (S1, n= 20) and 7.19 ± 1.09 fmol (mg tissue)−1 in the presence of EFS (S2, n= 20, P > 0.05). Therefore, conventional HPLC analysis in conjunction with HPLC fractional analysis showed that: (i) eADPR is formed from eNAD in primate colonic muscles, (ii) degradation of eNAD occurs rapidly within muscles, (iii) degradation of eNAD is greater per unit mass in isolated CM than WM, and (iv) no additional degradation could be resolved during nerve stimulation.
β-NAD+ is likely to be degraded by multiple enzymes in the murine colon
To determine the role of CD38 and possibly other enzymes in the degradation of β-NAD+ in the colon we next examined the degradation of eNAD and NGD in colonic preparations (tunica muscularis) isolated from strain-matched, wild-type (CD38+/+ mice) and from CD38−/− mice. We found equal amounts of eADO were generated from eNAD by muscles of wild-type and CD38−/− mice for 30 s contact with tissue (Fig. 2A and B). Likewise, the amounts of total e-product eADPR + eAMP + eADO were comparable in colons from wild-type and CD38−/− mice: 27.28 ± 2.43 (n= 4) and 24.2 ± 2.78 fmol (mg tissue)−1 (n= 6) in wild-type and CD38−/− mice, respectively (P > 0.05). Therefore, either NAD glycohydrolase activity is not attributed to CD38 in the murine colon or other enzymes, in addition to CD38, are also involved in the degradation of eNAD.
Figure 2. Degradation of eNAD in colon preparations isolated from wild-type and CD38−/− mice.
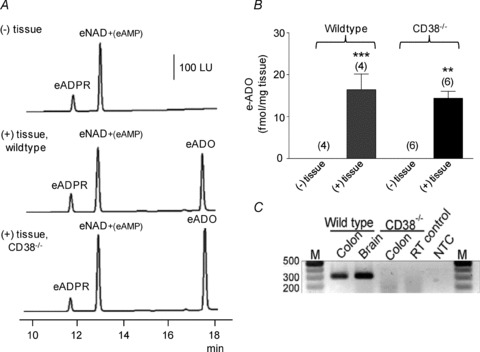
A, original chromatograms of eNAD (0.2 μm) in the absence, (–) tissue, and presence, (+) tissue, of wild-type and CD38−/− colons. Note the increase in eADO formation in the (+) tissue samples after 30 s contact of substrate with tissue. No noticeable changes were observed in the peak of eNAD at 12.5 min, because this peak also contains eAMP formed from eNAD. LU, luminescence units. B, graphic representation of eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. eADO was formed in the presence of tissues. Note that the formation of eADO was comparable in preparations isolated from CD38+/+ and CD38−/− mice. Asterisks denote significant differences from the amounts of eADO in (–) tissue samples (**P < 0.01, ***P < 0.001); number of experiments in parentheses. C, genotyping confirms absence of CD38 from CD38−/− mice. The presence of CD38 (301 bp) was confirmed in the colons of wild-type controls but was absent in CD38−/− mice. The presence of CD38 was also confirmed in the brains of wild-type controls. RT control represents reverse transcriptase control and NTC represents non-template control.
Importantly, eADO was also formed from eNAD after very brief contacts of the substrate eNAD with the tissue (i.e. for 1 s or 5 s, Fig. 3). Thus, in the absence of tissue eADO was not present (Fig. 3). However, in colonic preparations of wild-type mice eADO was 7.27 ± 1.76 (n= 6) and 10.49 ± 2.56 fmol (mg tissue)−1 (n= 6) after 1 s and 5 s contact of tissue with eNAD. Likewise, in colons isolated from CD38−/− mice the amount of eADO formed from eNAD after 1 s and 5 s of contact of tissues was 4.80 ± 1.2 (n= 6) and 6.92 ± 1.23 fmol (mg tissue)−1 (n= 6), respectively.
Figure 3. Degradation of eNAD after brief contacts with murine colon.
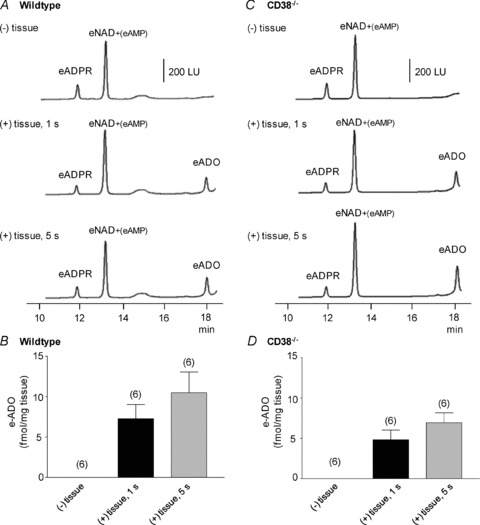
A and C, original chromatograms of 0.2 μm eNAD in the absence of tissue, (–) tissue, and after 1 s and 5 s contact of eNAD with colon muscles isolated from wild-type mice (A) and colons isolated from CD38−/− mice (C). Note the appearance of the end product, eADO, in tissue superfusates after these brief exposures to eNAD. LU, luminescence units. B and D, graphic representation of eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue in colonic preparations isolated from wild-type (B) and CD38−/− (D) mice; number of experiments in parentheses.
As in the monkey colons no further degradation of eNAD was observed during EFS in wild-type and CD38−/− mice: eADO was 16.4 ± 3.74 fmol (mg tissue)−1 in S1 and 23.6 ± 4.47 fmol (mg tissue)−1 in S2 in wild-type mice (n= 4, P > 0.05) and 14.34 ± 1.69 fmol (mg tissue)−1 in S1 and 17.42 ± 1.56 fmol (mg tissue)−1 in S2 (n= 6, P > 0.05) in CD38−/− mice. As shown in Fig. 2C RT-PCR analysis verified the lack of CD38 gene in the CD38−/− mice.
In contrast to eNAD, degradation of NGD to cGDPR occurred in colonic muscles from wild-type mice but not from CD38−/− mice (Fig. 4). These data demonstrate that the ADP-ribosyl cyclase activity in the murine colon is attributable to CD38.
Figure 4. Degradation of NGD in colon preparations isolated from wild-type and CD38−/− mice.
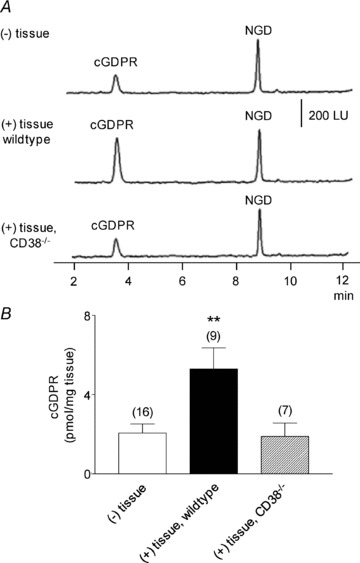
A, original chromatograms of 0.2 mm NGD in the absence (–) tissue and presence of wild-type and CD38−/− colons. Note the increase in cGDPR formation in the (+) tissue samples from colon preparations isolated from wild-type mice and lack of increase in cGDPR in colon preparations isolated from CD38−/− mice. LU, luminescence units. B, graphic representation of cGDPR formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Asterisks denote significant differences from the amounts of cGDPR in (–) tissue samples (**P < 0.01); number of experiments in parentheses.
Extracellular ATP is degraded in the colon
In a previous study we found that ATP was also released in colonic muscles during EFS, but ATP did not mimic the endogenous inhibitory neurotransmitter (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). In the present study we investigated the metabolism of ATP by WM and CM preparations of monkey colon and by colonic muscles from wild-type and CD38−/− mice. As shown in Fig. 5, eATP, prepared from commercial ATP obtained from Sigma-Aldrich, contained a small amount of eADP, but no eAMP or eADO (Fig. 5A and C, (–) tissue). However, when WM or CM was superfused with eATP, eADP appeared, along with eAMP and eADO (Fig. 5A, middle and bottom chromatograms), indicating that degradation of ATP is accomplished by colonic muscles. There was significant formation of eADO in both WM and CM preparations, and no significant difference in eATP metabolism was noted between WM and CM (Fig. 5A and B). eADO formed from eATP was comparable to eADO formed from eNAD. In WM eADO formed from eNAD was 4.39 ± 0.47 fmol (mg tissue)−1 (n= 14) and eADO formed from eATP was 7.13 ± 1.65 fmol (mg tissue)−1 (n= 15) (P > 0.05 comparison in eADO formation from eNAD and eATP). In CM eADO formed from eNAD was 7.37 ± 1.14 fmol (mg tissue)−1 (n= 20), and eADO formed from eATP was 10.57 ± 2.18 fmol (mg tissue)−1 (n= 22) (P > 0.05; comparison in eADO formation from eNAD and eATP).
Figure 5. Degradation of eATP in colon preparations isolated from monkey and murine large intestine.
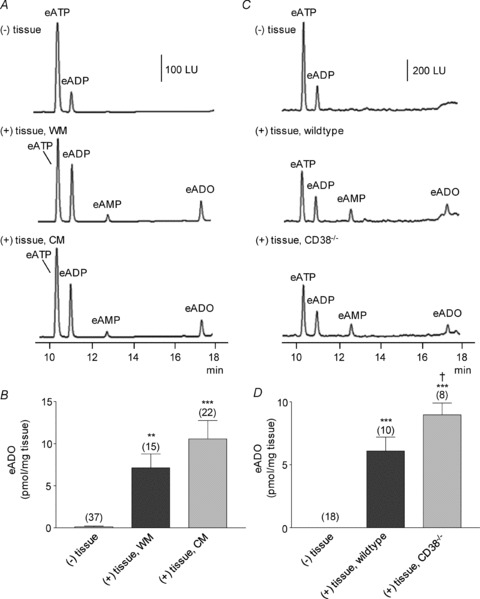
A, original chromatograms of eATP (0.05 μm) in the absence, (–) tissue, and presence, (+) tissue, of WM and CM of monkey colons. Note the increase in eADP, eAMP and eADO in the (+) tissue samples. LU, luminescence units. B, graphic representation of eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Asterisks denote significant differences vs. amounts in (–) tissue samples (**P < 0.01, ***P < 0.001); number of experiments in parentheses. C, original chromatograms of eATP (0.05 μm) in the absence, (–) tissue, and presence, (+) tissue, of colonic preparations isolated from wild-type and CD38−/− mice. eATP was decreased and eAMP and eADO were increased in both groups of preparations. LU, luminescence units. D, graphic representation of eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Asterisks denote significant differences from the amounts of eADO in (–) tissue samples (***P < 0.001). †Significant difference from CD38+/+ samples (P < 0.05); number of experiments in parentheses.
eATP was also metabolized by murine colon (Fig. 5C and D). Interestingly, eADO formation from eATP appeared greater in colon from CD38−/− mice than in colons from wild-type mice. Analysis of eADP + eAMP + eADO also showed greater product formation in preparations from CD38−/− than wild-type mice: 24.15 ± 4.00 (n= 18), 38.3 ± 4.14 (n= 10), and 47.55 ± 5.53 fmol (mg tissue)−1 (n= 8) in (–) tissue, wild-type and CD38−/− mice, respectively (P > 0.05 vs. (–) tissue in wild-type animals and P < 0.01 vs. (–) tissue in CD38−/− mice).
No further degradation of eATP occurred during EFS in all tissues studied. For example, the e-product (eADP + eAMP + eADO) formation was 47.85 ± 9.38 (n= 15) and 60.18 ± 12.16 fmol (mg tissue)−1 (n= 15) in the absence and presence of EFS in monkey WM, respectively (P > 0.05). The e-product was 53.06 ± 8.79 (n= 22) and 49.91 ± 8.45 fmol (mg tissue)−1 (n= 22) in the absence and presence of EFS in monkey CM (P > 0.05). Similar results were obtained using colonic muscles from wild-type and CD38−/− mice (n= 12 and n= 10, respectively, data not shown). Therefore, degradation of ATP occurs in monkey WM and CM colon and in the wild-type and CD38−/− murine colon; however, no ‘releasable’ metabolic activity was observed in colonic muscles.
EFS-induced IJPs are sensitive to apamin and P2Y1 receptor inhibition in the monkey circular smooth muscle cells
The rapid appearance of metabolites of putative purine neurotransmitters raised the question of whether the metabolites might contribute to postjunctional responses. Therefore, we investigated whether ADPR and/or ADP mimic responses to endogenous purine neurotransmitter(s) in monkey colonic preparations. In agreement with previous observations (Hwang et al. 2011), circular muscle cells of the monkey proximal colon had resting membrane potentials (i.e. most negative potentials between action potential complexes) averaging −49 ± 1 mV (n= 20). An ongoing noisy fluctuation in membrane potential, previously referred to as unitary potentials, was observed in these recordings (Beckett et al. 2004; Hirst et al. 2004). IJPs were evoked in monkey colonic muscles by electrical field stimulation (EFS; 1 pulse, 0.5 ms duration) and were reduced by apamin and the P2Y1 receptor antagonist MRS2500. Under control conditions (in the presence of atropine, 1 μm and l-NNA, 100 μm), IJPs were monophasic hyperpolarization responses, 24.3 ± 0.7 mV in amplitude and 0.77 ± 0.04 s in duration (n= 15). Apamin (0.3 μm) reduced the amplitudes of IJPs by 69 ± 4% (n= 7; P= 0.00001) and MRS2500 (1 μm) reduced the amplitude of the IJPs by 95 ± 2% (n= 8; P= 0.000001; Fig. 6A and B).
Figure 6. Inhibitory junction potentials and effects of ADPR and ADP on membrane potential of monkey colonic muscles.
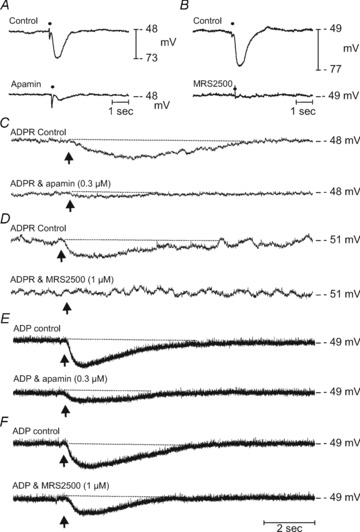
A and B, electrical field stimulation (single pulse 0.5 ms duration; point of stimulation indicated by •), performed in the presence of atropine (1 μm) and l-NNA (100 μm) of monkey colonic circular muscles produced large inhibitory junction potentials (IJPs, control in both A and B) that were greatly attenuated or inhibited by apamin (0.3 μm; A) or by MRS2500 (1 μm; B). C and D, picospritzed ADPR (10 mm loaded in a spritz pipette, arrow, upper traces) produced robust and sustained membrane hyperpolarizations that were inhibited by apamin (0.3 μm) and MRS2500, (1 μm) respectively (lower traces in both panels). E and F, membrane hyperpolarizations induced by ADP (10 mm loaded in a spritz pipette, arrows upper traces) were also reduced by apamin (0.3 μm) and to a lesser extent by MRS2500 (1 μm; lower traces in each panel).
Hyperpolarization responses to localized application of exogenous ADPR and ADP
ADPR and ADP (10 mm loaded into picospritz pipettes) were applied (10 ms pulses) to circular muscles and electrical responses were recorded to evaluate the effects of ADPR and ADP on membrane potential. ADPR caused hyperpolarization of circular muscle cells, averaging 8 ± 1 mV (n= 10) that was sustained for several seconds before the membrane potential recovered to control levels (Fig. 6C and D, Control). ADP spritzes caused similar hyperpolarization responses, averaging 10 ± 0.5 mV (n= 10), which also persisted for several seconds (Fig. 6E and F, Control). The response to ADPR was reduced 84 ± 3% (P= 0.006, n= 5) by apamin (0.3 μm) and 95 ± 3% (P= 0.0003; n= 5; bottom traces) by MRS2500 (1 μm). Examples are shown in Fig. 6C and D. Apamin reduced the response to ADP by 78 ± 6% (P= 0.0004; n= 5), but the hyperpolarization to MRS2500 was reduced by only 38 ± 7%, and this did not reach a level of significance (P > 0.05; n= 5; Fig. 6E and F, bottom traces). Therefore, the pharmacology of ADPR responses better mimicked postjunctional responses to the endogenous purine neurotransmitter than ADP.
It is possible that if ADP is more potent than ADPR that the responses to ADPR might be more fully antagonized than responses to ADP. Therefore, we performed an additional series of experiments to test the effectiveness of MRS2500 in blocking responses over the full range of the concentration–response relationships of ADP and ADPR. While maintaining single impalements, the pulse duration of spritzing was varied over the range of 10–100 ms and responses to ADPR and ADP were recorded before and after addition of MRS2500 (1 μm). Figure 7 shows that ADP and ADPR were approximately equally potent in producing hyperpolarization and that MRS2500 antagonized responses to ADPR to a much greater extent than to ADP.
Figure 7. Concentration–response relationship for ADPR and ADP on membrane hyperpolarizations in murine colon.
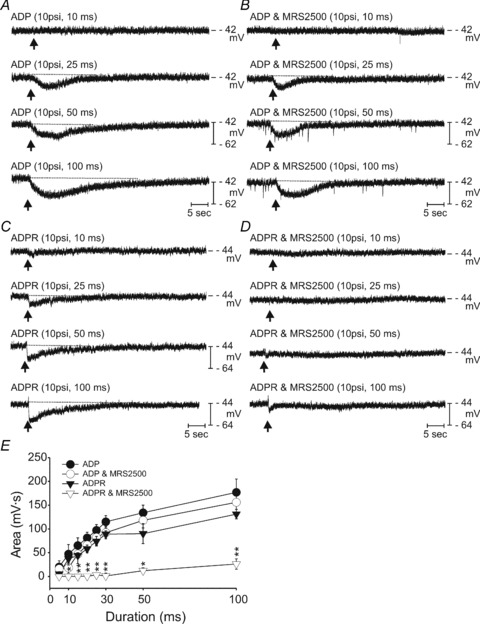
A, a series of responses to spritzes of ADP (10 mm in spritz pipette) using pulse durations from 10–100 ms. Note the increase in hyperpolarization response as the spritz pulse is increased. B, responses to ADP after addition of MRS2500 (1 μm) to the bath solution. This P2Y1 antagonist only slightly decreased the area of the hyperpolarization responses to ADP. The fast voltage transients superimposed upon the record in B are due to static electricity. C, spritz responses to ADPR (10 mm in spritz pipette) using pulse durations from 10 to 100 ms. D, blockade of responses to ADPR after addition of MRS2500. The records in panels A–D were recorded during a single continuous impalement. E, summary of the results from 5 experiments using this protocol. Hyperpolarization responses were tabulated as areas under the response curves (mV s). *P < 0.01; **P < 0.001.
Similar to previous results testing the effects of β-NAD+ (Mutafova-Yambolieva et al. 2007), ADPR also inhibited spontaneous contractions of colonic muscles cut parallel to the circular muscle fibres (Supplemental material, Fig. 1).
Electrophysiological evidence that degradation of β-NAD+ does not exclusively require CD38
We also tested whether postjunctional responses differ in colonic muscles isolated from wild-type and CD38−/− mice. In agreement with previous observations (Mutafova-Yambolieva et al. 2007), robust IJPs were evoked by nerve stimulation in colonic muscles of wild-type mice, and the IJPs were blocked by apamin, suramin, PPADS and MRS2500 (Fig. 8A–D and Table 2). Colonic muscles of wild-type mice were hyperpolarized by ADPR, and this response was also inhibited by apamin, suramin, PPADS and MRS2500 (Fig. 8E–H and Table 2). Picospritzing of ADP also caused hyperpolarization, and this response was significantly inhibited by apamin, but not by MRS2500 (Fig. 8I and J and Table 2).
Figure 8. Effects of ADPR and ADP on membrane potential and inhibitory junction potentials of colonic circular muscle cells from wild-type CD38+/+ mice.
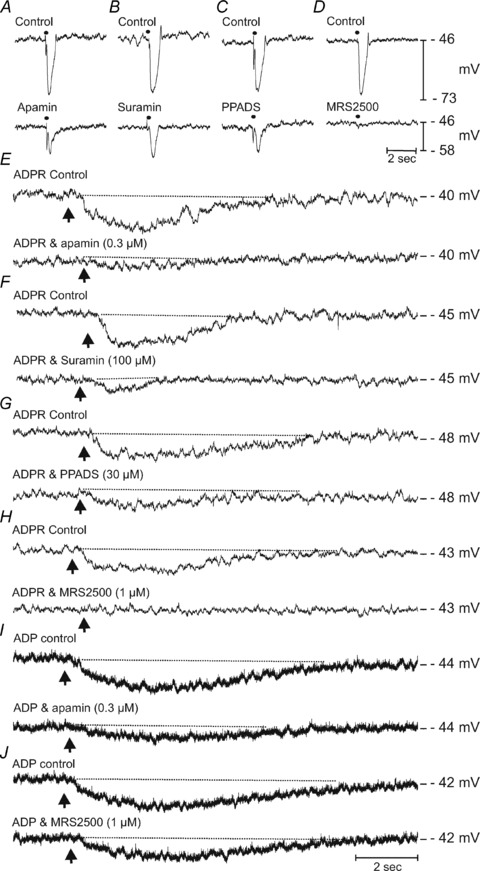
A–D show IJP evoked by EFS (single pulse 0.5 ms duration, •) in the presence of atropine (1 μm) and l-NNA (100 μm) (Control, upper traces in each panel) and after apamin (0.3 μm; A), suramin (100 μm; B), PPADS (30 μm; C) and MRS2500 (1 μm; D) (lower traces in each panel). Control responses were all recorded in the presence of atropine (1 μm) and l-NNA (100 μm). E–H shows the effects of ADPR (10 mm loaded in a spritz pipette, picospritzed onto circular muscles) at a time point indicated by arrow. Upper traces of each panel represent control responses recorded in the presence of atropine (1 μm) and l-NNA (100 μm). ADPR produced reproducible membrane hyperpolarizations which were sustained for several seconds before returning to pre-stimulus levels. ADPR-induced membrane hyperpolarizations were antagonized by apamin (0.3 μm; E), and the P2Y receptor antagonists, suramin (100 μm; F), PPADS (30 μm; G) and MRS2500 (1 μm; H). I and J shows the effects of ADP (10 mm, picospritzed) on murine colonic circular muscles before (upper traces) and in the presence of apamin (0.3 μm; I) and MRS2500 (1 μm; J). Apamin antagonized the membrane hyperpolarizations induced by ADP.
Table 2.
Effects of antagonists to neural responses and exogenous ADPR and ADP in colons isolated from wild-type CD38+/+ mice
| IJP | ADPR | ADP | ||||
|---|---|---|---|---|---|---|
| % inhibition | P value | % inhibition | P value | % inhibition | P value | |
| Apamin | 65 ± 7 (9) | 0.000001 | 90 ± 3 (5) | 0.001987 | 71 ± 6 (4) | 0.018600 |
| Suramin | 55 ± 6 (5) | 0.000915 | 78 ± 2 (5) | 0.000010 | — | — |
| PPADS | 65 ± 4 (5) | 0.000134 | 73 ± 4 (5) | 0.003722 | — | — |
| MRS2500 | 93 ± 2 (12) | 0.000001 | 96 ± 2 (6) | 0.006596 | 30 ± 6 (6) | 0.064000 |
In muscles from CD38−/− mice, robust IJPs were evoked by nerve stimulation, and these responses were blocked by apamin and MRS2500 (Fig. 9A and B). There was no significant difference in IJPs or the pharmacology of IJPs in wild-type and CD38−/− mice. Picospritzing β-NAD+ (Fig. 9C and D), ADPR (Fig. 9E and F), ATP (Fig. 9G and H), or ADP (Fig. 9I and J) caused hyperpolarization, but the hyperpolarization responses were differentially blocked by apamin and MRS2500. For example, responses to β-NAD+ and ADPR were significantly reduced by apamin and MRS2500, but hyperpolarizations to ATP and ADP, although significantly reduced by apamin, were not affected by MRS2500 (Fig. 9 and Table 3). It appears, therefore, that the responses to endogenous neurotransmitter(s) and exogenous β-NAD+ are similar in colonic muscles from wild-type and CD38−/− mice and, therefore, pathways other than CD38 appear to be responsible for the degradation of β-NAD+ to ADPR. Moreover, the responses to locally applied β-NAD+ and ADPR, but not ATP and ADP, were attenuated by the P2Y1 receptor antagonist in colonic muscles from CD38−/− mice as shown for monkey WM and CM (Fig. 6).
Figure 9. Effects of β-NAD+, ATP, ADPR and ADP on membrane potential and inhibitory junction potentials of colonic circular muscles from CD38−/− mice.
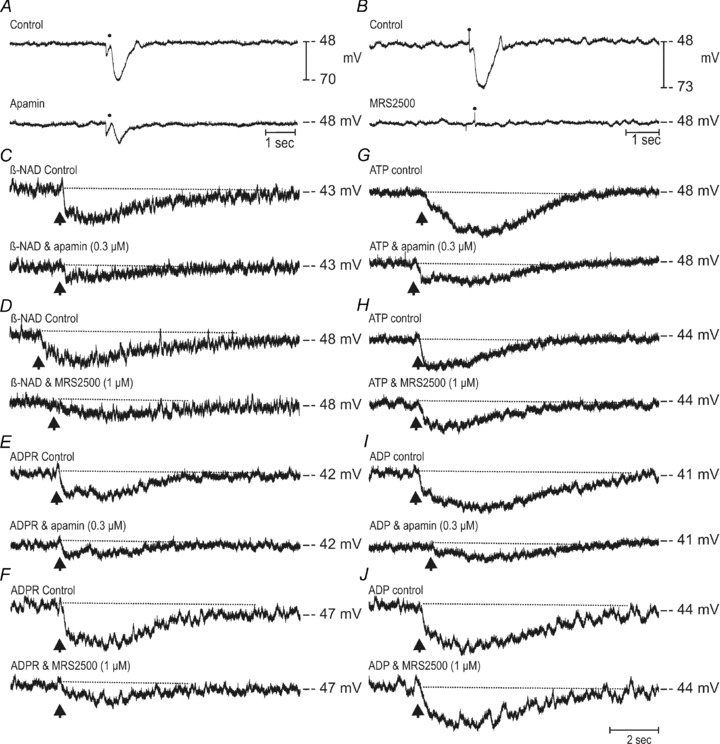
A and B, IJPs recorded from colonic circular muscles of CD38 null mice under control conditions (upper traces) and after apamin (A; lower trace) or MRS2500 (B; lower trace). Control conditions represent experiments that were performed in the presence of atropine (1 μm) and l-NNA (100 μm). C and D, β-NAD+ induced membrane hyperpolarizations (50 mm loaded in a spritz pipette, picospritzed at arrow) before (upper traces) and in the presence of apamin (0.3 μm; lower trace C) and MRS2500 (1 μm; lower trace D). E and F, membrane hyperpolarizations to ADPR (10 mm, picospritzed at arrow) before (upper traces) and in the presence of apamin (0.3 μm; lower trace E) or MRS2500 (1 μm; lower trace F). G and H, membrane hyperpolarizations in responses to picospritzed ATP (10 mm, upper traces in each panel) and in the presence of apamin (0.3 μm; lower trace G) or MRS2500 (1 μm; lower trace H). I and J, the effects of ADP on membrane potential under control conditions (10 mm, upper traces in each panel) and after apamin (0.3 μm; lower trace I) or MRS2500 (1 μm; lower trace J).
Table 3.
Effects of apamin and MRS2500 on neural responses and exogenous purines in colons isolated from CD38−/− mice
| IJP | β-NAD | ADPR | ATP | ADP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % inhibition | P value | % inhibition | P value | % inhibition | P value | % inhibition | P value | % inhibition | P value | |
| Apamin | 56 ± 3 (20) | 0.000001 | 82 ± 5 (5) | 0.00048 | 85 ± 4 (5) | 0.0013 | 66 ± 9 (5) | 0.0008 | 75 ± 8 (5) | 0.013 |
| MRS2500 | 99 ± 1 (21) | 0.000001 | 82 ± 3 (6) | 0.023 | 83 ± 4 (5) | 0.0161 | 17 ± 7 (5) | 0.3436 | 26 ± 4 (5) | 0.2063 |
Mechanisms for ADPR degradation are present in the colon
As shown in Figs 6–9, localized application of ADPR caused hyperpolarization responses similar to the endogenous purine neurotransmitter. We investigated whether exogenous ADPR can be degraded by monkey and murine colonic muscles. Superfusion of monkey WM or CM tissues with eADPR resulted in formation of eADO (Fig. 10A and B); eADO was 1.53 ± 0.24 (n= 10) and 3.53 ± 0.75 fmol (mg tissue)−1 (n= 19) in experiments with WM and CM, respectively. No eADO was resolved in superfusates in the absence of muscle tissues (P < 0.001 vs. superfusates exposed to muscles). Note that eADO formation was significantly greater per unit mass in CM preparations than in WM (P < 0.05). There was also significant formation of total products (eAMP + eADO) in WM (P < 0.05) and CM (P < 0.01), and total metabolite formation was greater per unit mass in CM than in WM, P < 0.05 (Fig. 10B).
Figure 10. Degradation of eADPR in colon preparations isolated from monkey and murine large intestine.
A, original chromatograms of eADPR (0.05 μm) in the absence, (–) tissue, and presence, (+) tissue, of WM and CM of monkey colons. Note the decrease in eADPR and increase in eADO in the (+) tissue samples. LU, luminescence units. B, graphic representation of eAMP + eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Note that the formation of eAMP + eADO product was greater in CM than in WM. Asterisks denote significant differences from the amounts of eAMP + eADO in (–) tissue samples (*P < 0.05, **P < 0.01). †Significant difference from WM (P < 0.05); number of experiments in parentheses. C, original chromatograms of 0.05 μm eADPR in the absence, (–) tissue, and presence, (+) tissue, of colonic preparations isolated from wild-type and CD38−/− mice. eADPR was decreased and eAMP + eADO was increased in both groups of preparations. LU, luminescence units. D, graphic representation of eAMP + eADO formation in superfusate samples collected in the absence (–) or presence (+) of tissue. Asterisks denote significant differences from the amounts of eAMP + eADO in (–) tissue samples (***P < 0.001); number of experiments in parentheses.
In wild-type and CD38−/− colonic muscles, significant formation of eAMP and eADO occurred after exposure of eADPR to muscle tissues (Fig. 10C and D). In the absence of tissue the amount of eAMP was 2.79 ± 1.4 fmol (mg tissue)−1 (n= 27) and no eADO was resolved. eAMP formed was 15.36 ± 1.46 (n= 14, P < 0.001) and 12.74 ± 1.47 fmol (mg tissue)−1 (n= 13, P < 0.001) in wild-type and CD38−/− colons, respectively. eADO was 9.92 ± 0.96 and 9.57 ± 1.9 fmol (mg tissue)−1 in wild-type and CD38−/−, respectively, P < 0.001 vs. no tissue control for both. The formation of total product (eAMP + eADO) was also significantly increased in the presence of wild-type and CD38−/− colons, respectively. Therefore, ADPR metabolism was similar in muscles of wild-type and CD38−/− mice.
Discussion
In the present study we investigated tissue metabolism of purine neurotransmitter candidates in colonic muscles of monkeys and mice and postjunctional responses to direct metabolites. In confirmation of previous work, we found that colon muscle superfusates contained not only primary transmitter substances, β-NAD+ and ATP, but also their direct metabolites ADPR and ADP, respectively (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). We provided evidence that ADPR and ADP are rapidly produced from β-NAD+ and ATP in colonic tunica muscularis and evaluated the role of CD38 in β-NAD+ degradation. We tested whether ADPR and ADP mimic the effects of the endogenous purine neurotransmitter. In both monkey and murine colons, local application of ADPR and ADP evoked membrane hyperpolarization similar to β-NAD+ and ATP. However, only ADPR, like its precursor β-NAD+ (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011), mimicked the pharmacology of the endogenous purine neurotransmitter. These findings demonstrate that ADPR, either generated by metabolism of β-NAD+ released by enteric inhibitory neurons or released as a primary neurotransmitter, may contribute to postjunctional purinergic responses in GI muscles.
A component of nerve-evoked inhibitory responses in GI smooth muscles is blocked by antagonists of P2 purine receptors, indicating that a purine nucleotide, released from enteric motor neurons, mediates part of the inhibitory response in the GI tract. Since ATP is a common bioactive compound and a principle agonist of numerous P2 receptors (reviewed in von Kugelgen, 2006), it has been assumed for many years that responses mediated by P2 receptors are elicited by ATP. In the context of GI physiology this concept has amounted to ‘assigning’ ATP to be the purine inhibitory neurotransmitter (Burnstock et al. 1970; Burnstock, 2008). However, ATP frequently failed to meet presynaptic criteria for a neurotransmitter substance or to mimic postjunctional responses elicited by the endogenous inhibitory neurotransmitter in GI muscles (Serio et al. 2003, Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). In contrast, β-NAD+, another endogenous purine nucleotide, is also released upon stimulation of enteric nerves and it mimics the effects and pharmacology of the inhibitory neurotransmitter in the murine and primate large intestine better than ATP (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). These studies, although unable to formally rule out ATP as a neurotransmitter, raise the possibility that inhibitory purinergic neurotransmission is more complex than typically considered. The present study adds to the complexity by showing, that ADPR, the primary metabolite of extracellular β-NAD+, also mimics postjunctional responses to the endogenous inhibitory neurotransmitter.
Postjunctional electrophysiological responses (i.e. IJPs) attributed to release of purine neurotransmitter(s) are inhibited by P2Y1 receptor antagonists and by the small conductance K+ channel blocker apamin (Gallego et al. 2006, 2008, 2011; Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). Similar to IJPs, hyperpolarization responses to either β-NAD+ or ATP are inhibited by apamin (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). β-NAD+ and ATP both bind to P2Y1 receptors (von Kugelgen, 2006; Mutafova-Yambolieva et al. 2007; Kurahashi et al. 2011); however, only hyperpolarization responses to β-NAD+, and not ATP, were inhibited by P2Y1 receptor antagonists (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). Therefore, in contrast to β-NAD+, the effects of ATP are mediated largely by receptors in addition to P2Y1. With this observation, it is hard to imagine how ATP (or its metabolites) could be the purine neurotransmitter in these muscles if transmitters are released or metabolites are generated ‘in volume’ within the interstitium. If this were the case, postjunctional responses (IJPs) would not be blocked by P2Y1 antagonists, just as responses to spritzed ATP and ADP are not blocked by P2Y1 antagonists. Our results, favour the concept that purine transmitters (or metabolites) are released (or generated) and bind receptors in a limited volume of the interstitium (e.g. in a synapse-like space), where P2Y1 receptors are the dominant receptors available for binding purines on postjunctional cells. In fact specialized cells, termed PDGFRα+ cells, have recently been described in colonic muscles. PDGFRα+ cells are closely associated with motor nerve terminals, form gap junctions with smooth muscle cells, and express the molecular apparatus to mediate P2Y1-dependent postjunctional responses (Kurahashi et al. 2011). If purine transmitter(s), either by concentration or physical constraints, are limited to a receptive field, such as that provided by PDGFRα+ cells, then it is possible that both ATP and β-NAD+ could participate as co-transmitters. ADPR might be generated after release of β-NAD from enteric neurons, or ADPR could be stored and released as a primary neurotransmitter. It is also possible that ADP could be generated within junctional spaces and participate in P2Y1-dependent responses. At present we cannot clearly distinguish between these possibilities, but our data demonstrate that once released, β-NAD can be rapidly metabolized. Finding that ADPR is bioactive in GI muscles suggests that multiple purines could contribute to postjunctional purinergic responses in the gut.
We compared the degradation of extracellular β-NAD+, ADPR and ATP in whole tunica muscularis and in circular muscles (CM), containing nerve processes but no ganglia, and we examined the role of CD38 in β-NAD+ metabolism. We first monitored metabolism of β-NAD+ and ATP in contact with muscles for 30–60 s to match neurotransmitter overflow studies performed with similar stimulation parameters (i.e. Mutafova-Yambolieva et al. 2007; Hwang et al. 2011). Degradation of β-NAD+ was greater in monkey CM than in WM preparations. The increase in degradation of β-NAD+ in CM was not due to differences in ADO metabolism in WM versus CM as ADO did not undergo additional degradation during 60 s incubation with colonic tissues. Similar to β-NAD+, ADPR was degraded more in CM than in WM of monkey colon. In contrast, degradation of ATP was similar in WM and CM preparations. Taken together these data suggest that the enzymatic activities responsible for the degradation of extracellular β-NAD+ and ADPR are more prominent in parts of the colon wall containing nerve processes and varicosities than in parts containing myenteric ganglia. Thus, β-NAD+ and ADPR may be degraded primarily near nerve terminals or close to the site of release. ATP in contrast may be degraded at sites other than near sites of neurotransmitter release.
We found that a proportion of eNAD was degraded to eADO within a few seconds of contact with colonic muscles. One might argue on the basis of kinetics that 1 s or more is still slow compared to the kinetics of an IJP. However, this argument is too limited because metabolism of an exogenous neurotransmitter substance might have quite different kinetics than metabolism of neurotransmitters released from nerve terminals. In the former case, the exogenous neurotransmitter must first reach the site of metabolism, which might be very close to sites of transmitter release. It is not currently possible to simulate the delivery of endogenous neurotransmitter by application of an exogenous substance. Analogous to this, responses to exogenous neurotransmitter substances develop with far slower kinetics than responses to neurotransmitters released from nerve terminals (Fig. 11). IJP hyperpolarization responses develop with a time constant that is nearly 10-fold less than the time constant of hyperpolarization responses to exogenous neurotransmitter (even applied locally with a spritz pipette). The same discrepancy might be expected in the kinetics of metabolism, and therefore, the breakdown in endogenous purines might occur 10-fold faster than metabolism of exogenous purines. It is likely that purinergic regulation of colonic muscles is the result of many IJPs, and previous studies have demonstrated tonic purinergic inhibition of colonic excitability (e.g. Spencer et al. 1998). Therefore, ample time exists for metabolism of purine neurotransmitters, and postjunctional responses are likely to be integrated responses to all primary transmitters and bioactive metabolites. The latter is also applicable to tissue pharmacology. Our results suggest that concentration–response studies of purines added to muscle baths are likely to be heavily contaminated by responses to bioactive metabolites. It should also be emphasized that quantitative analyses and comparisons of mechanisms and rates of degradation of purine nucleotides cannot be determined, at present, in whole tissue preparations due to the complexity of extracellular NAD+ metabolism (de Figueiredo et al. 2011), simultaneous processing by multiple enzymatic pathways, inability to simultaneously detect the formation of additional NAD metabolites (e.g. nicotinamide), and possible participation of other mechanisms in the elimination of β-NAD+ from the neuroeffector junction (e.g. neuronal uptake).
Figure 11. Superposition of inhibitory junction potential (IJP) and hyperpolarization responses to exogenous ATP and β-NAD spritzed near the site of recording in a murine colonic preparation.
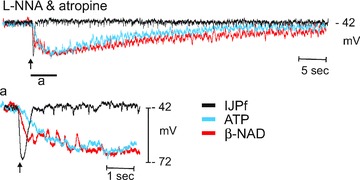
This experiment demonstrates the kinetic differences in responses to the endogenous purinergic neurotransmitter released from nerve terminals and hyperpolarization responses to exogenous transmitter candidates. Single pulses (0.5 ms pulse duration) of electrical field stimulation released neurotransmitter that resulted in fast inhibitory junction potentials (IJPs). The time constant of the upstroke of the IJPs, fitted by a single exponential (Clampfit; Molecular Devices, Sunnyvale, CA, USA), was 135 ± 8 ms (n= 41). In contrast responses to spritzed neurotransmitter candidates (10 psi; 25 ms pulses) developed more slowly (amplitudes scaled to approximate amplitude of the IJP); time constants for ATP (10 mm in spritz pipette) and β-NAD (50 mm in picospritz pipette) averaged 1217 ± 159 (n= 20) and 1001 ± 149 (n= 21), respectively. These data demonstrate that substantially more time is required for exogenous compounds to reach and bind receptors and for responses to develop than is required for the responses to neurotransmitters released from neurons. This implies that responses to neurotransmitters are transduced by postjunctional receptive fields quite close to the sites of release. These data also indicate that sites of neurotransmitter metabolism might be more accessible to transmitters released from neurons than to exogenous transmitter candidates. Thus, the kinetics of metabolism of exogenous substances may underestimate the kinetics of metabolism of endogenous transmitters by nearly an order of magnitude.
Since β-NAD+ meets pre- and postsynaptic criteria for a neurotransmitter in the colon better than ATP (Mutafova-Yambolieva et al. 2007; Hwang et al. 2011) and since the primary β-NAD+-metabolizing enzyme, CD38, is expressed in colon (Mutafova-Yambolieva et al. 2007), we examined the functional role of CD38 in the degradation of extracellular β-NAD+ in the colon. As expected, the ADP-ribosyl cyclase activity, converting part of β-NAD+ into cADPR, is provided by CD38. However, NAD appeared to be degraded to ADO with equal efficiency in colonic muscles of wild-type and CD38−/− mice. If CD38−/− colons lack the ADP-ribosyl cyclase activity they also probably lack the NAD-glycohydrolase activity that is associated with CD38, since the same active centre in the CD38 molecule carries both the ADP-ribosyl cyclase and NAD-glycohydrolase activities (Sauve et al. 1998). Therefore, enzymes other than CD38 are likely to be involved in the degradation of β-NAD+ in the colons isolated form CD38−/− mice. Moreover, amplitudes and durations of IJPs elicited by single stimuli were identical in colonic preparations isolated from wild-type and CD38−/− mice, in contrast to what would be expected if CD38 was the only enzyme degrading β-NAD+ released from motor neurons. If, on the other hand, ADPR contributed significantly to endogenous IJPs in the colon, reduced responses would be expected in CD38−/− mice, because ADPR would not be formed from β-NAD+ in the absence of CD38. Furthermore, responses to exogenous β-NAD+ should be reduced in CD38−/− mice as ADPR would not be formed. However, our results show that both IJPs and hyperpolarization responses to exogenous β-NAD+ and ADPR were comparable in the presence and absence CD38. These are surprising observations, since CD38 is considered to be the main protein carrying NAD-glycohydrolase activity in mammals (Lee, 2001) and was established to be present in nerve terminals in vascular smooth muscles (Smyth et al. 2006). The present study suggests, however, that additional enzymes with NAD-glycohydrolase activity are present in the GI muscles and might be upregulated to compensate for the CD38 absence as can occur in animals with congenital gene deletions. For example, the GPI-anchored protein CD157 shares several characteristics with CD38, including a similar amino acid sequence and enzymatic functions involved in the metabolism of β-NAD+ (Ortolan et al. 2002); no information is available about the potential role of CD157 in the degradation of β-NAD+ in the colon. Further studies are warranted to determine the protein(s) responsible for the degradation of extracellular β-NAD+ in the colon.
In non-GI visceral smooth muscles (e.g. guinea-pig vas deferens), it has been reported that degradation of ATP increases during nerve stimulation due to ‘releasable’ enzymatic activity (Todorov et al. 1997). We investigated whether a similar mechanism for terminating transmitter activity exists in colonic muscles. Soluble (i.e. releasable) enzymatic activity was not observed in our experiments, as nerve stimulation did not enhance degradation of ATP and β-NAD+. This is in agreement with a previous study which also failed to detect soluble nucleotidase activity in enteric nerves of the guinea-pig taenia coli (Westfall et al. 2000), suggesting that soluble enzymatic activity is not generally a property of enteric motor neurons. Our data suggest that degradation of extracellular purines and purine neurotransmitter(s) released by nerve stimulation occurs via membrane-bound, ‘ecto-’, enzymes.
In summary, the present study demonstrates that: (i) bioactive metabolites of β-NAD+ and ATP are rapidly formed in the tunica muscularis of the colon; (ii) ADPR, like its precursor β-NAD+, mimics the inhibitory purine neurotransmitter in the colon, (iii) more than a single purine substance may mediate purinergic neurotransmission in colonic muscles; (iv) degradation of extracellular β-NAD+ and ADPR is more pronounced in parts of the colon wall enriched in nerve terminals, and therefore, mechanisms exist to degrade purine neurotransmitters and possibly limit their duration of action; (v) degradation of β-NAD+ appears to be only partially associated with CD38; degradation of β-NAD+ seems to be accomplished by at least one additional pathway. Taken together, our data suggest that both primary neurotransmitters and metabolites are likely to contribute to purinergic signalling in GI muscles.
Acknowledgments
This work was supported by a NIH grant, PO1 DK41315 (S.M.W., K.M.S. and V.M.-Y.), and a research award from ANMS to S.J.H. The authors would like to acknowledge the excellent technical assistance of Lauren Peri and Deborah Russell.
Glossary
Abbreviations
- ADO
adenosine
- ADPR
adenosine 5′-diphosphate ribose
- AMP
adenosine 5′-monophosphate
- CM
circular muscle
- GI
gastrointestinal tract
- β-NAD+
β-nicotinamide adenine dinucleotide
- cADPR
cyclic ADPR
- cGDPR
cyclic guanosine diphosphate-ribose
- EFS
electrical field stimulation
- EHNA
erythro-9-(2-hydroxy-3-nonyl)-adenine
- IJP
inhibitory junction potential
- E-NPP
ecto-nucleotide pyrophosphatases/phosphodiasterase
- E-NTPDase
ecto-nucleotide triphosphate diphsophohydrolase
- NPP
nucleotide pyrophosphatase
- WM
whole muscle
Author contributions
L.D., S.J.H. and V.M.Y. carried out the experiments. L.D., S.M.W., K.M.S. and V.M.Y. designed the experiments and wrote the paper. L.D. and S.J.H. contributed equally to this study. The work was carried out at the Department of Physiology and Cell Biology, University of Nevada School of Medicine, Reno, NV. All authors approved the final version for publication. The authors declare no conflicts of interests.
References
- Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Agteresch HJ, Dagnelie PC, van den Berg JW, Wilson JH. Adenosine triphosphate: established and potential clinical applications. Drugs. 1999;58:211–232. doi: 10.2165/00003495-199958020-00002. [DOI] [PubMed] [Google Scholar]
- Beckett EA, Bayguinov YR, Sanders KM, Ward SM, Hirst GD. Properties of unitary potentials generated by intramuscular interstitial cells of Cajal in the murine and guinea-pig gastric fundus. J Physiol. 2004;559:259–269. doi: 10.1113/jphysiol.2004.063016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobalova J, Mutafova-Yambolieva VN. Co-release of endogenous ATP and noradrenaline from guinea-pig mesenteric veins exceeds co-release from mesenteric arteries. Clin Exp Pharmacol Physiol. 2001;28:397–401. doi: 10.1046/j.1440-1681.2001.03460.x. [DOI] [PubMed] [Google Scholar]
- Bobalova J, Mutafova-Yambolieva VN. Membrane-bound and releasable nucleotidase activities: differences in canine mesenteric artery and vein. Clin Exp Pharmacol Physiol. 2003;30:194–202. doi: 10.1046/j.1440-1681.2003.03808.x. [DOI] [PubMed] [Google Scholar]
- Bobalova J, Bobal P, Mutafova-Yambolieva VN. High-performance liquid chromatographic technique for detection of a fluorescent analogue of adp-ribose in isolated blood vessel preparations. Anal Biochem. 2002;305:269–276. doi: 10.1006/abio.2002.5667. [DOI] [PubMed] [Google Scholar]
- Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. [DOI] [PubMed] [Google Scholar]
- Burns AJ, Lomax AE, Torihashi S, Sanders KM, Ward SM. Interstitial cells of Cajal mediate inhibitory neurotransmission in the stomach. Proc Natl Acad Sci U S A. 1996;93:12008–12013. doi: 10.1073/pnas.93.21.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. The journey to establish purinergic signalling in the gut. Neurogastroenterol Motil. 2008;20(Suppl 1):8–19. doi: 10.1111/j.1365-2982.2008.01107.x. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Campbell G, Satchell D, Smythe A. Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br J Pharmacol. 1970;40:668–688. doi: 10.1111/j.1476-5381.1970.tb10646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo LF, Gossmann TI, Ziegler M, Schuster S. Pathway analysis of NAD+ metabolism. Biochem J. 2011;439:341–348. doi: 10.1042/BJ20110320. [DOI] [PubMed] [Google Scholar]
- Di Girolamo M, Lupi R, Silletta MG, Turacchio S, Iurisci C, Luini A, Corda D. Modulatory role of GTP-binding proteins in the endogenous ADP-ribosylation of cytosolic proteins. Adv Exp Med Biol. 1997;419:343–347. doi: 10.1007/978-1-4419-8632-0_45. [DOI] [PubMed] [Google Scholar]
- Gallego D, Hernandez P, Clave P, Jimenez M. P2Y1 receptors mediate inhibitory purinergic neuromuscular transmission in the human colon. Am J Physiol Gastrointest Liver Physiol. 2006;291:G584–G594. doi: 10.1152/ajpgi.00474.2005. [DOI] [PubMed] [Google Scholar]
- Gallego D, Gil V, Aleu J, Auli M, Clave P, Jimenez M. Purinergic and nitrergic junction potential in the human colon. Am J Physiol Gastrointest Liver Physiol. 2008;295:G522–G533. doi: 10.1152/ajpgi.00510.2007. [DOI] [PubMed] [Google Scholar]
- Gallego D, Gil V, Aleu J, Martinez-Cutillas M, Clave P, Jimenez M. Pharmacological characterization of purinergic inhibitory neuromuscular transmission in the human colon. Neurogastroenterol Motil. 2011;23:792–e338. doi: 10.1111/j.1365-2982.2011.01725.x. [DOI] [PubMed] [Google Scholar]
- Graeff R, Liu Q, Kriksunov IA, Kotaka M, Oppenheimer N, Hao Q, Lee HC. Mechanism of cyclizing NAD to cyclic ADP-ribose by ADP-ribosyl cyclase and CD38. J Biol Chem. 2009;284:27629–27636. doi: 10.1074/jbc.M109.030965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff RM, Franco L, De Flora A, Lee HC. Cyclic GMP-dependent and -independent effects on the synthesis of the calcium messengers cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate. J Biol Chem. 1998;273:118–125. doi: 10.1074/jbc.273.1.118. [DOI] [PubMed] [Google Scholar]
- Gustafsson AJ, Muraro L, Dahlberg C, Migaud M, Chevallier O, Khanh HN, Krishnan K, Li N, Islam MS. ADP ribose is an endogenous ligand for the purinergic P2Y1 receptor. Mol Cell Endocrinol. 2011;333:8–19. doi: 10.1016/j.mce.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Hirst GDS, Bywater RAR, Teramoto N, Edwards FR. An analysis of inhibitory junction potentials in the guinea-pig proximal colon. J Physiol. 2004;558:841–855. doi: 10.1113/jphysiol.2004.065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Durnin L, Dwyer L, Rhee PL, Ward SM, Koh SD, Sanders KM, Mutafova-Yambolieva VN. β-Nicotinamide adenine dinucleotide is an enteric inhibitory neurotransmitter in human and nonhuman primate colons. Gastroenterology. 2011;140:608–617. doi: 10.1053/j.gastro.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal Z, Afkham-Ebrahimi A, Saggerson ED. A novel assay for 5′-nucleotidase using 1,N6-etheno-AMP as substrate, and comments on the properties of the reaction product, ethenoadenosine. Biochem J. 1988;250:369–373. doi: 10.1042/bj2500369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato I, Yamamoto Y, Fujimura M, Noguchi N, Takasawa S, Okamoto H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J Biol Chem. 1999;274:1869–72. doi: 10.1074/jbc.274.4.1869. [DOI] [PubMed] [Google Scholar]
- Klein C, Grahnert A, Abdelrahman A, Muller CE, Hauschildt S. Extracellular NAD+ induces a rise in [Ca2+]i in activated human monocytes via engagement of P2Y1 and P2Y11 receptors. Cell Calcium. 2009;46:263–272. doi: 10.1016/j.ceca.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Kurahashi M, Zheng H, Dwyer L, Ward SM, Don KS, Sanders KM. A functional role for the ‘fibroblast-like cells’ in gastrointestinal smooth muscles. J Physiol. 2011;589:697–710. doi: 10.1113/jphysiol.2010.201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev Pharmacol Toxicol. 2001;41:317–345. doi: 10.1146/annurev.pharmtox.41.1.317. [DOI] [PubMed] [Google Scholar]
- Levitt B, Head RJ, Westfall DP. High-pressure liquid chromatographic-fluorometric detection of adenosine and adenine nucleotides: application to endogenous content and electrically induced release of adenyl purines in guinea pig vas deferens. Anal Biochem. 1984;137:93–100. doi: 10.1016/0003-2697(84)90352-x. [DOI] [PubMed] [Google Scholar]
- Mihaylova-Todorova S, Todorov LD, Westfall DP. Correlation between the release of the sympathetic neurotransmitter atp and soluble nucleotidases from the guinea pig vas deferens. J Pharmacol Exp Ther. 2001;296:64–70. [PubMed] [Google Scholar]
- Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC. Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J Biol Chem. 2000;275:21566–21571. doi: 10.1074/jbc.M909365199. [DOI] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM. β-Nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104:16359–16364. doi: 10.1073/pnas.0705510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortolan E, Vacca P, Capobianco A, Armando E, Crivellin F, Horenstein A, Malavasi F. CD157, the Janus of CD38 but with a unique personality. Cell Biochem Funct. 2002;20:309–322. doi: 10.1002/cbf.978. [DOI] [PubMed] [Google Scholar]
- Sanders KM, Ward SM. Nitric oxide as a mediator of nonadrenergic noncholinergic neurotransmission. Am J Physiol Gastrointest Liver Physiol. 1992;262:G379–G392. doi: 10.1152/ajpgi.1992.262.3.G379. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- Secrist JA, Barrio JR, Leonard NJ, Weber G. Fluorescent modification of adenosine-containing coenzymes. Biological activities and spectroscopic properties. Biochemistry. 1972;11:3499–3506. doi: 10.1021/bi00769a001. [DOI] [PubMed] [Google Scholar]
- Serio R, Alessandro M, Zizzo MG, Tamburello MP, Mule F. Neurotransmitters involved in the fast inhibitory junction potentials in mouse distal colon. Eur J Pharmacol. 2003;460:183–190. doi: 10.1016/s0014-2999(02)02923-0. [DOI] [PubMed] [Google Scholar]
- Smyth LM, Breen LT, Yamboliev IA, Mutafova-Yambolieva VN. Novel localization of CD38 in perivascular sympathetic nerve terminals. Neuroscience. 2006;139:1467–1477. doi: 10.1016/j.neuroscience.2006.01.043. [DOI] [PubMed] [Google Scholar]
- Smyth LM, Bobalova J, Mendoza MG, Lew C, Mutafova-Yambolieva VN. Release of β-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J Biol Chem. 2004;279:48893–48903. doi: 10.1074/jbc.M407266200. [DOI] [PubMed] [Google Scholar]
- Spencer NJ, Bywater RA, Holman ME, Taylor GS. Inhibitory neurotransmission in the circular muscle layer of mouse colon. J Auton Nerv Syst. 1998;70:10–14. doi: 10.1016/s0165-1838(98)00045-9. [DOI] [PubMed] [Google Scholar]
- Todorov LD, Mihaylova-Todorova S, Westfall TD, Sneddon P, Kennedy C, Bjur RA, Westfall DP. Neuronal release of soluble nucleotidases and their role in neurotransmitter inactivation. Nature. 1997;387:76–79. doi: 10.1038/387076a0. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110:415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Westfall TD, Sarkar S, Ramphir N, Westfall DP, Sneddon P, Kennedy C. Characterization of the ATPase released during sympathetic nerve stimulation of the guinea-pig isolated vas deferens. Br J Pharmacol. 2000;129:1684–1688. doi: 10.1038/sj.bjp.0703271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H, Braun N, Kegel B, Heine P. New insights into molecular structure and function of ectonucleotidases in the nervous system. Neurochem Int. 1998;32:421–425. doi: 10.1016/s0197-0186(97)00126-5. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]