

Abstract
Flurbiprofen (FP) is a phenyl alkanoic acid derivative and a family of non–steroidal anti-inflammatory drug used in the treatment of arthritis. The aim of this study was to prepare a new parenteral formulation for FP that can prolong the biologic half-life of the drug, improve its therapeutic efficacy, and reduce its associated side effects targeting the inflammation due to arthritis. PEG-anchored (stealth) and non–PEG-anchored liposomes were prepared by thin film hydration technique followed by extrusion cycle and characterized for in vitro and in vivo. Stealth liposomes (SLs) exhibited increase in percent encapsulation efficiency (68%) and percent drug retention during release studies in 24 h (71%) with good stability for a period of 1 month at –20°C and 4°C (refrigerated temperature) compared with other liposomes. The maximum percent edema inhibition (58%) and significant analgesic effect of 13 s were determined for SLs. The pharmacokinetic parameters after i.v. administration to arthritis induced rats were determined and compared with non-SLs. The marked differences produced for SLs over those of non-SL (conventional) formulations with an increase in area under plasma concentration time curve, t1/2, mean residence time, and reduced clearance. The drug localization in liver, spleen, and kidney were significantly higher for non-PEGylated liposomes than the SLs. Nearly 3-fold increase in drug concentration was measured in arthritic paw when compared with the other liposome formulations. Thus SLs may help to increase the therapeutic efficacy of FP by increasing the targeting potential at the site of action.
Keywords: DSPC, flurbiprofen, in vitro, in vivo, PE 18:0/18:0-PEG 2000, stability
INTRODUCTION
Liposomes are spherical vesicles of different sizes consisting of a lipid bilayer and aqueous center compartment that are generated in vitro.[1] These are popular in terms of biocompatibility, biodegradability, low toxicity, and can control biodistribution of drug by altering the size, composition of lipids, and hence the characteristics.[2] These are the carriers that are suitable for encapsulation of drugs with different lipophilicities, such as strongly lipophilic drugs, strongly hydrophilic drugs, and drugs with intermediate log P.[3] Liposomes can protect the encapsulated drug or drugs and can target the organ or tissue passively.[3] But it was found that conventional liposomes suffer with 2 major drawbacks as sustained as well as targeted release system for drugs in vivo. First one is its attraction toward reticuloendothelial system (RES), which will cause the removal of drug from blood stream as well as will result in adverse effects on the host defense system[4] and will decrease the availability of entrapped drug to the other tissues. The next is recognition of conventional liposomes by RES leads to nonlinear pharmacokinetics for the carrier, which makes calculating the amount of entrapped drug required to attain therapeutic drug dose difficult.[5–8] In addition, conventional liposome formulations containing saturated phospholipids and cholesterol are more prone to the influence of plasma proteins and other biologic fluids in vivo, which leads to rapid removal of drug contents.[9–11] To avoid the above-mentioned difficulties, especially to avoid the RES uptake of the vesicles it is necessary to have previous administration of empty liposomes. Moreover, small unilamellar vesicles have the drawback of low aqueous entrapment volume; the use of charged liposomes could be toxic. Thus, mechanical or electrostatic stabilization cannot improve the long circulation of liposomes in biologic systems.[12] Further attempts to alter the biodistribution of liposomes resulted in the generation of new liposomal formulations called as stealth liposomes (SLs), which have considerably reduced RES uptake, and remain in circulation for long period of time[13,14] with dose-independent pharmacokinetics[8] and have reduced susceptibility to protein-induced leakage.[15,16]
Flurbiprofen (FP) is a hydrophobic and potent acidic non- steroidal anti-inflammatory drug which widely used for the treatment of rheumatoid arthritis in humans.[17,18] FP is 99% bound to human serum albumin and it may bind to red blood cells at therapeutic concentration.[19] Area under plasma concentration time curve (AUC) of plasma drug concentration Vs time curve increases with increasing dose of drug administration.[20] The most frequently reported side effects upon oral administration are abdominal discomfort and other gastrointestinal side effects. Its high log P value of 4.16 and short elimination half-life of 3.9 h necessitates the drug to be administered frequently. To avoid the possible side effects and by considering the fact that FP is generally used for a prolonged periods to maintain the therapeutic activity, numerous delivery systems have been designed, such as transdermal drug delivery system, microspheres, microsponges, niosomes, and so on. But there is no evidence of liposomes, by which the drug can be targeted directly into arthritic inflammation through the gaps formed between the endothelial cells of vasculature. Hence reduced systemic availability and increased accumulation of drug in inflammatory area is expected to reduce the side effects and increase the therapeutic effect. The authors have already studied and reported the effect of drug–lipid ratio and lipid composition on the development of FP liposomes.[21] The present work was aimed at increasing the circulation time of liposomes by grafting polyethylene glycol (PEG)-2000 to the lipid bilayer surface (SLs) and understanding the pharmacodynamics, pharmacokinetics, and biodistribution profile of the drug.
MATERIALS AND METHODS
Materials
FP was gifted by FDH Ltd, Mumbai. Hydrogenated soy phosphatidyl choline (SPC), distearoyl phosphatidyl choline (DSPC), PE 18:0/18:0-PEG2000 (PE-PEG) were donated by Lipoid, Germany. High purity cholesterol was purchased from Sigma–Aldrich (St. Louis, MO, USA). All the chemicals and solvents used in the present study were of analytic grade and were supplied by Sigma Chemicals Co. (St. Louis, MO, USA), Himedia Laboratories Ltd, Mumbai, India, and S.D. Fine Chemicals Pvt. Ltd. (Mumbai, India). Deionized water was used throughout the experiment.
Preparation of liposomes
Three batches of liposomes were prepared with SPC: CH - 4:1 (FL), DSPC: CH - 4:1 (DSPCL) and DSPC: CH: PE-PEG - 4:1:0.2 (SL) molar compositions by thin film hydration technique,[22,23] using rotary evaporator (HS-3001NS). SPC, cholesterol, and drug were weighed accurately and then dissolved in organic phase, that is, chloroform and methanol (2:1, v/v) in a 250 mL round bottom flask. This was attached to rotary evaporator and organic phase was removed by evaporating at 45°C ± 2°C for FL and 55°C ± 2°C for DSPCL and SL, which led to the formation of the film on the wall of the flask. The other processing conditions, such as rotational speed of evaporating flask (120 rpm), vacuum (250 mmHg) were maintained constant. The round bottom flask containing thin lipid film was left in vacuum desiccator overnight to remove the solvent residuals if any. Then it was hydrated with phosphate-buffered saline (PBS) pH 7.4 using vortex mixture for about 2 min to form conventional liposomes. This liposomal suspension was allowed to stand at room temperature for about 2 h to achieve complete swelling. The resultant suspension was sonicated for 8 min in probe sonicator (ultrasonic 3000) to obtain small and homogenous vesicles and extruded through polycarbonate membrane of 0.2 μm pore size. In the same manner as described above FP SLs were prepared using DSPC, PE 18:0/18:0-PEG 2000, and cholesterol. These formulations were used on the same day for further studies.
Separation of unentrapped drug
Unentrapped free FP was separated from conventional and SLs by centrifugation at 10,000 g at 4°C in refrigerated centrifuge for 2 cycles of 30 min with 10 min interval. The pellet obtained was washed with 10 mL of PBS pH 7.4 2 times and recentrifuged.
Determination of percentage encapsulation efficiency
The percentage encapsulation efficiency (%EE) was determined by centrifuging 10 mL of liposomal suspension at 10,000 g at 4°C twice each for 30 min duration with 10 min gap. Thus the pellet obtained was collected and lysed in absolute alcohol and sonicated for 10 min. The concentration of FP was determined after diluting suitably with alcohol at 247 nm using UV–vis spectrophotometer.[24,25] %EE was calculated using the following formula:
%EE = Amount of drug in pellet /Total drug × 100
This was calculated for 3 formulations of each formulation code and average was tabulated.
Fourier transform infrared study
All the excipients, such as DSPC, cholesterol, PE 18:0/18:0-PEG2000, pure drug FP individually, and physical mixture of drug and excipients, were mixed with infrared (IR) grade KBr in the ratio of 1:100 and corresponding pellets were prepared by applying 15,000 lb of pressure in a hydraulic press. The pellets were scanned in an inert atmosphere over a wave number range of 4000–400 cm–1 in Magna IR 750 series II (Nicolet, USA) Fourier transform infrared (FTIR) instrument.
Determination of vesicle size distribution and zeta potential
Average diameter of the liposome vesicles, size distribution, and zeta potential were determined by Zeta master apparatus (Malvern Instruments, Malvern, UK) at a temperature of 25°C±0.1°C after appropriate dilution with 0.45 μm membrane filtered water to avoid opalescence. Then the selected formulations were studied with the help of scanning electron microscopy (SEM) for their morphology.[26,27]
Zeta potential was determined by taking suitable amount of sample (50–100 mL) and diluting it with 5 mL of 0.45 μm membrane filtered water and injected in the electrophoretic cell of the instrument, where a potential of ±150 mV was set. The zeta-potential value was calculated by the instrument software, using Helmholtz–Smoluchosky equation. All the measurements were taken at 25°C.
Stability studies
The stability of FP-encapsulated SL was evaluated by storing the liposomal suspension at –20°C, 4°C, and 25°C for a month. During the studied period, liposomes were kept in sealed vials of 10 mL capacity. Drug content was determined every 10 days as given in percent encapsulation efficiency determination.[2]
Freeze drying (lyophilization)
For freeze drying, liposomal suspension was prepared with cryoprotectant (lactose; 1:5 lipid–carbohydrate ratio). The freshly prepared liposomal suspension was enriched with lactose solution and quickly frozen with iced acetone, stored at -80°C overnight and lyophilized for 48 h using freeze dryer. Before measurements the lyophilized samples were resuspended in double distilled water. Rehydration process is completed in 5 min by vortexing.
Differential scanning calorimetric analysis
Differential scanning calorimetric (DSC) studies were carried out using differential scanning calorimeter (Model number TA – 60, Shimadzu, Japan) for the samples, such as pure drug FP, DSPC, cholesterol, PE-PEG, and FP loaded and unloaded vesicles. For this purpose 2 mg of samples of each were sealed thermatically in standard aluminum pans. Thermograms were obtained at a scanning rate of 5°C/min. Each sample was scanned between 25°C and 225°C using nitrogen as the purge gas. For calibrating enthalpy, indium was sealed in aluminum pan with sealed empty pan as a reference. DSC studies were carried out for drug loaded and unloaded vesicles after freeze drying using lactose as a cryoprotectant.
In vitro drug release
Modified USP XXI dissolution rate model was used for the determination of drug release from liposomic preparation. This model consists of a beaker (250 mL) and a plastic tube of diameter 17.5 mm opened from both the ends. Sigma membrane (Sigma 12000 MW cutoff) was tied at one end of the tube and the other end left free. This assembly was dipped into the beaker containing 90 mL of dissolution medium. The temperature was maintained at 37°C ± 1°C. Ten milliliters of liposomal suspension was added to the tube and a paddle-type stirrer was placed in the center of the beaker. The speed of the stirrer was maintained at 100 rpm. Dissolution sample of 1 mL was withdrawn periodically every 1 h up to 24 h and analyzed spectrophotometrically at 247 nm.[28] With the help of the standard curve prepared earlier, drug concentration was measured.
Assessment of anti-inflammatory activity
Carrageenan-induced rat paw edema method was used to study the in vivo performance of the prepared liposomes and the study was approved by Animal Ethical Committee of Malla Reddy College of Pharmacy, Hyderabad (CPCSEA/MRCP/1217).
Adult male albino Wistar rats of each weighing 200 ± 5 g were divided into 5 groups with each group comprising 6 animals. All the animals were marked on the right hind paw just behind the tibia–tarsal junction to ensure constant paw volume up to the fixed mark whenever dipped into the plethysmograph. Initial paw volume of the rats was measured by dipping rat paw into the electrolyte column just before carrageenan administration. A 0.1 mL of 1%w/v of carrageenan (in 0.9% normal saline) was injected in the subplantar region of the right hind paw of rats. Group I (ST-saline treated) (control group) animals were injected with 0.9% normal saline. Group II (FS) animals were administered intravenously with flurbiprofen solution (FS) in the dose of 2.5 mg/kg body weight through tail vein. Group III (FL), Group IV (DSPCL), and Group V (SL) were given intravenously FP-loaded conventional liposomes, liposomes made up of long alkyl chain lipid, DSPC, and SLs, respectively, in the dose of 2.5 mg/kg body weight. Paw volumes of rats were measured every 15 min up to 1 h then followed by 2, 3, 4, 5, 6, 7, and 8 h. The edema rate and inhibition rate of each group were calculated as follows:
Edema rate (E%) = ((Vt – V0)/V0) × 100
Inhibition rate (I%) = ((Ec – Et)/Ec) × 100
where,
V0 - paw volume before carrageenan injection (mL)
Vt - paw volume after carrageenan injection (mL)
Ec - edema rate of control group
Et - edema rate of treated group.[29]
Assessment of analgesic activity
Hot plate test
Animals (mice) were divided into 5 groups with each group comprising 5 animals and were intraperitoneally administered with 0.9% saline, FS, conventional liposomes, liposomes made up of long alkyl chain lipid, DSPC, and SLs, respectively. The hot plate test was performed by placing the mice on aluminum hot plate at a temperature of 62°C±0.5°C for a maximum time of 30 s.[30] The reaction time was noted when the rats were licking their fore and hind paws or jumped at 0, 0.25, 0.5, 0.75, 1, 2, 3, 4, 5, 6, 7, 8, and 24 h.
Tail flick test
Tail flick test was performed on mice of 5 groups, each group comprising 5 animals by focusing radiant heat on the dorsal surface of tail. Latency or the time it took for the mice to withdraw tail from a noxious thermal stimulus was measured using a tail flick meter. To minimize tissue damage, a maximum latency of 30 s was imposed. The nichrome wire was about 1/8 below the tail. Each mouse was thus tested at 0, 0.25, 0.5, 0.75, 1, 2, 3, 4, 5, 6, 7, 8, and 24 h after the administration of saline for Group I (ST), FS for Group II (FS), FL, DSPCL, and SL formulations, respectively, for Group III, IV, and V intraperitoneally.
Pharmacokinetics and biodistribution
Male Wistar rats weighing 200 ± 10 g were selected for this study. The subplantar region of right hind paw of rats under study were intradermaly injected with 0.25 mL of Freund's complete adjuvant.[31] The difference in paw volume was measured for assessing arthritis. Difference in thickness of the right and left paw was found to be around 6.83 ± 0.26 mm, after 3 weeks of injection. These rats were used for biodistribution studies.
Arthritic rats were divided into 4 groups. Group I with free FP solution at a dose 2.5 mg/kg and Group II (FL), Group III (DSPCL), and Group IV (SL) were given conventional liposomes (FL), liposome preparation with long alkyl chain synthetic lipid DSPC (DSPCL) and SLs, respectively, at the same dose via tail vein intravenously. Blood samples were collected from retro orbit puncture at specified time intervals. Rats were sacrificed and various tissues, such as liver, spleen, kidney, and paw were removed, dried, weighed, and stored at –20°C until further analysis. Required quantity of methanol was added to each organ, homogenized for few minutes in ice, and centrifuged at 4500 rpm. Supernatant was collected and assayed for drug by high performance liquid chromatography (HPLC; Alliance HPLC system, Waters, USA) as described by Taro et al. in 1989.[32] Blank plasma of 60 μL was spiked with 40 μL of standard solutions of FP in a microcentrifuge and 1 mL of ethanol was added. The tubes were vortexed and then centrifuged at 4000 rpm for 5 min, after centrifugation, an aliquot 20 μL supernatant solution was injected to HPLC. The procedure was repeated to prepare resultant concentrations of 200–1000 ng. Kromasil C18 (250 nm × 4.6 mm i.d. μL) column was used as stationary phase and acetonitrile: 20 mM disodium hydrogen phosphate and 20 mM monosodium hydrogen ortho phosphate (pH adjusted to 3.5 with phosphoric acid) in the ratio of 65:35 v/v as mobile phase. The mobile phase was filtered through 0.45 μm membrane filter and degassed before analysis. The flow rate was 1 mL/min and the column effluent was monitored at 254 nm. Blank plasma of 60 μL was spiked with 40 μL of standard solutions of FP in a microcentrifuge and 1 mL of ethanol was added. The tubes were vortexed and then centrifuged at 4000 rpm for 5 min, after centrifugation, an aliquot 20 μL supernatant solution was injected to HPLC. Pharmacokinetic parameters, such as AUC, elimination half-life, clearance, and mean residence time were calculated using NCOMP-A Windows-based computer program for noncompartmental analysis. FP was estimated in tissue homogenate by taking 100 μL of tissue homogenate in a microcentrifuge tube and 1 mL of ethanol was added. The tubes were vortexed, then centrifuged at 4000 rpm for 5 min. After centrifugation, an aliquot 20 μL supernatant solution was injected to HPLC.
RESULTS AND DISCUSSION
Preparation and characterization of liposomes
%EE of liposomal batches were found to be between 51.54% and 68.32%. It was found that %EE was increased for the liposomes prepared with synthetic long alkyl chain lipid DSPC (16 alkyl chain length) comparatively with natural phospholipid, SPC (3 alkyl chain length). Thus the former lipid was identified as the lipid of choice for formulating SLs [Table 1].
Table 1.
Physicochemical characterization of prepared liposomal batches
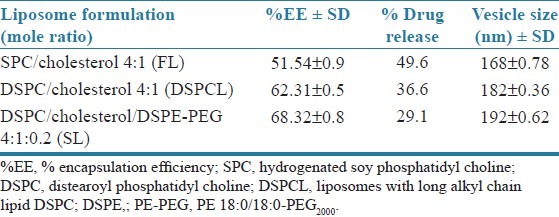
Drug–excipients interaction was studied before developing the formulation by using FTIR-spectroscopy, which is one of the most important analyses to describe the stability of formulation, presence of drug, and drug release [Figure 1]. FTIR spectrum of physical mixture of excipients shows minor shifting of some peaks compared with FTIR spectrum of individual excipients and pure drug like O–H stretching of aliphatic acid from 3422.91 to 3426.74, C–H stretching of aliphatic methyl group from 2918.34 to 2918.43, C=O stretching of acid from 1740.48 to 1740.61, C–H bending of methyl group from 1468.42 to 1468.52, C–O stretching of acid from 1236.17 to 1235.05. These minor shifts observed may be due to the formation of hydrogen bonds, van der Waals attractive forces or dipole moment, which are weak forces seen in the polar functional groups of drugs and excipients. The shifts seen due to the above mentioned interaction may, however, support the formation of favorable vesicle shape, structure with good stability, and sustained drug release.
Figure 1.
(a) Fourier transform infrared (FTIR) spectrum of distearoyl phosphatidyl choline, (b) FTIR spectrum of cholesterol, (c) FTIR spectrum of PE 18:0/18:0-PEG, (d) FTIR spectrum of flurbiprofen, (e) FTIR spectrum of physical mixture of flurbiprofen and excipients
Vesicles were found to be spherical in shape when the liposomal dispersion was observed through an optical microscope. The mean vesicle size of conventional liposomes (FL) was 168 ± 0.78 nm, which was increased to 182 ± 0.36 nm when lipid DSPC was used to prepare the liposomes (DSPCLs) and the vesicle size was found to be 192 ± 0.62 nm for PEG grafted/ SLs [Table 1, Figure 2].
Figure 2.
Scanning electron microscope picture of liposomal formulation stealth liposomes
Zeta potential for SLs was found to be –24.3 ± 0.00 and –24.88 ± 3.72 at 0 and 24 h, respectively, with good result quality. This showed that SL was found to be appreciably stable.
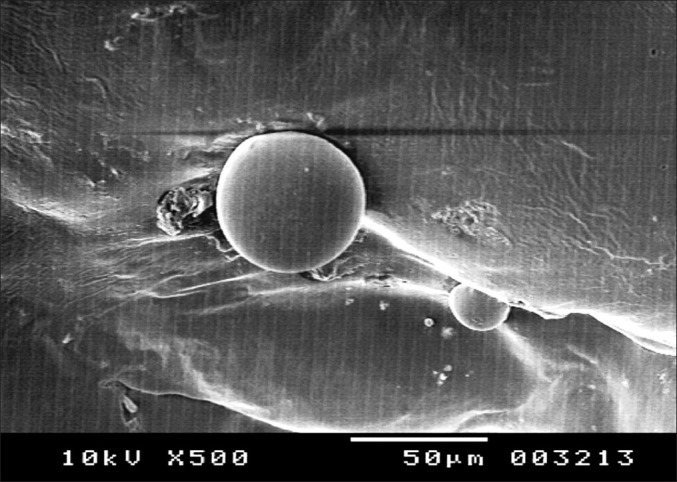
The selected PEGylated liposome was evaluated for physical and chemical stability by storing the liposomal formulation for 1 month at 4 different temperatures as previously described. There are no significant changes in %EE for the formulations stored at –20°C and 4°C. But it was found that there was a considerable reduction in %EE of the liposomes stored at room temperature as well as 37°C ± 2°C [Figure 3].
Figure 3.
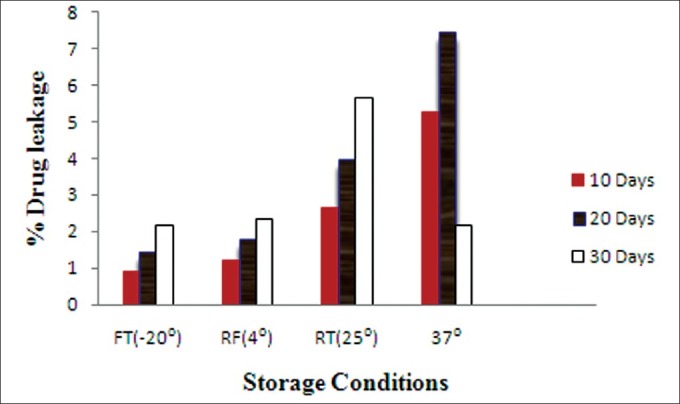
Extent of drug leakage from SLs at different storage temperatures. (FT, freezer temperature (–20°C); RT, room temperature; RF, refrigerated temperature; SLs, stealth liposomes)
The 3 months comparative stability studies of lyophilized form and colloidal liposomes at refrigerated temperature showed that there are negligible drug loss and changes in vesicle size without any change in color in both the forms of liposomes at the end of first month. At the end of the second month, percent drug loss was increased (25.73%) for colloidal liposome compared with freeze dried form (1.58%) with a marked increase in vesicle size (0.230 μm and 0.323 μm, respectively, for lyophilized form and colloidal liposomes). After the third month, the colloidal liposome has shown drastic increase in drug loss (39.56%) and vesicle size (0.823 μm), whereas the freeze dried form showed only 4.83% of drug loss with vesicle size of 0.289 μm. Thus the freeze dried form of liposomes was found to be stable for nearly 3 months. There were no noticeable color changes observed for both colloidal liposome and freeze-dried form of liposome at the end of the 3 months.
The DSC analysis showed a single sharp peak that corresponds to phase transition (54.9°C ± 0.1°C for DSPC, 150.5°C ± 0.1°C for cholesterol, and 56.1°C ± 0.1°C for PE-PEG2000). FP showed a sharp peak at 121°C ± 0.1°C corresponding to the melting point of drug in crystalline form. DSC curves for FP-loaded and -unloaded liposomes observed at 67°C ± 0.1°C and 71°C ± 0.1°C, respectively. The peak of DSPC was found to be shifted from 54.9°C to 67°C signifying that DSPC and cholesterol interacted with each other to a great extent while forming the lipid bilayer. The absence of FP peak and shifting of components peak to 71°C suggested that there was significant intercalation of FP with bilayer leading to enhanced entrapment of drug and decreased rate of release. The peaks corresponding to FP-loaded and -unloaded liposomes were found to be above 40°C, being the prerequisite for liposomes to maintain, which is necessary to remain in the solid state at body temperature. The peak for FP-loaded liposomes was at lower temperature than the peak for unloaded liposomes, which allowed the anticipation of solublization of FP in the lipid matrix [Figure 4].
Figure 4.
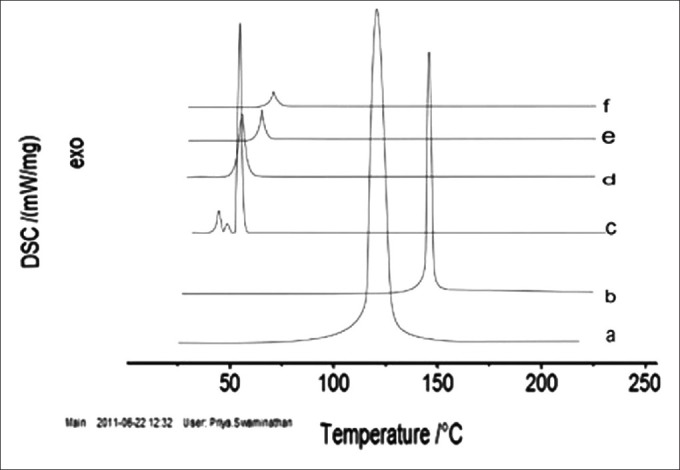
Differential scanning calorimetric (DSC) thermograms of pure drug flurbiprofen (FP) (a) cholesterol (b) distearoyl phosphatidyl choline (DSPC) (c) PE 18:0/18:0-PEG2000 (PE-PEG2000 (d) FP-loaded liposomes (e ) FP-unloaded liposomes (f)
In vitro release performance of the formulations was found to be in the following order SL < DSPCL < FL. This may be because increase in alkyl chain length of lipids increases the phase transition temperature, due to stronger van der Waals interaction between the lipid chains. Thus higher energy is required to disrupt the ordered pack.[33] On the other hand, great lipidic area of longer alkyl chain lipids may enhance the drug binding with lipid bilayer resulting slower or sustained drug release. When compared with the release profile of DSPCLs, the SLs prolonged further the drug release. This slower release in SLs could be because of the fast hydration process of PEG presence on the surface of the vesicle [Table 1, Figure 5].
Figure 5.
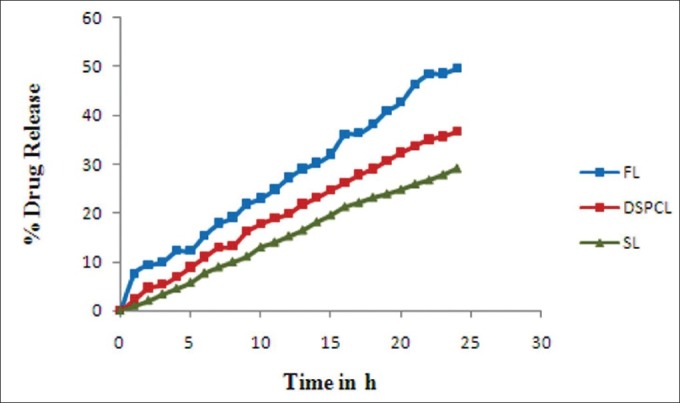
Plots of in vitro cumulative percentage drug released versus time for various flurbiprofen liposomes
Assessment of anti-inflammatory activity
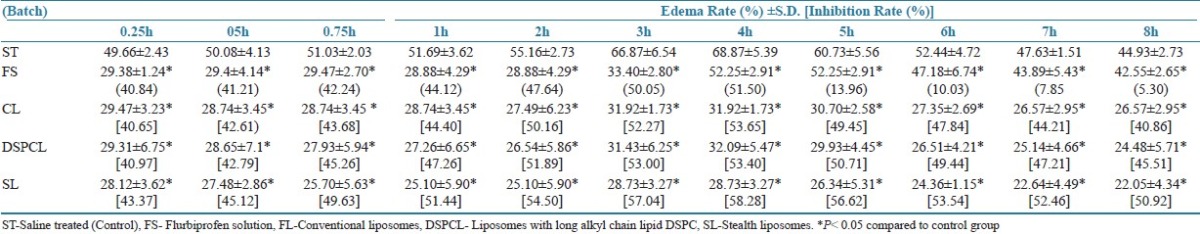
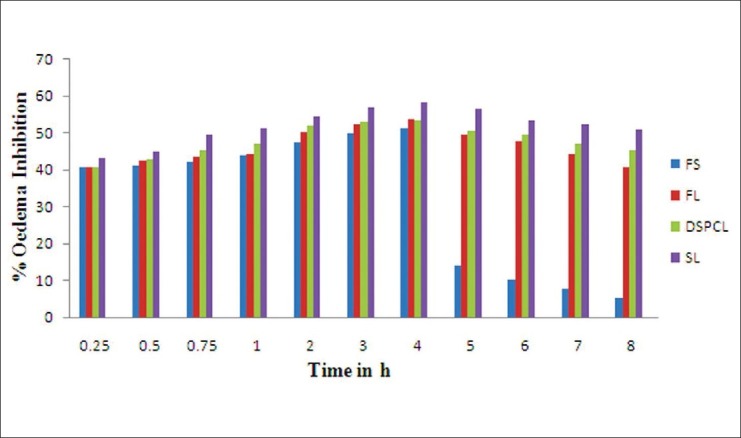
The in vivo performance of the liposomal group, such as FL, DSPCL, and SL were carried out in carrageenan-induced rat paw edema model in comparison with group treated with free drug solution and 0.9% saline (ST; control). It was found that all the liposomal batches reduced the inflammation to the larger magnitude and also sustained the magnitude. Both FLs and DSPCLs have shown statistically significant anti- inflammatory effect with slight increase in inhibitory effect for DSPCLs. In both the cases maximum inhibitory effect was observed at 4 h and more than 40% of the anti-inflammatory effect was maintained at 8 h. But in case of SLs, maximum inhibition of 58.28% was observed at 4 h and above 50% was maintained at even 8 h. However, in case of free drug solution maximum inhibitory effect was found to be at 4 h with the magnitude of 51.50% and just after 4 h it reduced drastically and at 8 h only 5% of the effect was observed. Thus SLs resulted in larger as well as sustained inhibitory effect throughout the studied period. This was attributed to the longevity of SLs in blood stream and enhanced accumulation of drug in inflammatory area [Table 2].
Table 2.
The Anti-inflammatory Activity of conventional and stealth liposomes of flurbiprofen compared to the control and free drug solution. Presented values are means of 6 determinations ± SD
Data were expressed as mean ± SD. The data were analyzed using one-way analysis of variance (ANOVA) followed by Dunnett's t-test. P < 0.05 was considered significant.
Assessment of analgesic activity
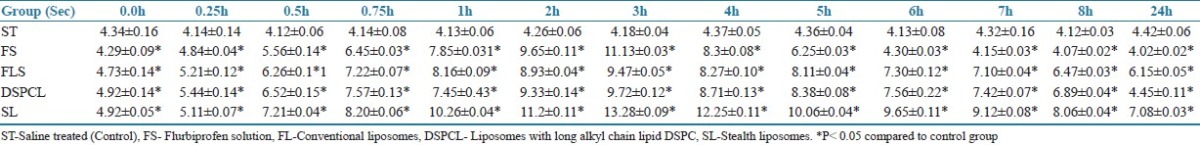
Administration of FP in liposomes (FL, DSPCL, and SL) increased tail flick and hot plate latency when compared with control (ST). There was no change in latent period of control group animals in both hot plate and tail flick tests. The SL has shown maximum analgesic response of 13 s at 3 h and the response of 7 s was maintained at even 24 h. For FL and DSPCL the responses were found to be lesser than SLs. However, they were also able to maintain 4 s of response at 24 h. In drug suspension analgesic, response peaked at 3 h, that is, 11 s, after that it reduced drastically and minimum response was observed at 24 h [Table 3].
Table 3.
The latency time for mice subjected to tail flick Test. Presented values are means of 6 determinations ± SD
Pharmacokinetic study
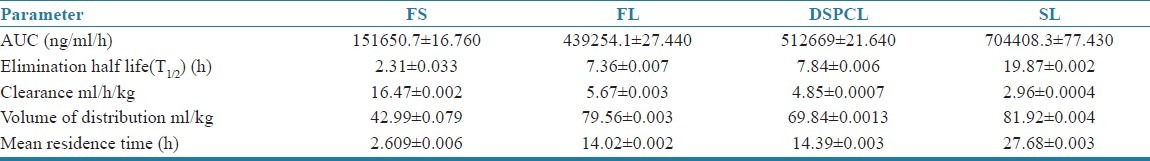
The decrease in plasma FP concentration with time for the drug solution and various liposomes followed a biexponential clearance model. After i.v. bolus administration of liposome formulations to arthritis induced rats, a fall in plasma drug level was found up to 8 h followed by a pattern of slow clearance up to 24 h [Figure 6]. The biphasic clearance was more pronounced in case of both the non-PEGylated liposomes than SLs. This may be due to the slow release of drug from PEGylated (stealth) liposomes and could be explained by better stability (in vivo) achieved by PEG-grafting, which might have decreased the drug release from liposomes and interaction with plasma components, and thus reduced the clearance of the drug. The pharmacokinetic parameters derived from plasma concentration time profile are summarized in Table 4. There was a marked increase in area under FP concentration Vs time curve of SLs, that is, 7,04,408.3 ng/mL/h than the other liposomes, such as FL and DSPCL. The AUC of FS was significantly lower than all the liposome formulations (1,51,650.7 ng/ml/h). It denotes the slow and steady clearance of the drug when intercalated in liposomes and the clearance further slowed down when encapsulated in PEGylated (stealth) liposomes. Elimination half-life and mean residence time (MRT) were found to be increased for liposome formulations, such as FL and DSPCL, compared with drug solution and these parameters were found to be increased enormously for long circulating liposomes [Table 4]. The clearance was 16.47 mL/h/kg for FS and it was 5.67, 4.85, and 2.96 mL/h/kg for CL, DSPCL, and SL formulations. Thus increase in AUC and reduction in clearance rate with SLs signifies the extended bioavailability of the drug in blood for longer period of time. Data were expressed as mean ± SD and statistically assessed by ANOVA followed by Dunnett's t test. P < 0.05 was considered significant.
Figure 6.
Plasma concentration–time profile of various liposomes and free drug solution
Table 4.
Pharmacokinetic parameters of free flurbiprofen solution (FS), Conventional liposomes (FL), DSPC liposomes (DSPCL), Stealth Liposomes (SL) in arthritic rats. Dose:2.5mg/kg. (n=3; P< 0.05)
Biodistribution study
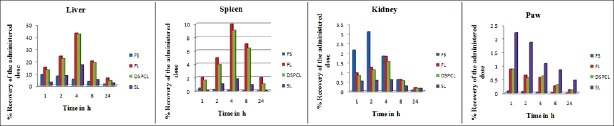
Biodistribution study of liposome formulations in comparison with free drug solution was performed up to 24 h. Conventional liposomes (FL) and DSPC liposomes (DSPCL) were found to be distributed mainly in liver within 1 h post administration. This may be due to the attraction of the liposomes toward reticuloendothelial system and to be cleared off from the blood. It was observed that the percentage of injected drug localized in liver was increased up to 4 h to a maximum of 42.98% and 42.05% for FL and DSPCL, respectively. But only 16.95% of injected dose was found to be localized in liver at 4 h for SLs. In case of free drug solution, interestingly only 9.205% was found to be mounted maximum at 1 h. Maximum drug was mounted in spleen also at 4 h for all liposome formulations; 9.98% and 9.06% of injected dose were found to be in spleen for both FL and DSPCL, respectively. It scored only 1.8% in case of SLs. and 1.82% and 1.55% of injected dose, respectively, were mounted in kidney at 4 h for FL and DSPCL. This was only 0.61% in case of SLs at a tmax of 4 h. In contrast to this Cmax was achieved within 1 h in case of free drug solution.
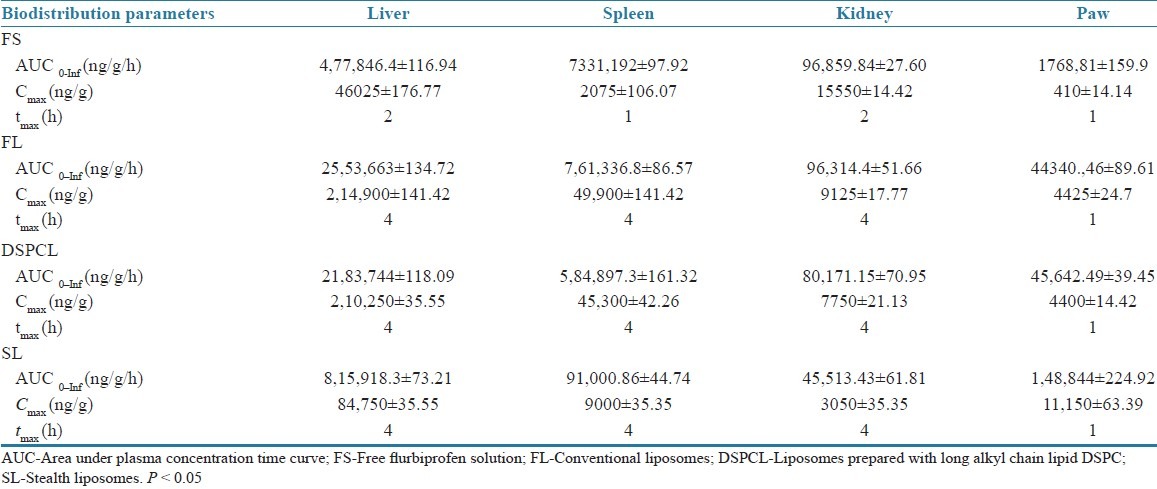
The main goal of entrapping FP into SLs is to reduce the RES uptake and increase the therapeutic availability of the drug at the inflammatory site of arthritic joints. Therefore, drug concentration in arthritic joint was measured up to 24 h. Maximum drug levels were attained in paw after 1 h of administration for all the formulations. In paw only 0.08% of drug was found 1 h after administration of free drug, whereas 0.88% of administered drug was found for FL and DSPCL groups. However, 2.23% of drug was localized in paw for SLs. This may be because of PEG coating of SLs and steric hindrance on its surface, which sufficiently prevent opsonization of liposomes with plasma components, resulting in enhanced passive targeting into inflamed region. Biodistribution profile of the drug as percent recovery of administered dose in liver, spleen, kidney, and paw with respect to time are shown in Figure 7 for various liposome formulations and free drug solution. A comparative profile of biodistribution parameters, such as AUC, Cmax (μg/g) and tmax of different liposomes and free drug in various organs, such as liver, spleen, kidney, and paw are reported in Table 5.
Figure 7.
% Recovery of administered dose–time profile of flurbiprofen-free drug solution and various liposomes after i.v. administration
Table 5.
Biodistribution parameters of various flurbiprofen liposome formulations in arthritis-induced rats at a dose of 2.5mg/kg (n = 3; P < 0.05)
Overall, conventional liposomes and DSPCLs were accumulated intensely in the liver and in the spleen, although the size of liposomes was only 168 and 182 nm, respectively. It indicated that SLs could remain in blood for prolonged time, reduce the uptake of liposomes to RES, and be less concentrated in kidney, thereby reducing the possibility of the risk of toxicity to RES and renal organs and increase the accumulation of drug in inflammatory area by passive targeting.
CONCLUSION
In conclusion, our study indicates that FP-loaded liposomes show favorable pharmacokinetics and biodistribution from the free drug. Targeting potential of sterically stabilized liposomes was increased 3 times than conventional and liposomes prepared with long alkyl chain synthetic lipid, DSPC. Furthermore, the results show that SLs may serve as suitable parenteral dosage form and replace oral therapy, which has poor bioavailability, short half-life, and require frequent dosing. This mode of targeting of FP at the sites of inflammation may also reduce the associated side effects.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Bangam AD, Standish MM, Miller N. Cation permeability of phospholipid model membranes: Effect of narcotics. Nature. 1965;208:1295–7. doi: 10.1038/2081295a0. [DOI] [PubMed] [Google Scholar]
- 2.Panwar P, Pandey B, Lakhera PC, Sing KP. Preparation, characterization and in vitro release study of albendazole-encapsulated nano size liposomes. Int J Nanomedicine. 2010;5:101–8. doi: 10.2147/ijn.s8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Immordino ML, Dosio F, Cattel L. Stealth liposomes: Review of the basic science, rationale and clinical applications existing and potential. Int J Nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 4.Allen TM. Toxicity of drug carriers to the mononuclear phagocyte system. Adv Drug Del Rev. 1988;2:55–67. [Google Scholar]
- 5.Mauk MR, Gamble RE. Stability of lipid vesicles in tissues of the mouse. A gamma-ray perturbed angular correlation study. Proc Natl Acad Sci USA. 1979;76:765–9. doi: 10.1073/pnas.76.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abra RM, Hunt CA. Liposome distribution in vivo: III Dose and vesicle- size effects. Biochim Biophys Acta. 1981;666:493–503. doi: 10.1016/0005-2760(81)90311-8. [DOI] [PubMed] [Google Scholar]
- 7.Kunie V, Maeda F, Harashima H, Kiwada H. Saturable, non-Michaelis- Menten uptake of liposomes by the reticuloendothelial system. J Pharm Pharmacol. 1990;43:162–6. doi: 10.1111/j.2042-7158.1991.tb06658.x. [DOI] [PubMed] [Google Scholar]
- 8.Allen TM, Hansen C. Pharmacokinetics of stealth and non-stealth liposomes: Effect of dose. Biochim Biophys Acta. 1991;1068:133–41. doi: 10.1016/0005-2736(91)90201-i. [DOI] [PubMed] [Google Scholar]
- 9.Gregoriadis G, Senior J. The phospholipid component of small unilamellar liposomes controls the rate of clearance of entrapped solutes from the circulation. FEBS Lett. 1980;119:43–6. doi: 10.1016/0014-5793(80)80994-x. [DOI] [PubMed] [Google Scholar]
- 10.Allen TM, Cleland LG. Serum-induced leakage of liposomes contents. Biochim Biophys Acta. 1980;597:410–26. doi: 10.1016/0005-2736(80)90118-2. [DOI] [PubMed] [Google Scholar]
- 11.Scherphof GL, Damen J, Wilschut J. Interactions of liposomes with plasma proteins. In: Gregoriadis G, editor. Liposome technology. Vol. 3. Boca Raton FL: CRC Press; 1984. pp. 205–24. [Google Scholar]
- 12.Danilo DL. Liposomes. Sci Med. 1996;3:34–43. [Google Scholar]
- 13.Allen TM, Chonn A. Large unilamellar liposome with low uptake by the reticuloendothelial system. FEBS Lett. 1987;223:42–6. doi: 10.1016/0014-5793(87)80506-9. [DOI] [PubMed] [Google Scholar]
- 14.Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85:6949–53. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen TM, Hansen C, Rutledge J. Liposomes with prolonged circulation times: Factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta. 1989;981:27–35. doi: 10.1016/0005-2736(89)90078-3. [DOI] [PubMed] [Google Scholar]
- 16.Allen TM, Ryan JL, Papahadjopoulos D. Gangliosides reduce leakage of aqueous space markers from liposomes in the presence of human plasma. Biochim Biophys Acta. 1985;818:205–10. doi: 10.1016/0005-2736(85)90571-1. [DOI] [PubMed] [Google Scholar]
- 17.Orlu M, Cevher E, Araman A. Design and evaluation of colon specific drug delivery system containing flurbiprofen microsponges. Int J Pharm. 2006;318:103–17. doi: 10.1016/j.ijpharm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Brogden RN, Heel RC, Speight TM, Avery GC. Flurbiprofen: A review of its pharmacological properties and therapeutic use in rheumatic diseases. Drugs. 1979;18:417–38. doi: 10.2165/00003495-197918060-00001. [DOI] [PubMed] [Google Scholar]
- 19.Charoo NA, Shamsher AA, Kohli K, Pillai K, Rahman Z. Improvement in bioavailability of transdermally applied flurbiprofen using tulsi (Ocimum sanctum) and turpentine oil. Colloids Surf B Biointerfaces. 2008;65:300–7. doi: 10.1016/j.colsurfb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Cardoe N, Risdall JJ, Glass RC. Serum concentrations of flurbiprofen in rheumatoid patients receiving flurbiprofen over long periods of time. Curr Med Res Opin. 1977;5:21–5. doi: 10.1185/03007997709108971. [DOI] [PubMed] [Google Scholar]
- 21.Begum MY, Abbulu K, Sudhakar M. Design and evaluation of flurbiprofen Liposomes. J Pharm Res. 2011;4:653–5. [Google Scholar]
- 22.Umrethia M, Ghosh PK, Majithya R, Murthy RS. 6-Mercaptopurine (6-MP) entrapped stealth liposomes for improvement of leukemic treatment without hepatotoxicity and nephrotoxicity. Cancer Invest. 2007;25:117–23. doi: 10.1080/07357900701224862. [DOI] [PubMed] [Google Scholar]
- 23.Ramana MV, Choudhary AD. An approach to minimize pseudomembranous colitis caused by clindamycin through liposomal formulation. Indian J Pharm Sci. 2007;69:390–3. [Google Scholar]
- 24.Hathout RM, Mansour S, Mortada ND, Guinedi AS. Liposomes as an ocular delivery system for acetazolamide: In vitro and in vivo studies. AAPS PharmSciTech. 2007;8:E1–12. doi: 10.1208/pt0804080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonepally CR, Challa SR, Yellu NR, Akkaladevi M, Aukunuru JV. Preparation and characterization of piperine multilamellar vesicular liposomes intended for testing anticonvulsant activity in rats. Indian Drugs. 2007;44:458–65. [Google Scholar]
- 26.Kumar R, Katare OP. Tamoxifen in topical liposomes: Development, characterization and in vitro evaluation. J Pharm Pharm Sci. 2004;7:252–9. [PubMed] [Google Scholar]
- 27.Buddhadev L, Biswajit M. Tamoxifen citrate encapsulated sustained release liposomes: Preparation and evaluation of physico chemical properties. Sci Pharm. 2010;76:507–15. doi: 10.3797/scipharm.0911-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rathode S, Deshpande SG. Design and evaluation of liposomal formulation of pilocarpine nitrate. Indian J Pharm Sci. 2010;72:155–60. doi: 10.4103/0250-474X.65014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasr M, Mansour S, Mortada ND, El Shamy AA. Lipospheres as carriers for topical delivery of aceclofenac: Preparation, characterization and in vivo evaluation. AAPS PharmSciTech. 2008;9:154–62. doi: 10.1208/s12249-007-9028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ustündağ Okur N, Apaydın S, Karabay Yavaşoğlu NÜ, Yavaşoğlu A, Karasulu HY. Evaluation of skin permeation and anti-inflammatory and analgesic effects of new naproxen microemulsion formulations. Int J Pharm. 2011;416:136–44. doi: 10.1016/j.ijpharm.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Graeme NL, Fabry E, Sigg EB. Myocardial adjuant periarthritis in rodents and its modification by anti-inflammatory agents. J Pharmacol Exp Ther. 1966;153:373–80. [Google Scholar]
- 32.Taro O, Yoshimasa I, Masahiro I, Hidehiko A. A pharmacokinetic model for the percutaneous absorption of indomethacin and the prediction of drug disposition kinetics. J Pharm Sci. 1989;78:319–23. doi: 10.1002/jps.2600780412. [DOI] [PubMed] [Google Scholar]
- 33.Mohammed AR, Weston N, Coombes AG, Fitzgerald M, Perrie Y. Liposome formulation of poorly water soluble drugs: Optimization of drug loading and ESEM analysis of stability. Int J Pharm. 2004;285:23–34. doi: 10.1016/j.ijpharm.2004.07.010. [DOI] [PubMed] [Google Scholar]