

Abstract
Introduction
Estrogen receptor (ER) signaling and its interaction with epidermal growth factor receptor (EGFR) is a potential therapeutic target in non-small cell lung cancer (NSCLC). To explore cross-communication between ER and EGFR, we have correlated ER pathway gene and protein expression profiles and examined effects of antiestrogens with or without EGFR inhibitors in preclinical models of human NSCLC.
Methods
We evaluated 54 NSCLC cell lines for growth inhibition with EGFR inhibitors, antiestrogen treatment or the combination. Each line was evaluated for baseline ER pathway protein expression. The majority were also evaluated for baseline ER pathway gene expression. Human NSCLC xenografts were evaluated for effects of inhibition of each pathway either individually or in combination.
Results
The specific antiestrogen fulvestrant has modest single agent activity in vitro, but in many lines fulvestrant adds to effects of EGFR inhibitors, including synergy in the EGFR mutant, erlotinib-resistant H1975 line. ERα, ERβ, progesterone receptor (PR)-A, PR-B and aromatase proteins are expressed in all lines to varying degrees, with trends towards lower aromatase in more sensitive cell lines. Sensitivity to fulvestrant correlates with greater baseline ERα gene expression. Tumor stability is achieved in human tumor xenografts with either fulvestrant or EGFR inhibitors, but tumors regress significantly when both pathways are inhibited.
Conclusions
These data provide a rationale for further investigation of the antitumor activity of combined therapy with antiestrogen and anti-EGFR agents in the clinic. Future work should also evaluate dual ER and EGFR inhibition in the setting of secondary resistance to EGFR inhibition.
Keywords: epidermal growth factor receptor, estrogen, estrogen receptor, lung cancer, fulvestrant
Introduction
Lung cancer is the leading cause of cancer death among both men and women in the United States.1 A great deal of work has focused on the role of EGFR in NSCLC.2, 3 Erlotinib is the only FDA-approved EGFR tyrosine kinase inhibitor (TKI), inducing objective tumor responses in approximately 10% of unselected NSCLC patients.4 Patient selection is clearly important, with greatest benefit from erlotinib seen in patients whose tumors harbor EGFR mutations.2, 3, 5 Even in this population, responses are often quite transient and resistance eventually emerges. Although there are several different mechanisms by which EGFR mutant NSCLC eventually become resistant to EGFR TKIs, resistance in approximately half of the cases is caused by a secondary mutation in exon 20 at position 790 (T790M).6
The ER pathway is among the most well studied pathways in breast cancer,7 but its role in NSCLC has only been explored more recently as lung cancer rates among women have increased in recent decades.8 New data from randomized, prospective clinical trials show a significant impact of hormone replacement therapy (HRT) in lung cancer. In the Women’s Health Initiative, over 16,000 post-menopausal women with an intact uterus and no breast cancer history were randomly allocated to supplemental estrogen and progesterone or no HRT. After 5.6 years of study and 2.4 years of follow-up, the hazard ratio (HR) for lung cancer incidence in the HRT group was 1.28 (p = 0.12), with HR for death in NSCLC patients and death specifically from NSCLC of 1.61 (p = 0.02) and 1.87 (p = 0.004) respectively.9 Further, a prospective cohort study confirmed an increased, dose-dependent lung cancer risk among women who received HRT.10
Antiestrogens demonstrate impressive efficacy in breast cancer when tumors express estrogen receptors, with relatively modest toxicity.11 Accumulating evidence now supports a potential stimulatory role for ER pathway in NSCLC pathogenesis and for ER inhibition in NSCLC suppression.12–17 Significant ER expression is seen in lung cancer tissue and cell lines, particularly adenocarcinoma.14, 18 Estrogens stimulate proliferation in vitro and progression in vivo in lung cancer models.19 Data from mouse models shows that inhibition of this pathway can reduce the development of lung cancer.20 In a retrospective analysis of a European database, a reduced risk of lung cancer death was seen in women with breast cancer if they received antiestrogen therapy, tamoxifen in most cases.21 Molecular details of the interaction between ER and EGFR are emerging, and ER appears to be an important locus for signal convergence.22–24 Clinical evaluation to date includes a phase I study showing safety and potential anti-tumor activity of gefitinib and fulvestrant in postmenopausal women,25 and data demonstrating safety of erlotinib combined with fulvestrant.26–28
Materials and Methods
Cell lines, cell cultures and reagents
Erlotinib and fulvestrant were studied in 54 NSCLC cell lines in vitro (A427, A549, Calu-1, Calu-3, Calu-6, H23, H226, H358, H441, H460, H520, H522, H596, H661, H810, H838, H1155, H1299, H1385, H1435, H1437, H1563, H1568, H1581, H1623, H1651, H1666, H1703, H1734, H1755, H1793, H1836, H1838, H1869, H1944, H1975, H2023, H2030, H2073, H2106, H2110, H2122, H2126, H2135, H2172, H2228, H2286, H2291, H2342, H2405, H2444, HCC827, SHP-77, SK-LU-1). The breast cancer line MCF-7 was evaluated in additional analyses. All lines were obtained from ATCC and identity was confirmed by genomic DNA, with comparison to the ATCC database, within 3 months of the described experiments.
A-549 was cultured in HAM’s F12 (ATCC) supplemented with 5% heat-inactivated fetal bovine serum (FBS), 1% penicillin G-streptomycin-fungizone solution (PSF, Irvine Scientific, Santa Ana, CA), 5 mg/ml insulin (Sigma, Saint Louis, MO) and 1 mg/ml hydrocortisone (Sigma, Saint Louis, MO). Calu-3 and SK-LU-1 were grown in EMEM (ATCC) 10% FBS/PSF. H1155, H1435, H1581, H2286 and H2405 were grown in ACL-4 10% FBS/PSF. H1836, H2342, and H810 were grown in HITES 10% FBS/PSF. Remaining cell lines were cultured in RPMI 1640 (Cellgro, Manassas, VA) 10% FBS/PSF and 2 mmol/L glutamine (Invitrogen, Carlsbad, CA). Each cell line was also grown in a second proliferation experiment using phenol red-free media with dextran-coated charcoal-stripped (DCC) FBS (same concentration as above). F-12K and EMEM were replaced by RPMI for these experiments and estrogen was not added to HITES, but the type of media was otherwise unchanged.
HCC827 harbors an EGFR mutation in exon 19, and H1975 harbors both an L858R mutation and a T790M mutation in EGFR. All other cell lines evaluated harbored wildtype EGFR genes. KRAS was mutant in the following lines: A427, A549, Calu-1, Calu-6, H23, H358, H441, H460, H1155, H1385, H1944, H2030, H2122, H2291, H2444, SHP77 and SK-LU-1 (sequenome data was unavailable for H1836). ATCC classifies the following cells as adenocarcinoma: Calu-3, H23, H522, H838, H1437, H1563, H1568, H1651, H1734, H1755, H1793, H1838, H1944, H1975, H2030, H2122, H2228, H2291, H2342, H2405, SK-LU-1; bronchoalveolar: H358, H441, H1666; large cell: H460, H661, H810, H1155, H1299, H1581; adenosquamous: H596, H1703; squamous: H226 and H520. Calu-6 is classified as anaplastic, SHP-77 is classified as neuroendocrine and the other cell lines do not have a specific histology allocated.
Western blots
Cells growing in log-phase were washed in ice-cold PBS and lysed at 4°C in lysis buffer. Insoluble material was cleared by centrifugation at 10,000g for 10 min. Protein was quantitated using BCA (Pierce Biochemicals, Rockford, IL, USA), resolved by SDS-PAGE, and transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). Expression was detected by ERα (62A3) Ab: #2512 (Cell Signaling, Danvers, MA), ERβ Ab: #5513 (Cell Signaling), PR–A and PR–B (6A1) Ab: #3172 (Cell Signaling), Aromatase Ab: Abcam #ab18995 (Abcam, Cambridge, MA), and α-Tubulin antibody #2144 (Cell Signaling).
Microarray analysis of cell lines
Agilent microarray analysis was performed to assess baseline gene expression as previously described.29 Baseline microarrays were performed on Agilent Human 1A V2 chips with individual cell lines (labeled with cyanine-5) characterized by comparison to a NSCLC cell line mixed reference pool29 (labeled with cyanine-3) on a single slide. Slides were read using an Agilent Scanner, and gene expression values were calculated using Agilent Feature Extraction software version 7.5. Extracted data was imported into Rosetta Resolver 5.1 to create expression profiles. Heat maps evaluated cell lines across probes corresponding to genes for ERα, ERβ, PR, and aromatase.
Proliferation assays
For experiments looking at erlotinib and fulvestrant individually, cells were seeded in duplicate at 5,000–10,000 cells per well. The day after plating, drug was added with a starting concentration of 10 µM with 2-fold dilutions over 12 concentrations. Cells were harvested by trypsinization and counted immediately using a Coulter Z2 particle counter (Beckman Coulter Inc.) Cells were counted on the day drug was added and five days later with percent growth inhibition defined as 100 × (1 – [generations in treated wells/generations in untreated controls]). IC50 was calculated using a four parameter logistic model. For synergy assays, the process was the same with both drugs added at a 1:1 ratio at a starting concentration of 10 µM with 2-fold dilutions over 6 concentrations. Combination index was calculated based on method C as described elsewhere (Calcusyn, Biosoft).30 For surveying the entire panel with both drugs, in duplicate experiments, fulvestrant was added with a starting concentration of 10 µM with 2-fold dilutions over 6 concentrations in the presence of erlotinib 1 µM.
Apoptosis analysis
Effects of erlotinib, fulvestrant and the combination on apoptosis were assessed in duplicate experiments using Nim-4', 6-diamidino-2-phenylindole (DAPI) staining. Cells were plated evenly in control and experimental wells and allowed to grow to log-phase, then treated with 1 μM erlotinib, 200 nM fulvestrant or the combination for 72 hours. Cells were washed with PBS, and trypsin was applied, and then centrifuged at 3,000 rpm for 5 minutes. Supernatant was aspirated and cells were resuspended in 100 μL of Nim-DAPI (NPE Systems) and gently vortexed. Cells were analyzed with UV using a Cell Lab Quanta SC flow cytometer (Beckman-Coulter).
In vivo analysis of EGFR TKIs plus fulvestrant
A549 cells (5×107 cells/mouse) were injected subcutaneously (SQ) in ovariectomized nude mice with estradiol-17β supplements in a biodegradable binder (1.7 mg/pellet; Innovative Research) as before.31 When tumors reached 50–75 mm3, mice were randomized to treatment groups of 6–8 mice and administered vehicle control, gefitinib (160 mg/kg daily), fulvestrant 5 mg SQ weekly or a combination of gefitinib and fulvestrant at the doses above for 21 days. The experiment was continued to day 28. An independent experiment employed an identical design, but erlotinib (25 mg/kg by oral gavage daily for 21 days), replaced gefitinib, and the experiment was continued until tumors achieved a limiting volume of 500 mm3 as before.32 Data are presented as mean ± SEM for tumor volumes measured in mm3. Tumor volumes of mice in the single treatment and combination treatment arms were compared to controls and to each other. Data were analyzed using ANOVA and student’s t-tests.
Results
Sensitivity to erlotinib and fulvestrant differ among human NSCLC cell lines
Significant differences were found in the anti-proliferative effects of erlotinib across the cell lines in our panel, with IC50 < 1 µM in 6 lines, including the extremely sensitive EGFR mutant line HCC827, a line with a known deletion in exon 19 of the EGFR gene (Figure 1A). Seven lines had IC50 < 5 µM for fulvestrant in standard media (Figure 1B), and 8 had IC50 < 5µM in phenol red-free media with DCC-FBS with a trend toward greater sensitivity across the cell panel in this estrogen-depleted media (Figure 1C). The breast cancer cell line MCF-7 was used as a positive control, and in phenol red-free media with DCC-FBS, the IC50 was below the lowest dose (325 nM) tested (data not shown). To assess combined effects of fulvestrant with erlotinib, sensitivity to fulvestrant in the setting of 1 µM erlotinib was evaluated across the cell line panel. Assessing the panel at 2.5 µM of fulvestrant plus 1 µM erlotinib demonstrated several lines in which fulvestrant added significantly to the inhibition seen with erlotinib alone. This was shown particularly in lines with greater sensitivity to fulvestrant as a single agent. (Figure 1D). The changes with lower doses of fulvestrant showed similar results with slightly less inhibition (Supplemental Figure)
Figure 1. In vitro sensitivity to erlotinib, fulvestrant and the combination of both agents.

54 NSCLC cell lines with IC50 for (A) erlotinib and (B) fulvestrant using ATCC-recommended media are presented in µM. (C) IC50 values for fulvestrant in lines grown in phenol red-free media with DCC-FBS are shown. (D) In phenol red-free media with DCC-FBS, sensitivity to combined erlotinib 1 µM and fulvestrant 2.5 µM is shown, with fulvestrant (white bars), erlotinib (gray bars) and the combination (black bars). Lines are arranged in descending order of the difference in IC50 between combination treatment and erlotinib, with boxes around cell lines with IC50 for fulvestrant < 5 µM. Error bars indicate standard error based on duplicate experiments.
Western blot and microarray evaluation of NSCLC cell lines identify potential biomarkers for efficacy of ER inhibition in NSCLC
To assess baseline levels of relevant pathway genes and proteins, the cell line panel was evaluated by microarray analysis using a hormonal gene set and Western blots for ERα, ERβ, aromatase, PR–A and PR–B as compared to a tubulin control. No clear association was seen between sensitivity to fulvestrant and protein levels of ERα, ERβ, PR–A or PR–B; however, there was a trend toward lower aromatase levels in cell lines with greater sensitivity to fulvestrant (Figure 2A, B). Baseline gene expression data was available for 44 of the 54 lines. Microarray analysis demonstrated low variability across the panel in comparison to a similar analysis in our group’s breast cancer panel.33 There was a greater sensitivity in cell lines with higher ESR1 gene expression, the gene for ERα. One group of cell lines, including 20 of the 44 lines for which we had baseline expression data, were defined by elevated ESR1 expression. This group included all 4 of the lines with IC50 < 5 µM (p < 0.022) using standard media and 4 of 5 lines with IC50 < 5 µM (p < 0.1) in phenol red-free media with DCC-FBS (Figure 2C).
Figure 2. Baseline expression of hormone receptor genes and proteins.
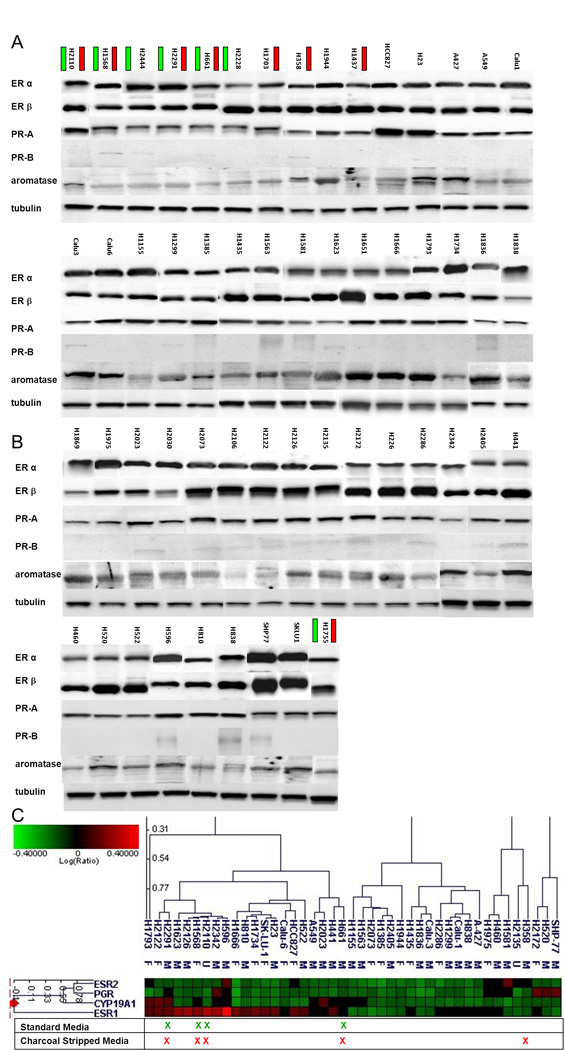
For each cell line, protein levels of ERα, ERβ, PR–A, PR–B, aromatase and tubulin were evaluated by established Western Blot methods (A and B). Green bars by the cell line name indicate an IC50 < 5 µM in the ATCC recommended media, while red bars by the cell line name indicate an IC50 < 5 µM in phenol red-free media with DCC-FBS. In a subset of evaluated cell lines, baseline gene expression data was available, and expression of a hormonal gene set is shown with an X marking lines with an IC50 < 5 µM (C).
In vivo evaluation of the combination of inhibition of EGFR and ER pathways demonstrates significant and sustained tumor inhibition
Mice harboring tumors from human NSCLC A549 line were randomly assigned to vehicle control, gefitinib 160 mg/kg daily; fulvestrant 5 mg SQ weekly; or gefitinib plus fulvestrant for a total of 21 days. Gefitinib alone delayed tumor growth, while fulvestrant alone led to tumor stabilization. Treatment with both gefitinib and fulvestrant elicited significantly reduced tumor volume as compared to all other treatments (p<0.001), with no visible tumors after several weeks of therapy (Figure 3A). When a similar study was conducted with erlotinib as the EGFR TKI as previously reported,31 tumor volumes of mice in the combination arm (fulvestrant and erlotinib) were significantly different from all other treatments (p<0.001). We have now followed animals treated on the combination arm for 60 days, and tumor recurrence was not seen over that time (Figure 3B).
Figure 3. In vivo evaluation of gefitinib, fulvestrant and the combination of both drugs in human A-549 xenografts in nude mice.
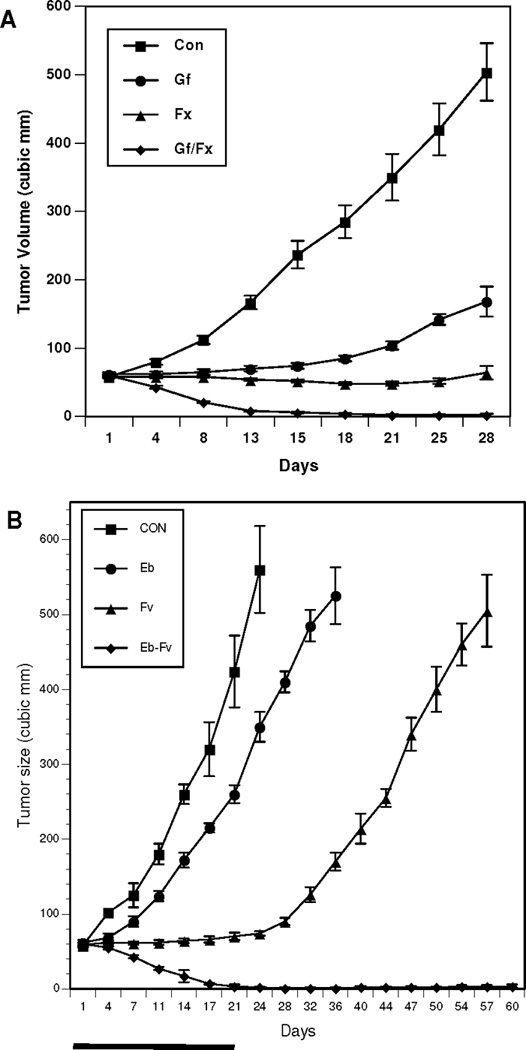
(A) Ovariectomized nude mice with estradiol supplements and A-549 tumors received 21 days of vehicle control (squares); gefitinib (circles) 160 mg daily; fulvestrant (triangles) 5 mg SQ weekly; or the combination of gefitinib and fulvestrant (diamonds). (B) The same model was assessed with erlotinib (circles) as the EGFR TKI. Mice were treated with assigned therapy for 21 days, and then followed for 60 days to assess outcomes.
Combination of erlotinib and fulvestrant is more effective than either agent alone in a model harboring a secondary EGFR mutation
To assess whether antiestrogen therapy can overcome secondary EGFR TKI resistance, dose response curves for the combination of erlotinib and fulvestrant were generated for the H1975 line. This line is resistant to EGFR TKIs and harbors two mutations in the EGFR gene, the sensitizing L858R mutation and the T790M mutation which is associated with EGFR TKIs resistance. In this model, the combination of erlotinib and fulvestrant demonstrated synergy (Figure 4A). When evaluating apoptosis in the H1975 cell line, low doses of fulvestrant (200 nM) led to increased apoptosis at 72 hours compared to control. A slight increase in apoptosis at 72 hours was seen in response to erlotinib 1 µM, but the combination of erlotinib and fulvestrant led to a large increase in apoptosis (Figure 4B).
Figure 4. In vitro effects of erlotinib, fulvestrant or the combination of both agents on EGFR-TKI resistant, EGFR-mutant lung cancer cell proliferation and viability.


Growth curves of the H1975 line after 6 days of treatment with erlotinib (diamonds), fulvestrant (squares), or the combination of the two drugs (triangles) are shown at the concentrations listed. Combination index (CI) for the combination at constant ratio is shown (A). Apoptosis at 72 hours (B) was assessed in H1975 cells with no therapy (white), erlotinib 1 µM (stripes), fulvestrant 200 nM (black) or the combination (checked). Error bars indicate standard error based on duplicate experiments.
Discussion
The role of ER signaling in NSCLC is an emerging area of interest based on laboratory discoveries and clinical data. We demonstrate that components of the hormone receptor signaling pathway (including ERα, ERβ, PR–A, PR–B and aromatase) are present ubiquitously, but to varying degrees, throughout a large panel of human NSCLC cell lines. To our knowledge, this is the most comprehensive evaluation to date of human NSCLC cell lines for gene and protein expression in this signaling pathway.
Fulvestrant was selected for use in these studies because it is generally regarded as a pure antiestrogen that selectively inhibits estrogen receptor signaling in target cells.34, 35 Further, fulvestrant was shown to exhibit significant antitumor activity in NSCLC cells in vitro and in vivo, and the drug showed evidence of significant interactions with EGFR TKIs.14, 31 In the present studies, single agent antitumor activity with fulvestrant in vitro was modest, but was observed more frequently in NSCLC lines with increased ESR1 gene expression. The results achieved statistical significance for cells maintained in standard media and a trend in phenol red-free media with DCC-FBS. We note that the higher frequency of drug inhibitory activity was not directly related to the level of ESR1 RNA, but instead correlated with the “ESR1 high” group. This finding is considered hypothesis–generating. This data contrasts with some, but not all, data suggesting that ERβ is more relevant in NSCLC, but the relative contribution of different components of this pathway remains an area of active investigation.16 Patients with higher levels of estrogenic stimulus would be a group for whom there is a great clinical need, as a review of survival by estrogen levels among several Southwest Oncology Group trials showed lower survival in women with higher estrogen levels.36 Similarly, Olivo-Marsten et al report a significant association of increased serum estrogen levels with poorer survival among male and female lung cancer patients.37 In addition, our findings show a trend toward increased fulvestrant sensitivity in lines with lower levels of aromatase protein as reported by others,16 although this trend is difficult to quantitate and cannot be considered definitive data on its own. Estrogen levels in lines with lower aromatase levels should be lower, thus allowing fulvestrant to compete with less endogenous estrogens for ER binding.
For proliferation assays, two sets of experiments were conducted. Cells were grown in either ATCC-recommended media or in phenol red-free media with DCC-FBS with a trend toward greater sensitivity in the latter analysis. As a potential screening tool, we also evaluated the entire panel for sensitivity to fulvestrant in the presence of 1 µM of erlotinib. Although dose-response curves were generated across the panel, it was more representative to graph percent inhibition at a single point across the cell line panel. The combination data correlated well with sensitivity to erlotinib, but in cell lines in which the dual therapy significantly surpassed the response to erlotinib alone, the sensitivity to fulvestrant was generally high, as was the level of ESR1 gene expression. Of note, there are some lines for which the dual treatment appeared to have antagonistic effects. There is some clinical precedence for such an effect, in which the addition of an EGFR TKI elicits worse outcomes.38 Further characterization of individual patients that may potentially experience harm will be important as clinical evaluation of this combination treatment regimen proceeds. It is possible that, in some subsets of NSCLC, erlotinib is not an appropriate partner for antiestrogen therapy. However, as reported previously in breast cancer, the synergy seen in some lung cancer cell lines with dual antiestrogen and EGFR/HER kinase inhibitors could well be attributable to biologic interactions between estrogen and EGFR signaling pathways.35, 39 Indeed, significant molecular interactions of estrogen and EGFR signaling pathways in non-small cell lung cancer cells have now been reported in several independent investigations.14, 16, 21, 23, 24
In an attempt to correlate sensitivity with protein levels of ER pathway genes, we evaluated the entire panel by Western Blot. For each protein, we selected a single antibody. The antibodies used have not been clinically validated, and in the context of the ongoing clinical trial, we will use clinically validated antibodies. We also evaluated most of the cell lines (all of the lines for which we have baseline gene expression data) for ER pathway genes, using the Agilent platform which compares a sample to a reference, namely pooled NSCLC cell lines. By definition, there will be cell lines with lower and higher expression than the pooled average, but the magnitude of differences was narrow when compared to a similar breast cancer panel in our laboratory. The evaluation of cell lines against a control of pooled cell lines could be another factor responsible for differences between our observations and those of others with respect to the relative importance of ERα and ERβ. Our analysis doesn’t assess differences between normal tissues and cancerous tissue, but instead assesses differences among different cancer models. This approach is designed to identify subgroups within a given histology in which disproportionate benefit would be expected.40, 41
In patients managed with EGFR inhibitors, drug resistance eventually occurs. Of patients with EGFR mutations (e.g. exon 19 deletions or the L858R mutation in exon 21), significant initial response to erlotinib is observed, but about half develop T790M mutations at the time of tumor progression. The H1975 cell line was chosen for further investigation based on molecular characteristics consistent with this clinical scenario. In the H1975 cell line, which has a T790M mutation, dual treatment with erlotinib and fulvestrant in vitro leads to significantly greater inhibition and induction of apoptosis than either agent given alone. The mechanism of this increased sensitivity to EGFR inhibition after the addition of an antiestrogen is not clear. Recent work by Xu et al demonstrates that estrogen can increase phoshpo-EGFR, and that this effect can be inhibited by gefitinb or fulvestrant, but it is inhibited to a greater degree when both agents are used.42 We are planning further experiments to elucidate potential molecular mechanisms responsible for this effect. Such work will include evaluation of antiestrogens in the setting of cell lines that are initially sensitive to EGFR TKIs, but develop acquired resistance after exposure to erlotinib, rather than the de novo resistant H1975 cell lines.
In a NSCLC cell line for which neither erlotinib nor fulvestrant had appreciable single agent in vitro activity in these experiments, namely A549 cells, the combination of EGFR inhibition and ER inhibition led to significant tumor resolution in an in vivo setting. These results are striking and were not predicted by in vitro drug sensitivity or baseline expression levels of ER pathway protein or gene expression. The selection of the A549 cell line for in vivo work was made on the basis of data showing expression and activity of estrogen receptors among initially surveyed NSCLC cell lines.15 It is notable that H23 lung tumor cells also demonstrate significant additive antitumor effects in vivo when fulvestrant is combined with a different EGFR kinase inhibitor, 43 despite an antagonistic effect in vitro The fact that synergy between antiestrogens and EGFR kinase inhibitors is significant in vivo but less significant in lung tumor cells used during in vitro assays of synergy poses interesting questions to be addressed more fully in future work. For example, in this study we explored alternative in vitro assay conditions (e.g. with or without steroid-depleted culture medium) to find if the in vitro assay may be less sensitive than the in vivo system due to competition for fulvestrant binding to tumor cell ER. As shown in the results, this may have been a small factor influencing antiestrogen activity. Another factor may be that the tumor microenvironment and neighboring cells (absent in the in vitro assay) may play a role in modulating either estrogen or EGFR signaling. In studies on the action of antiestrogens in preventing lung carcingenesis in vivo, Stabile et al. similarly report that the tumor niche may have an important effect on the in vivo activity of antiestrogens.17 Finally, some actions of estrogens in lung in vivo may be due to interaction with subpopulations of tumor progenitor cells or to stimulation of tumor-associated angiogenesis,44 actions that may be better assessed using in vivo models of tumor progression. One or more of these factors may explain differences observed between in vitro and in vivo assays of estrogen and EGFR signaling.
In addition to independent evidence on significant interactions of estrogen and EGFR signaling in NSCLC20, 45 results described in this manuscript helped to develop and launch an ongoing, randomized phase II clinical trial of dual erlotinib and fulvestrant therapy as compared to erlotinib alone in previously-treated NSCLC patients. All patients participating in the clinical trial were required to provide a tissue sample, and we plan to analyze these specimens for EGFR mutation status and expression of ER and aromatase, with a focus on identification of biomarkers that may identify an optimal patient population for further development of antiestrogen therapy in NSCLC.
Supplementary Material
Supplemental Figure: In vitro sensitivity to fulvestrant in the setting of fixed dose erlotinib. In phenol red-free media with DCC-FBS, sensitivity to combined erlotinib 1 µM and fulvestrant 1.25 µM (A) and combined erlotinib 1 µM and fulvestrant 0.625 µM (B) are shown, with fulvestrant (white bars), erlotinib (gray bars) and the combination (black bars). Error bars indicate standard error based on duplicate experiments.
Acknowledgements
Financial Support: 1K23CA149079 (Garon), Wolfen Family Clinical/Translational Lung Cancer Research Program, One Ball Matt Memorial Golf Tournament, SPORE CDA FDP-NIH CA090388 (Dubinett), National Lung Cancer Partnership, UCLA Lung Cancer SPORE Program, Stiles Program in Oncology, CDMRP DOD Lung Cancer Research Program, NIH CTSI UL1TR000124
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: No conflicts of interest related to this work
References
- 1.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 7.Pietras RJ, Marquez-Garban DC. Membrane-associated estrogen receptor signaling pathways in human cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:4672–4676. doi: 10.1158/1078-0432.CCR-07-1373. [DOI] [PubMed] [Google Scholar]
- 8.Donington JS, Colson YL. Sex and gender differences in non-small cell lung cancer. Seminars in thoracic and cardiovascular surgery. 2011;23:137–145. doi: 10.1053/j.semtcvs.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Chlebowski RT, Schwartz AG, Wakelee H, et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women's Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet. 2009;374:1243–1251. doi: 10.1016/S0140-6736(09)61526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slatore CG, Chien JW, Au DH, et al. Lung cancer and hormone replacement therapy: association in the vitamins and lifestyle study. J Clin Oncol. 2010;28:1540–1546. doi: 10.1200/JCO.2009.25.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burstein HJ, Prestrud AA, Seidenfeld J, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol. 2010;28:3784–3796. doi: 10.1200/JCO.2009.26.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stabile LP, Siegfried JM. Estrogen receptor pathways in lung cancer. Curr Oncol Rep. 2004;6:259–267. doi: 10.1007/s11912-004-0033-2. [DOI] [PubMed] [Google Scholar]
- 13.Patel JD, Bach PB, Kris MG. Lung cancer in US women: a contemporary epidemic. JAMA. 2004;291:1763–1768. doi: 10.1001/jama.291.14.1763. [DOI] [PubMed] [Google Scholar]
- 14.Stabile LP, Davis AL, Gubish CT, et al. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer research. 2002;62:2141–2150. [PubMed] [Google Scholar]
- 15.Weinberg OK, Marquez-Garban DC, Fishbein MC, et al. Aromatase inhibitors in human lung cancer therapy. Cancer research. 2005;65:11287–11291. doi: 10.1158/0008-5472.CAN-05-2737. [DOI] [PubMed] [Google Scholar]
- 16.Miki Y, Abe K, Suzuki S, et al. Suppression of estrogen actions in human lung cancer. Molecular and cellular endocrinology. 2011;340:168–174. doi: 10.1016/j.mce.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Stabile LP, Rothstein ME, Cunningham DE, et al. Prevention of tobacco carcinogen-induced lung cancer in female mice using antiestrogens. Carcinogenesis. 2012 doi: 10.1093/carcin/bgs260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niikawa H, Suzuki T, Miki Y, et al. Intratumoral estrogens and estrogen receptors in human non-small cell lung carcinoma. Clin Cancer Res. 2008;14:4417–4426. doi: 10.1158/1078-0432.CCR-07-1950. [DOI] [PubMed] [Google Scholar]
- 19.Hershberger PA, Stabile LP, Kanterewicz B, et al. Estrogen receptor beta (ERbeta) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells. J Steroid Biochem Mol Biol. 2009;116:102–109. doi: 10.1016/j.jsbmb.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siegfried JM, Gubish CT, Rothstein ME, et al. Combining the multitargeted tyrosine kinase inhibitor vandetanib with the antiestrogen fulvestrant enhances its antitumor effect in non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:485–495. doi: 10.1097/JTO.0b013e31824177ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouchardy C, Benhamou S, Schaffar R, et al. Lung cancer mortality risk among breast cancer patients treated with anti-estrogens. Cancer. 2011;117:1288–1295. doi: 10.1002/cncr.25638. [DOI] [PubMed] [Google Scholar]
- 22.Pietras RJ, Marquez DC, Chen HW, et al. Estrogen and growth factor receptor interactions in human breast and non-small cell lung cancer cells. Steroids. 2005;70:372–381. doi: 10.1016/j.steroids.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Stabile LP, Lyker JS, Gubish CT, et al. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer research. 2005;65:1459–1470. doi: 10.1158/0008-5472.CAN-04-1872. [DOI] [PubMed] [Google Scholar]
- 24.Marquez DC, Lee J, Lin T, et al. Epidermal growth factor receptor and tyrosine phosphorylation of estrogen receptor. Endocrine. 2001;16:73–81. doi: 10.1385/ENDO:16:2:073. [DOI] [PubMed] [Google Scholar]
- 25.Traynor A, Schiller J, Stabile L, Kolesar J, Belani C, Hoang T, Dubey S, Eickhoff J, Marcotte S, Siegfried J. Combination therapy with gefitinib and fulvestrant (G/F) for women with non-small cell lung cancer (NSCLC) ASCO. 2005;23:676S. Available at Macintosh HD:Users:Diani:Documents:Grants:Grants. [Google Scholar]
- 26.Garon EBSS, Kabbinavar F, Reckamp KL, Marquez-Garban DC, Stabile LP, Goodglick L, Dubinett SM, Siegfried JM, Pietras RJ. Interim safety analysis of a phase II study of erlotinib (E) alone or combined with fulvestrant (F) in previously treated patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2008;26:2008. (May 20 suppl; abstr 19091). [Google Scholar]
- 27.Garon EBDS, Hosmer W, Reckamp KL, Kabbinavar FF, Goodglick L, Marquez-Garban DC, Stabile LP, Siegfried J, Pietras RJ. Randomized phase II study of erlotinib (E) alone or combined with fulvestrant (F) in previously treated patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2010;28 15s:suppl;abstr TPS295. [Google Scholar]
- 28.Garon EBDS, Kabbinavar FF, Reckamp KL, Marquez-Garban DC, Goodglick L, Sharma S, Stabile LP, Siegfried J, Pietras RJ. Randomized, multicenter phase II study of erlotinib (E) or E plus fulvestrant (F) in previously treated advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011 29:suppl; abstr TPS216. [Google Scholar]
- 29.Garon EB, Finn RS, Hosmer W, et al. Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY- 142886) in human breast cancer and non-small cell lung cancer cell lines. Mol Cancer Ther. 2010;9:1985–1994. doi: 10.1158/1535-7163.MCT-10-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 31.Marquez-Garban DC, Chen HW, Fishbein MC, et al. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72:135–143. doi: 10.1016/j.steroids.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Williams JI, Pietras RJ. Squalamine and cisplatin block angiogenesis and growth of human ovarian cancer cells with or without HER-2 gene overexpression. Oncogene. 2002;21:2805–2814. doi: 10.1038/sj.onc.1205410. [DOI] [PubMed] [Google Scholar]
- 33.Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris C, Wakeling A. Fulvestrant ('Faslodex')--a new treatment option for patients progressing on prior endocrine therapy. Endocrine-related cancer. 2002;9:267–276. doi: 10.1677/erc.0.0090267. [DOI] [PubMed] [Google Scholar]
- 35.Hutcheson IR, Knowlden JM, Madden TA, et al. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast cancer research and treatment. 2003;81:81–93. doi: 10.1023/A:1025484908380. [DOI] [PubMed] [Google Scholar]
- 36.Albain KSUJ, Gotay CC, Davies AM, Edelman M, Herbst RS, Kelly K, Williamson S, Wozniak AJ, Gandara DR. Toxicity and survival by sex in patients with advanced non-small cell lung carcinoma (NSCLC) on modern Southwest Oncology Group (SWOG) trials. Journal of Clinical Oncology; ASCO Annual Meeting Proceedings Part I; 2007. p. 7549. (June 20 Supplement) [Google Scholar]
- 37.Olivo-Marston SE, Mechanic LE, Mollerup S, et al. Serum estrogen and tumor-positive estrogen receptor-alpha are strong prognostic classifiers of non-small-cell lung cancer survival in both men and women. Carcinogenesis. 2010;31:1778–1786. doi: 10.1093/carcin/bgq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly K, Chansky K, Gaspar LE, et al. Phase III trial of maintenance gefitinib or placebo after concurrent chemoradiotherapy and docetaxel consolidation in inoperable stage III non-small-cell lung cancer: SWOG S0023. J Clin Oncol. 2008;26:2450–2456. doi: 10.1200/JCO.2007.14.4824. [DOI] [PubMed] [Google Scholar]
- 39.Pietras RJ, Arboleda J, Reese DM, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- 40.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 41.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 42.Xu R, Shen H, Guo R, et al. Combine therapy of gefitinib and fulvestrant enhances antitumor effects on NSCLC cell lines with acquired resistance to gefitinib. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2012;66:384–389. doi: 10.1016/j.biopha.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Marquez-Garban DC, Chen HW, Goodglick L, et al. Targeting aromatase and estrogen signaling in human non-small cell lung cancer. Annals of the New York Academy of Sciences. 2009;1155:194–205. doi: 10.1111/j.1749-6632.2009.04116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marquez-Garban DC, Mah V, Alavi M, et al. Progesterone and estrogen receptor expression and activity in human non-small cell lung cancer. Steroids. 2011;76:910–920. doi: 10.1016/j.steroids.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raso MG, Behrens C, Herynk MH, et al. Immunohistochemical expression of estrogen and progesterone receptors identifies a subset of NSCLCs and correlates with EGFR mutation. Clin Cancer Res. 2009;15:5359–5368. doi: 10.1158/1078-0432.CCR-09-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure: In vitro sensitivity to fulvestrant in the setting of fixed dose erlotinib. In phenol red-free media with DCC-FBS, sensitivity to combined erlotinib 1 µM and fulvestrant 1.25 µM (A) and combined erlotinib 1 µM and fulvestrant 0.625 µM (B) are shown, with fulvestrant (white bars), erlotinib (gray bars) and the combination (black bars). Error bars indicate standard error based on duplicate experiments.