

Abstract
Experiment and theory favour a model of C–H borylation where significant proton transfer character exists in the transition state.
In the decade following its inception,1 Ir-catalyzed borylation of C–H bonds has emerged as a versatile methodology for functionalizing aromatic and heteroaromatic hydrocarbons.2 Despite the interest that C–H borylation has attracted, what governs reactivities and selectivities is not fully understood. For example, even though steric effects largely dictate regioselectivities for arenes, pronounced electronic effects on relative reactivities of arenes have been noted since the earliest reports.3 In contrast to arenes, the regioselectivities for aromatic heterocycles depend on the position and hybridizations of the heteroatoms they contain, and they are typically more reactive than their arene counterparts.4
Several mechanistic and computational studies of C–H borylations have been reported,2,5 but a full understanding of reactivity and selectivity in these reactions has been elusive. Because the development and control of new catalytic systems depends on this understanding, we have initiated a unified experimental and computational investigation of the Ir-mediated processes. Herein, we describe the initial results from this endeavour.
Taking a cue from an earlier study of monosubstituted benzenes, we probed electronic effects on regioselectivity by examining C–H borylations of anisole. When these were carried out with HBpin (pin = pinacolate) using the dmpe/(η5-indenyl)Ir(cod) ligand/precatalyst system (dmpe = 1,2-bis(dimethylphosphino)ethane, cod=1,5-cyclooctadiene), the para: meta: ortho isomer ratio is 19: 76: 5. The para: meta ratio of 1: 4 deviates significantly from the 1: 2 ratio expected absent electronic effects, and it is interesting that ortho-borylation is observed, as a minor product.
The favoured mechanism for this reaction involves a 5-coordinate intermediate as the key species responsible for C–H activation. We were curious about whether calculations based on this mechanism could predict the regioselectivities. In the computational model (dmpe)Ir(Beg)3 (1a, eg = ethyleneglycolate), the lowest energy structure is a square pyramid with two phosphorus atoms from dmpe and two boryl boron atoms in the basal plane. A third boryl ligand occupies the apical site. Transition structures for C–H activation by 1a are analogous to those in Sakaki’s earlier studies.6 The arene approaches trans to the apical boron and C–H scission occurs via the multi-centre interaction between H, C, Ir, and B atoms. For the reaction of 1a with anisole, multiple transition-state conformers were located for each regioselectivity and the lowest-energy structures are shown in Fig. 1. Free-energy barriers of ~30 kcal mol−1 were predicted in mPW1K/BS2//B3LYP/BS17 calculations, fitting well with overall reaction rates in contrast to previous calculations.6 The Boltzmann-weighted free energies of the various anisole transition states lead to a predicted para: meta: ortho ratio of 23: 73: 4, in striking agreement with experiment.
Fig. 1.

Transition structures for C–H activation of anisole by 1a at the ortho, meta, and para-positions. Distances are in Å and barriers are mPW1K/BS2//B3LYP/BS1 at 150 °C.
From Fig. 1a, borylation at the ortho position of anisole is sterically disfavoured, so electronic effects must be responsible for its formation. From an electronic perspective, the transition state can be viewed as falling on a continuum bounded by two extremes—proton transfer from the arene to the Ir–B bond vs. hydride transfer to an “empty” boron p-orbital of the boryl ligand (Fig. 2).
Fig. 2.

Limiting descriptions of the C–H activation transition states.
In catalysis, theory can shed light on the reactivity of intermediates that often go undetected. Recently, 5-coordinate analogues of 1a have been prepared and were shown to borylate arenes directly at room temperature.8 Thus, theory and experiment are perfectly poised to probe issues like those posed in Fig. 2.
Toward this end, stoichiometric reactions of the isolable 5-coordinate trisboryl complex (dippe)Ir(Bpin)3 (2, dippe = 1,2-bis(diisopropylphosphino)ethane) with veratrole (3) and benzodioxole (4) were investigated. As shown in Fig. 3, the ortho C–H bonds in 4 are more accessible than those in 3, separating steric from electronic effects. In addition, reactions catalyzed by 2, as well as stoichiometric and catalytic reactions employing the dipyridyl precatalyst (dtbpy)Ir(Bpin)3(η2-coe) (5, dtbpy = 4,4′-tert-butyl-2,2′-dipyridyl) were also examined. The results are shown in Table 1.
Fig. 3.
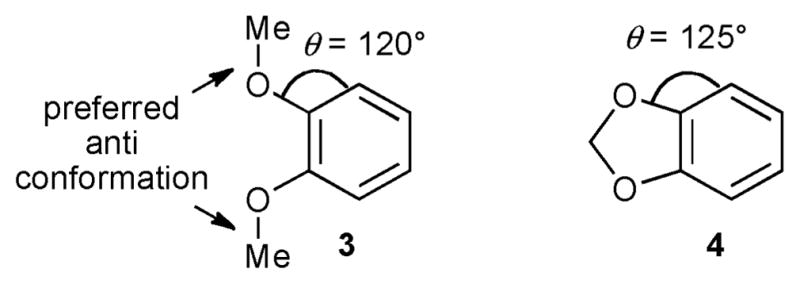
Structures of veratrole (3) and benzodioxole (4) highlighting the relative accessibilities of the ortho-C–H bonds.
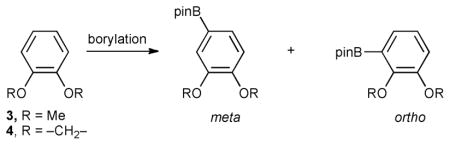
Table 1.
Borylations of veratrole and benzodioxole under stoichiometric and catalytic conditions
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ir complex | Substratea | Time | Temp/°C | meta (%) | ortho (%) |
| 1 | 2 | 3 | 4 h | 100 | >99 | <1 |
| 2 | 2 mol% 2b | 3 | 4 h | 130 | 98 | 2 |
| 3 | 5 | 3 | 1 h | rt | 99 | 1 |
| 4 | 2 mol%5 | 3 | 9 h | rt | 98 | 2 |
| 5 | 2 | 4 | 1 h | 100 | 2 | 98 |
| 6 | 2 mol%2 | 4 | 1 h | 130 | 4 | 96 |
| 7 | 5 | 4 | 15 min | rt | 5 | 95 |
| 8 | 2 mol% 5c | 4 | 1 h | rt | 3 | 91 |
8 equiv. of 3 or 4 were used to minimize diborylation.
Generated from 2 mol% (η5-indenyl)Ir(cod) and 2 mol% dippe.
6% of the product is 3,6-diborylated.
The striking result from this study is that in contrast to veratrole, which afforded the expected meta-borylated regio-isomer, benzodioxole yields almost exclusively the ortho-functionalized product! ortho-Borylation could arise from coordination of an oxygen atom of benzodioxole to a boron atom in one of the boryl groups, but computational studies do not support this. The lowest-energy transition structures for reactions of 4 with 1a and (bpy)Ir(Beg)3 (6a), a 5-coordinate model of the intermediate that arises from 5, are shown in Fig. 4. In both cases, O···B distances in the transition states are much longer than those in O → B adducts. These results firmly support a transition state with proton-transfer character and substantiate inferences that boryl nucleophilicity facilitates C–H borylation.9
Fig. 4.
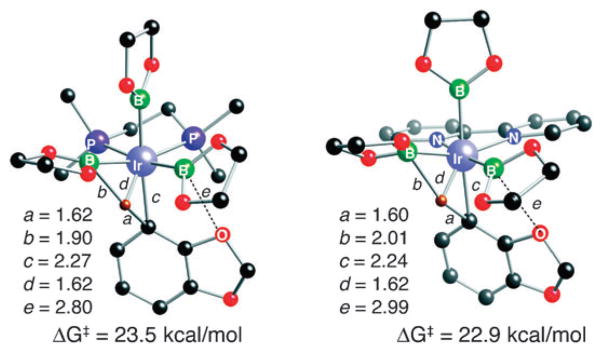
Transition structures for borylation of benzodioxole by 1a (left) and 6a (right) (distances in Å). The barriers are calculated at 25 °C.
Contributions from C–H acidity in the transition state could also explain the enhanced reactivity of pinacolate reagents over catecholate analogues in catalytic C–H borylations. Hartwig has recently shown that 5 is significantly more reactive than a catecholate analogue.10 Unfortunately, this difference could not be definitively linked to the C–H activation because coe dissociation, which Bpin should favour relative to Bcat, precedes this step.
(dippe)Ir(Bcat‡)3 (7, cat‡ = 3,5-tert-butylcatecholate) and (dippe)Ir(Bcat)3 (8, cat = catecholate) were prepared to assess effects of boron substitution on C–H activation.‡ Reactions of 7 and 8 with arenes required elevated temperatures, where decomposition preceded borylation. This problem was solved by using 2-methylthiophene, a more reactive substrate. Compounds 7 and 8 reacted at ambient temperatures, and borylated thiophene products formed as the boryl complexes were consumed. For 7 and 8, t½ is approximately 3.5 h at 30 °C. In contrast, t½ is less than 5min for the reaction of 2-methylthiophene with 2. Because Bpin is more electron rich than Bcat,10,11 our model predicts that 2 should be more reactive than 7 or 8, as is indeed the case.
We returned to theory for a more general understanding of these reactions. To avoid the ambiguity associated with interpretations of the partial bonding in transition states, we considered whether reactivity patterns could be understood from ground-state structures of intermediates. From the Hammond–Leffler postulate12 this seemed plausible because the transition states for C–H activation are late. To test this hypothesis, energies for transition states (9) and intermediates (10)13 in Scheme 1 were calculated for a series of 23 reactions involving four 5-coordinate Ir complexes [1a, (dmpe)Ir(Bcat)3 (1b), 6a, and (bpy)Ir(Bcat)3 (6b)] and electronically diverse substrates (benzene, 3, 4, 2-methylthiophene, pyrrole, m-xylene, m-dibromobenzene and m-bis(trifluoromethyl)benzene). Fig. 5a shows a plot of the activation barriers (ΔE‡) vs. the energy differences (ΔE) between the reactants and products in Scheme 1. The strong correlation (R2 = 0.96) shows that ΔE is an excellent predictor of ΔE‡. Thus, structures 10 will mirror transition states 9.
Scheme 1.
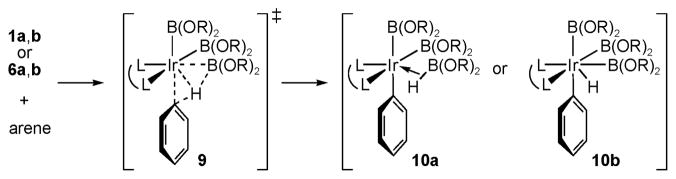
Transition states (9) and intermediates (10) for C–H activation.
Fig. 5.

(a) Correlation of calculated barriers (ΔE‡) for C–H activation (E + zpe, B3LYP/BS1) vs. the energy of the resulting intermediates (ΔE). (b) Correlation of ΔE with the charge developed on the aryl group with linear fits for 6- and 5-membered substrates. Sterically hindered arenes (blue) were omitted from the arene fit.
From detailed analysis of intermediates 10, a relationship that provides remarkable insight into reactivity emerges. Fig. 5b shows a plot of the energies vs. the total natural population analysis (NPA) charge on the aryl and heteroaryl groups in 10. For C–H activations at unhindered positions of benzene derivatives, including ortho sites on 4, energies of 10 correlate strongly with the arene charge (R2 = 0.95). Our interpretation is that this line defines the electronic effect on the energies of the intermediates (and, from the first correlation, the preceding transition states). Changes in the arene, boryl group, or bidentate ligand that promote transfer of negative charge to the arene in 10 favour its formation. This is the key electronic factor that governs C–H activation. As a corollary, the greater reactivities with bpy over dmpe or with Beg over Bcat result from greater donation to the Ir. This effect has been previously suggested,11 but the results here establish its dominant role in determining reactivity.
The outliers in Fig. 5b are enlightening. ortho-Substitution in anisole and veratrole lie above the arene line and reflect diminished reactivity due to unfavourable sterics in the intermediates (and the preceding transition states). The energies of intermediates arising from 5-membered heterocycles lie below the line defined by the arenes, and a separate linear fit gives a similar slope (R2 = 0.66). This is consistent with experiment and may reflect decreased sterics from ring contraction.
These experimental and computational results provide a clear picture of the underlying electronic effects in C–H borylation, where proton transfer character contributes to the transition state (structure A in Fig. 2). This model provides a simple explanation of other results from the literature. For example, coordination prior to C–H activation has been suggested to account for accelerated borylation rates for thiophene and furan.14 Likewise, heteroatom coordination has been offered to account for the selective functionalizations at the C–H positions flanking the heteroatoms in indoles and benzofurans.15 In both cases, the pKas of these C–H bonds are the lowest in the respective substrates.16 Hence, they are more reactive. Lastly, we note that while some of the reactivity and selectivity patterns of Ir-catalyzed C–H borylations mirror those of main group organometallic compounds like alkyl lithium reagents, significant differences are seen for certain substrates.17 Studies of these exceptions are underway.
Supplementary Material
Acknowledgments
We thank the Michigan Economic Development Corp. and the NIH (GM63188 to M. R. S. and GM45617 to D. A. S.) for financial support and BASF for a gift of HBPin.
Footnotes
Electronic supplementary information (ESI) available: Spectral data for all new compounds, general experimental procedures, as well as computational details. CCDC 782157 and 782158. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02041a
Crystal data for 7·0.5C8H18. C60H100O6B3P2Ir, M = 1203.97, triclinic, P1̄, a = 14.715(2), b = 14.882(2), c = 17.249(2) Å, α = 78.098(2)1, β = 85.375(2)°, γ = 65.636(2)°, V = 3366.76(8) Å3, T = 173(2) K, Z = 2, 25 455 reflections measured, 11 719 unique (Rint = 0.0792), which were used in all calculations. The final R1 and wR2 were 0.0670 and 0.11740. Crystal data for 8. C32H44O6B3P2Ir, M = 811.24, monoclinic, P21/c, a = 11.5773(3), b = 19.1172(6), c = 15.8919(5) Å, β = 99.009(2)°, V = 3473.89(18) Å3, T = 173(2) K, Z = 4, 30 124 reflections measured, 6364 unique (Rint=0.0667), which were used in all calculations. The final R1 and wR2 were 0.0409 and 0.0921.
Contributor Information
Daniel A. Singleton, Email: singleton@mail.chem.tamu.edu.
Milton R. Smith, III, Email: smithmil@msu.edu.
References
- 1.Iverson CN, Smith MR., III J Am Chem Soc. 1999;121:7696–7697. [Google Scholar]
- 2.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 3.Cho JY, Iverson CN, Smith MR., III J Am Chem Soc. 2000;122:12868–12869. [Google Scholar]
- 4.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651. [Google Scholar]
- 5.Wei CS, Jimenez-Hoyos CA, Videa MF, Hartwig JF, Hall MB. J Am Chem Soc. 2010;132:3078–3091. doi: 10.1021/ja909453g. [DOI] [PubMed] [Google Scholar]; Sawyer KR, Cahoon JF, Shanoski JE, Glascoe EA, Kling MF, Schlegel JP, Zoerb MC, Hapke M, Hartwig JF, Webster CE, Harris CB. J Am Chem Soc. 2010;132:1848–1859. doi: 10.1021/ja906438a. [DOI] [PubMed] [Google Scholar]
- 6.Tamura H, Yamazaki H, Sato H, Sakaki S. J Am Chem Soc. 2003;125:16114–16126. doi: 10.1021/ja0302937. [DOI] [PubMed] [Google Scholar]
- 7.BS1 used an SDD basis set and core potential on Ir and 6-31G** on other atoms. BS2 augmented the SDD basis with three f-functions and a separated d function (as a 3111 contraction instead of the normal 411), along with a 6-31+G** basis on the remaining atoms.
- 8.Chotana GA, Vanchura BA, II, Tse MK, Staples RJ, Maleczka RE, Jr, Smith MR., III Chem Commun. 2009:5731–5733. doi: 10.1039/b914736e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webster CE, Fan YB, Hall MB, Kunz D, Hartwig JF. J Am Chem Soc. 2003;125:858–859. doi: 10.1021/ja028394c. [DOI] [PubMed] [Google Scholar]; Dang L, Lin ZY, Marder TB. Chem Commun. 2009:3987–3995. doi: 10.1039/b903098k. [DOI] [PubMed] [Google Scholar]; Ess DH, Nielsen RJ, Goddard WA, Periana RA. J Am Chem Soc. 2009;131:11686–11688. doi: 10.1021/ja902748c. [DOI] [PubMed] [Google Scholar]
- 10.Liskey CW, Wei CS, Pahls DR, Hartwig JF. Chem Commun. 2009:5603–5605. doi: 10.1039/b913949d. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Lin ZY, Marder TB. Inorg Chem. 2005;44:9384–9390. doi: 10.1021/ic0513641. [DOI] [PubMed] [Google Scholar]
- 12.Hammond GS. J Am Chem Soc. 1955;77:334–338. [Google Scholar]; Leffler JE. Science. 1953;117:340–341. doi: 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]
- 13.The dmpe intermediates are better described as agostic B–H complexes of IrIII (10a). The bipy intermediates have significant IrV character (10b), consistent with Sakaki’s earlier studies6.
- 14.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 15.Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RE, Jr, Smith MR., III J Am Chem Soc. 2006;128:15552–15553. doi: 10.1021/ja0631652. [DOI] [PubMed] [Google Scholar]
- 16.Shen K, Fu Y, Li JN, Liu L, Guo QX. Tetrahedron. 2007;63:1568–1576. [Google Scholar]
- 17.While this manuscript was under review, a comparison between Ir C–H borylation and DoM was published: Hurst TE, Macklin TK, Becker M, Hartmann E, Kuegel W, Parisienne-La Salle J-C, Batsanov AS, Marder TB, Snieckus V. Chem–Eur J. 2010;16:8155–8161. doi: 10.1002/chem.201000401.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.