

Abstract
The number of elderly patients seeking clinical treatment for memory problems will rise sharply in coming years as our population ages. These patients present a challenge for diagnosis and prognosis since cognitive problems in older patients can arise from many etiologies, some of which are curable. With the development of clinically available biomarkers for detecting Alzheimer’s disease pathology in living patients, evaluation of cognitively impaired elderly patients is about to undergo a major paradigm shift. This article describes the two classes of biomarkers available for assessing Alzheimer’s disease risk: those that indicate presence of amyloid pathology and those that provide evidence of neuronal injury and neurodegeneration. We argue that, currently, incorporation of biomarkers of neurodegeneration can help in patient prognosis whereas tests for amyloid, if used in isolation, have potential for harm. Amyloid tests are clinically useful only when evidence suggests progressive cognitive decline or neurodegeneration.
Keywords: Alzheimer’s disease, amyloid imaging, biomarker, florbetapir, MCI, mild cognitive impairment, MRI, PET
Clinical evaluation of older patients with memory problems is about to undergo a major paradigm shift. Biomarkers for detecting neuronal injury and Alzheimer’s disease (AD) pathology are becoming increasingly available. The recent development of radiopharmaceuticals with high affinity for fibrillar amyloid has made it possible to test for elevated amyloid deposition, one of the defining pathological features of AD, in the living human brain [1–4]. One such radio-pharmaceutical, florbetapir (Amyvid™ Avid Radiopharmaceuticals, Eli Lilly, PA, USA), has recently received approval from the US FDA for clinical use in detecting brain amyloid deposition. Physicians now have a new test at their disposal to help determine the underlying cause of cognitive symptoms in older patients presenting with memory complaints. Given the excitement about this new technology, there will be strong temptation to incorporate amyloid imaging into the evaluation of elderly patients with memory complaints, and to interpret positive findings as diagnostic indicators of AD – but are they? And should amyloid imaging be routinely used in the clinical work-up of elderly patients with memory concerns? We argue here that the answer to both questions is “No”. To support this argument, we briefly review the current state of knowledge of AD pathophysiology and biomarkers for assessing AD risk in patients. We then present different clinical scenarios where amyloid testing may be considered and discuss the potential harms and benefits of such testing. We conclude with a clinical evaluation strategy that incorporates tests of neurodegeneration when physicians believe that additional information may be beneficial for patient management. Given the current state of knowledge, amyloid testing is not recommended in the diagnostic work-up of patients lacking evidence of neurodegeneration. However, if amyloid-modifying therapies become available, amyloid biomarkers may become essential in risk–benefit determination.
The changing clinical landscape
Due to increased longevity and the aging of population worldwide, the number of individuals aged 65 years or older will rise dramatically in coming years [5]. In 2010 there were 40.2 million people in the USA aged 65 years or older. That number is expected to more than double (to 88.5 million) by 2050 [6]. Clinicians can thus expect to see growing numbers of patients presenting with aging-related disorders, including memory problems. Older patients with cognitive complaints present a particular challenge for diagnosis and prognosis since cognitive problems can arise from many etiologies.
One of the most common and most feared causes of cognitive impairment in the elderly is AD. Approximately 1 in 8 adults (13%) over 65 years of age suffers from AD; that number approaches one in two (43%) by 85 years of age [7]. Despite its disturbingly high prevalence, not all cognitive impairment in the elderly arises from AD. Many treatable conditions impact on cognition, including medication side effects, sleep disorders, thyroid deficiency, depression and anxiety. Other less common but equally devastating neurodegenerative disorders, such as Lewy body disease, frontotemporal dementia, or hippocampal sclerosis must also be considered. Accurate identification of the underlying cause of cognitive symptoms in older patients is imperative for ensuring appropriate care. Recent development of biomarkers for detection and prediction of AD can aid in patient diagnosis and prognosis. Before discussing potential pitfalls and recommendations for use of these bio-markers in the clinical setting, we briefly review AD neuropathology and describe research results using the currently available biomarkers for predicting AD dementia.
AD clinical expression & neuropathology
AD is a progressive, ultimately fatal disorder with insidious onset. Initial subtle cognitive impairment, usually involving memory, slowly progresses to the point that other cognitive domains are affected and activities of daily living can no longer be performed independently. When the patient reaches this threshold of dementia, a clinical diagnosis of probable or possible AD is given, dependent on whether the clinical presentation is typical or atypical [8]. Mild cognitive impairment (MCI) has been introduced as a diagnostic category to capture the transitional stage between normal aging and dementia [9]. Patients with MCI have subjectively noted and objectively verified cognitive impairment that is insufficient to interfere with daily function, and are at elevated risk for developing AD [9].
AD is characterized by extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs). Amyloid deposition first appears in the basal neocortex and then spreads throughout the association cortex, with primary sensory and motor areas being affected at the latest stages; although there is substantial variation across individuals in the extent and distribution of these plaques [10]. The pathological phosphorylation of tau proteins leads to a sequence of events that result in intracellular formation of NFTs and eventual dystrophic changes and death of the affected neuron. In contrast to amyloid deposition, NFT progression proceeds in an orderly fashion, with NFTs first appearing in the transentorhinal region in the medial temporal cortex, prior to the onset of clinical symptoms, then spreading through limbic cortex as the disease becomes manifest, then throughout association cortex and finally into primary cortex with increasing disease severity [10]. The density and distribution of amyloid plaques and NFTs have been used in the definitive diagnosis of AD at autopsy in individuals with dementia [11]. Recognizing that AD develops over a prolonged, symptom-free period, and that a substantial proportion of cognitively intact individuals at time of death meet neuropathological criteria for AD [12–14], recently revised criteria for detection of AD neuropathologic changes have been broadened to apply to all individuals, regardless of clinical status at time of death [15].
Despite intense, ongoing research into the pathogenesis of AD, its underlying cause remains elusive. The most popular theory, the amyloid cascade hypothesis, posits that the aggregation of amyloid into plaques is the initiating event that leads, years, potentially decades, later to the development of NFTs, which then leads to synaptic dysfunction, brain atrophy and dementia [16,17]. Although there are considerable genetic, biochemical and animal modeling data to support this hypothesis [18], other evidence calls it into question, including: presence of similar amyloid burden in cognitively healthy elderly individuals as in AD patients [12,13]; lack of correlation between amyloid burden and disease stage or duration [19,20]; findings that NFTs precede amyloid pathology [21]; failure of anti-amyloid agents in AD clinical trials to alter the course of the disease [22]; and identification of other potential disease triggers [23,24], such as oxidative stress [25], neuroinflammation [26] and lipid dyshomeo-stasis [27]. Thus, AD pathogenesis remains the subject of vigorous debate [18,22,25].
Although the initial disease trigger is not known, animal and human neuroimaging data suggest a synergistic relationship between tau and amyloid pathology whereby tau is necessary for, and mediates, amyloid-related neurotoxicity [28,29]. This implies that amyloid pathology alone is insufficient to cause neurodegeneration. This is further supported by evidence showing that in AD, NFT density and distribution correlates with disease stage, disease duration, neuronal loss [19,20], and degree of atrophy observable with structural MRI [30], whereas amyloid burden does not.
Research results on biomarkers of AD
Propelled in part by large-scale studies such as the ground-breaking Alzheimer’s Disease Neuroimaging Initiative [31], recent years have witnessed enormous advance in the understanding of AD biomarkers that indicate presence of AD pathology or AD-related neuronal injury in patients with dementia or MCI, and in cognitively healthy individuals. AD biomarkers can be categorized into those that indicate the presence of brain amyloid pathology and those that reflect neuronal injury (see Box 1). The two biomarkers of amyloid pathology, PET imaging of amyloid deposition and cerebrospinal fluid (CSF) levels of the major constituent of amyloid plaques, amyloid-β42, (Aβ42) are similarly able to detect amyloid pathology [32–34], with elevated amyloid deposition in PET imaging corresponding to low CSF Aβ42 levels [35]. Both biomarkers provide evidence of amyloid pathology that aligns with post-mortem results [36–38]. Numerous studies have shown that these amyloid biomarkers are highly sensitive for discriminating AD patients from healthy controls [39–42], and that many patients with MCI test positive for amyloid pathology [40,42]. Consistent with neuropathological studies, biomarker studies also find evidence of amyloid pathology in approximately 30% of cognitively healthy individuals [40,42–45], with frequency of positive findings increasing with age [42,46]. One study reported that 65% of cognitively healthy individuals over 80 years of age showed elevated amyloid deposition [46].
Box 1. Biomarkers of Alzheimer’s disease.
A number of biomarkers have been developed that indicate the presence of Alzheimer’s disease (AD) pathology and AD-associated neuronal injury in vivo. These biomarkers have strong potential for clinical use in etiological determination, predictive prognosis, monitoring disease progression, and for serving as outcome measures in clinical trials of potential disease-modifying therapies.
Biomarkers of brain amyloid pathology: amyloid imaging
The first amyloid-sensitive radiotracer to be developed, Pittsburgh compound B binds to insoluble fibrillary amyloid in the brain, a major constituent of the amyloid plaques that are one of the hallmark pathological features of AD [1]. Pittsburgh compound B is used widely in research studies but has limited clinical potential since its short half-life restricts its availability to clinics with a cyclotron on site. More recently developed 18F-labeled tracers, flutemetamol, florbetaben and florbetapir, show similar high affinity for fibrillary amyloid, but have longer half-lives, allowing for central production and distribution, rendering them more amenable to widespread clinical use [4]. Florbetapir has recently received approval for clinical use by the US FDA to indicate presence of amyloid in the brain.
Cerebrospinal fluid Aβ42 levels
Cerebrospinal fluid (CSF) levels of Aβ42, the major constituent of amyloid plaques can be obtained from lumbar puncture. CSF Aβ42 levels decrease as plaque levels increase, suggesting that Aβ42 becomes sequestered in plaques, leaving less to diffuse into the CSF [86]. Decreased CSF Aβ42 levels can thus be used to infer presence of amyloid plaques in the brain.
Biomarkers of neuronal injury: CSF tau & phosphorylated tau levels
Elevated levels of tau in the CSF are a nonspecific reflection of neuronal injury. Phosphorylated tau is a more specific reflection of the phosphorylated state of tau, and reflects neurofibrillary tangle formation in the brain [87]; levels of both are increased in AD. The ratio of CSF tau or phosphorylated tau to CSF Aβ42 shows greater sensitivity to AD than any single CSF measure [58]. Commercial CSF analysis services (e.g., Athena Diagnostics, MA, USA) can indicate whether CSF biomarkers levels are consistent with AD.
Volumetric MRI
AD is associated with widespread brain atrophy, even in prodromal stages, with prominent involvement of the hippocampus, a medial temporal lobe structure important for memory [88]. The presence of atrophy in medial temporal structures, which can be visually rated or more precisely quantified using FDA-approved automated medical device image analysis software (e.g., NeuroQuant ®, CorTechs Labs, Inc., CA, USA), is associated with a high risk of imminent decline to dementia [60,61].
Fluorodeoxyglucose-PET
Synaptic dysfunction, neuronal injury and neuron loss leads to hypometabolism in parietal and temporal areas detectable with FDG-PET [82]. FDG-PET is currently approved for clinical use to distinguish AD from frontotemporal dementia, which is characterized by hypometabolism in frontal rather than in posterior brain regions.
Emerging evidence suggests that amyloid pathology in asymptomatic older adults may be associated with subtle cognitive decline and structural brain changes [29,47–51]. It is important to note that we are still in the early stages of understanding the relation of amyloid bio-markers in healthy older adults to the development of dementia. It is not known whether individuals who test positive for amyloid are in a preclinical stage of AD and will progress to dementia if they live long enough, or whether these individuals may be resistant to AD pathology due to cognitive reserve [52], genetic factors [53] or environmental influences. Accumulating data from ongoing longitudinal studies can be expected to inform on these important issues in coming years.
Research studies have shown, however, that biomarker evidence of amyloid pathology in symptomatic patients (i.e., those meeting diagnostic criteria for MCI [9]) is associated with elevated risk for developing dementia relative to MCI patients who test negative for amyloid [54–57]. Several studies have reported that the predictive ability of amyloid biomarkers is enhanced when combined with biomarkers of neuronal injury (such as CSF tau or atrophy on MRI) [57–60]. Since presence of neuronal injury is more proximal to the development of dementia than amyloid pathology [17], indicators of neuronal injury, such as medial temporal lobe atrophy on MRI, have been found to be more predictive of impending cognitive decline than presence of amyloid pathology [60,61].
The volume of the hippocampus, as assessed with structural MRI (Figure 1), has long been known to be sensitive to the progressive neuro-degeneration in AD. The development of automated methods to quantify atrophy across the cortex has revealed that widespread atrophy is apparent prior to the onset of dementia [62,63] and that the degree of atrophy in brain regions, characteristically affected early in AD is predictive of dementia [59,64,65] even at the level of the individual patient[66].
Figure 1. Coronal section of a T1-weighted volumetric MRI from an Alzheimer’s Disease Neuroimaging Initiative mild cognitive impairment patient.
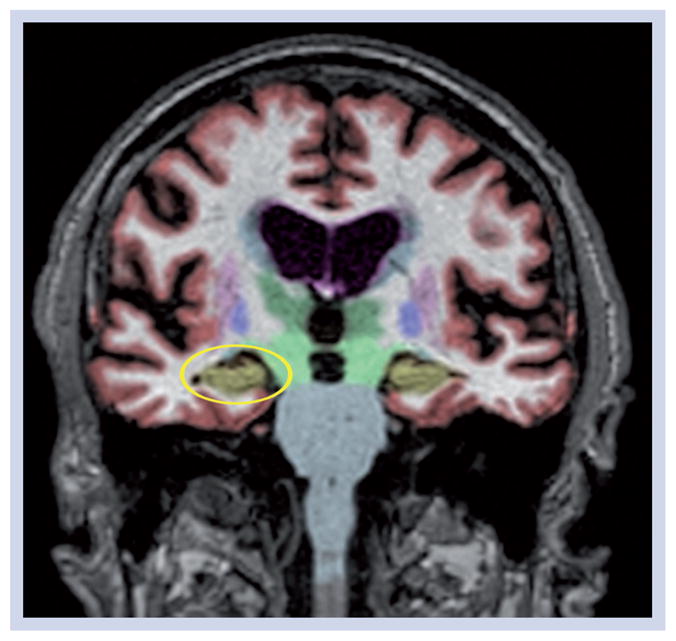
Data have been automatically segmented into different tissue types and brain regions using NeuroQuant® software [101]. The hippocampus, which is highly vulnerable to Alzheimer’s disease, but also affected in other neurodegenerative disorders, is shown in gold. This mild cognitive impairment patient, despite testing positive for cerebrospinal fluid Aβ42 had age-appropriate hippocampal and inferior lateral ventricle volumes (circled in yellow on the left) at baseline and at all follow-ups. This patient retained the mild cognitive impairment diagnosis through 3 years of follow-up.
In a recent study, we examined the relative ability of clinically available CSF and MRI biomarkers to predict risk of dementia in the Alzheimer’s Disease Neuroimaging Initiative’s highly selected MCI population [60]. Although CSF evidence of amyloid pathology was associated with increased risk of developing dementia (hazard ratio [HR] = 3.4), the combination of CSF Aβ42 with CSF tau, a marker of neuronal injury, provided better prediction (HR = 4.1; see Figure 2). Medial temporal lobe atrophy, defined as the ratio of hippocampal volume to the sum of the hippocampal and inferior lateral ventricle volumes was associated with similar risk (HR = 3.9), but patients with atrophy showed a faster rate of clinical decline than those with a positive CSF tau Aβ42 ratio. Median dementia-free survival time was 15 months for individuals at risk owing to temporal lobe atrophy, relative to 20–28 months for individuals classified as at risk on the basis of one or both CSF biomarkers (Figure 2). Stratifying patients on the basis of amyloid and atrophy risk substantially enhanced risk prediction. Very few patients who tested negative for both biomarkers developed dementia over 3 years, whereas those testing positive for both showed very high risk (HR = 14.3). Individuals who tested positive for medial temporal atrophy but negative for amyloid pathology showed a similarly high risk of developing dementia (HR = 9.8; relative to those testing negative for both biomarkers; see Figure 2). Other studies have also noted conversion to dementia in patients who test negative for amyloid [55,67]. It is not clear whether this represents false-negative biomarker results or whether these individuals suffered from vascular dementia or another non-AD dementia. Nevertheless, it is clear that absence of amyloid pathology in the presence of medial temporal atrophy does not imply a benign clinical course.
Figure 2. Risk of developing dementia in patients with mild cognitive impairment as a function of individual and combined biomarker status.

(A–C) Red shows risk of developing AD in MCI patients testing positive for cerebrospinal fluid (CSF) Aβ42 for the CSF tau/A ratio and for medial temporal atrophy (HOC), respectively. Blue lines indicate negative results on these tests. (D) Mild cognitive impairment patients are stratified on the basis of CSF A β42 and atrophy risk. Those testing negative for both (green line) are at very low risk of developing dementia within 3 years, those testing positive for both (red line) are at very high risk. Those testing positive for atrophy, even in the presence of a negative CSF Aβ42 test (purple line), are also at very high risk of developing Alzhemer’s disease.
HOC: Hippocampal occupancy score.
Adapted with permission from [60].
Patients without evidence of medial temporal atrophy but with evidence of amyloid pathology were also at elevated risk of developing dementia relative to those without either biomarker (HR = 4.9), though risk was not as high as when atrophy was present. Given lack of histopathological confirmation, it is unclear whether these cases were due to false negatives, atypical AD or whether other diseases contributed to the dementia.
Revised research & clinical criteria for diagnosis of AD & MCI
With the enormous advance in knowledge of AD biomarkers that has accumulated since the clinical criteria for AD diagnosis were introduced in 1984, there have been calls to revise the diagnostic criteria to incorporate biomarkers [68]. Working groups formed by the National Institute on Aging and the Alzheimer’s Association recently published guidelines for revised diagnostic criteria for AD and MCI that incorporate biomarkers (summarized in Box 2) [69,70]. To aid in research on the long preclinical phase of AD, these groups also proposed criteria for detecting preclinical AD in research participants who show little or no signs of cognitive impairment. They cautioned that in such asymptomatic individuals, the proposed criteria have no clinical or diagnostic utility at the present time [71]. For AD and MCI, however, these groups indicated that clinical incorporation of biomarkers can increase certainty that AD pathophysiology underlies the clinical syndrome, and aid in prognosis. The guidelines warn that widespread clinical use of biomarkers is premature given limited research to date in unselected patient populations and the need for further biomarker standardization and validation. However, with increasing clinical availability of biomarker tests, and informed patient populations, physicians are likely to face growing demand for such tests. Thus, it is timely and important to consider the potential harms as well as the potential benefits that may arise from biomarker use in clinical settings.
Box 2. Revised diagnostic criteria for Alzheimer’s disease and mild cognitive impairment.
Alzheimer’s disease
According to the revised criteria [69], the core clinical criteria for Alzheimer’s disease (AD) dementia will continue to be used for routine clinical diagnosis of probable and possible AD dementia. When deemed necessary by the clinician, and where tests are available, biomarker evidence can be used to determine the likelihood that AD pathophysiology underlies the clinical syndrome. The highest level of likelihood arises when both an amyloid and an injury biomarker is positive (although this does not rule out the possibility of a mixed dementia). AD etiology is considered to be of intermediate likelihood if only one class of biomarker evidence is positive, and the other is unavailable. When both classes of biomarkers are negative, dementia is unlikely to be due to AD. Biomarker evidence is considered uninformative on etiology if amyloid and injury biomarkers are in conflict.
Mild cognitive impairment
According to the new recommendations [70], the core clinical criteria for diagnosing mild cognitive impairment (MCI) include concern about change in cognitive function from the patient, an informant or a physician; objective evidence of impairment in one or more cognitive domains, but of insufficient severity to warrant a diagnosis of dementia; and maintained independence in functional abilities. Biomarkers to establish the likelihood that MCI is due to AD are currently recommended only for research studies and in academic medical settings, during to the dearth of research on use of biomarkers in typical clinical populations and the need for further standardization and validation. In these settings, biomarker evidence can be used to establish likelihood that MCI arises from AD pathophysiology, with three levels of certainly: MCI is highly likely to be due to AD when both classes of biomarker evidence are positive; MCI has intermediate likelihood of underlying AD when only one class of evidence is positive and the other is unavailable; and is unlikely to be due to AD when both classes are negative. Conflicting biomarker evidence is considered uninformative on etiology.
Amyloid biomarkers in clinical practice: potential for harm
Given the high prevalence of AD and its devastating effects, there is a lot of anxiety among older individuals about developing this disorder, especially among those with relatives with the disease. Thus, minor slips in memory function, including those that are normal in healthy aging, can become an obsession, generating a vicious cycle in which a patient notices a slip in memory, becomes more attuned to additional slips, and develops increasing anxiety about memory function, which itself may interfere with memory and memory testing. It is not uncommon to see cognitively unimpaired and, often, highly educated elderly patients presenting to the physician’s office debilitated by fear that they are developing dementia. In some cases, no amount of reassurance can assuage this fear, even when the patient is performing cognitively well above his or her peers.
Imagine, then, adding to this patient’s clinical evaluation an assessment for amyloid pathology, with the hope that the patient will be one of the approximate 35–80% (dependent on age [46]) of cognitively healthy older individuals with a negative test. A negative test would relieve the patient’s fear of AD, since an absence of amyloid is inconsistent with a diagnosis of AD. However, this would not rule out other neurodegenerative disorders. A positive test would be even harder to interpret, since 20–65% (dependent on age) of cognitively healthy individuals can be expected to test positive for amyloid [46].
Given that elevated amyloid deposition is thought to precede development of cognitive impairment by more than a decade, we believe that findings of amyloid positivity in the absence of objective cognitive impairment would be irrelevant, and possibly harmful to the well-being of the patient. Even if future research were to demonstrate that all healthy older individuals with elevated amyloid eventually develop AD, an amyloid test cannot yet tell whether the patient will decline in the coming year or even the coming decade; a positive test gives no indication of the phase of this slowly developing disease. For elderly patients especially, a warning sign loses all relevance if it can only suggest that cognitive impairment is likely to develop sometime in the next 10–20 years. Thus, knowing a cognitively intact patient’s amyloid status, in the absence of an indicator of neuronal injury, is not clinically helpful.
So what about the use of amyloid biomarkers in the setting of objective memory impairment? Some might argue that the presence of objective memory impairment concurrent with a positive amyloid test would provide sufficient evidence that the patient has entered the neurodegenerative phase of AD and is likely to progress to dementia within 1–5 years. Evidence of ‘insidiously progressive’ memory impairment would bolster the case, but could take more than a year to confirm. Although individuals with memory impairment and a positive amyloid test are at higher risk for developing dementia than those who test negative, amyloid testing in these patients also has potential for harm. Consider, for example, an elderly patient with objective memory impairment, but who also has depression, sleep problems or uses medications that interfere with memory. A positive amyloid test might lead the physician to mistakenly attribute the patient’s memory impairment to AD, minimizing attention to treatable causes of memory impairment. Beyond the psychological harm of being given a dire prognosis, the patient suffers from the diagnostic label in that care providers tend not to expect, thus tend not to strive for, full recovery of cognitive abilities in a patient with ‘AD’.
The high prevalence of amyloid positivity in the elderly and the long preclinical phase of the disease mean that a positive amyloid test in isolation, or even in the setting of objective memory impairment, which may arise from other causes, is insufficient to arrive at a diagnosis of AD. It indicates the presence of amyloid pathology but is uninformative on whether neuronal injury associated with AD underlies the cognitive complaint. Would a negative amyloid test be more informative in the setting of objective memory impairment? Such a finding would strongly suggest that the patient does not have AD and this may appropriately spur the search for treatable memory complaints. As discussed above, however, a negative amyloid test does not assure a benign prognosis. Synucleinopathies, tauopathies, and ubiquitinopathies confer equally devastating prognoses, so ruling out AD is not necessarily reassuring. Thus, even in patients with objective memory impairment, an amyloid test is insufficient to inform near-term prognosis and to guide clinical management.
An analogy to an existing, widely used clinical test, the fasting cholesterol test, may be helpful. Most agree that the cholesterol test is accurate and that high cholesterol is associated with cardiovascular disease. Similarly, biomarkers for amyloid are accurate and high cerebral amyloid is a hallmark of AD. The benefit of cholesterol-lowering therapies in preventing heart disease has been questioned, as has the (potential) benefit of amyloid-lowering therapies in preventing AD. The synthesis and removal of these disease-associated molecules are viable targets for risk reduction in their respective diseases, since each develop over decades before causing symptoms. Yet, when a patient presents to the hospital with chest pain, the information that guides near-term management is not the cholesterol level, but whether the heart tissue is being damaged. One would never use a cholesterol test to determine if the patient was in the midst of a heart attack. Instead, there are blood biomarkers for myocardial damage, creatinine kinase-MB and troponin, with which to rule-in cardiac damage.
Similarly, when a patient presents to the clinic with memory impairment, the operative information is whether or not the impairment is neuro-degenerative. If the etiology is not neurodegenerative, then diagnosis and treatment might be curative. If neurodegenerative, then finding that the patient’s neurodegeneration is due to AD and not other causes, though helpful in management and medication selection, currently has little effect on the patient’s prognosis. Thus, the most informative and useful biomarker in patients with cognitive complaints is one that detects neurodegeneration. Such a test may benefit from broadly capturing all neurodegenerative causes, rather than being selective for a particular disease. No blood test exists that mirrors the specificity of creatinine kinase-MB or troponin. CSF testing for tau protein remains a promising candidate, but, as of yet, no biofluid marker can fill this void. Direct visualization of brain structure coupled with quantification of atrophy in AD-vulnerable structures, such as the hippocampus, can provide this information.
Structural MRI in clinical practice
Structural MRI is already recommended for use in clinical assessment of older patients with cognitive impairment to rule out potentially treatable etiologies, such as tumors or hematomas. With minor modifications to the imaging protocol, appropriate images can be obtained to allow visual rating of degree of medial temporal lobe atrophy, or automated segmentation and quantification of medial temporal lobe structures. Visual rating scales, in which a ranking, usually between 0 and 4, is given to indicate degree of atrophy have proven useful for detecting atrophy in patients with cognitive impairment and for predicting development of dementia [72,73]. However, visual rating scales have lower reliability and lack the sensitivity of automated quantification methods [73,74].
Several software algorithms have been developed for use in research studies to automatically quantify hippocampal volume on structural MRIs [75–78]. For use in clinical settings, software must first receive regulatory approval. One method that has been cleared by the US FDA, and validated against manual segmentation [79,80] is NeuroQuant® (CorTechs, Labs, Inc, Ca, USA [101]). Similar to the widely used FreeSurfer software for research studies [75,81], NeuroQuant® uses a probabilistic atlas-based method to quantify volumes of whole brain, ventricles and several other brain structures, including the hippocampus. Unlike FreeSurfer, however, the method is integrated with clinical image archiving systems, is fully automated and generates quantitative reports within 10 min (an example report is presented in Figure 3). Automated algorithms are important in the clinical setting since they allow identical quantification procedures to be performed on large, publicly available imaging datasets acquired across all major scanner vendors to enable generation of normative databases for regional brain volumes across age, gender and intracranial volume with which to compare individual patient measurements.
Figure 3. Example NeuroQuant® report for one Alzheimer’s Disease Neuroimaging Initiative mild cognitive impairment patient.

This patient tested positive for cerebrospinal fluid Aβ42 and showed smaller hippocampal volumes and larger inferior lateral ventricle than expected for his age at baseline. Follow-up scans indicated progressive neurodegeneration. This patient was diagnosed with dementia at the 24-month follow-up visit.
Recommended clinical evaluation strategy for elderly patients with cognitive complaints
In clinical practice, assessment of the elderly patient with a cognitive complaint must begin with efforts to objectively confirm impaired cognitive function. A thorough history and brief cognitive testing performed during the clinic visit can help confirm whether the patient indeed has a cognitive problem. If no cognitive problem can be objectively confirmed through history, bedside testing or further neurocognitive testing, we believe that no biomarker studies should be pursued. Despite the high prevalence of AD in the elderly and the caveat that one can never rule out future development of AD, we feel that reassurance and patient education is the appropriate approach in cases where a cognitive complaint cannot be linked to clinical confirmation of a cognitive problem. Although there is some merit to the argument that objective biomarker testing might provide the greatest reassurance to the patient and serve as a baseline against which to compare future measures, we feel that the potential downside of biomarker testing in such cases, as discussed above, probably outweighs potential benefits.
If cognitive problems are confirmed, clinical judgment and the particular aspects of the case will guide the physician in the decision of whether to pursue additional biomarker testing. Assuming that further information is desired to guide management, we believe that the choice of biomarker to use in the next step is guided by the need to inform near-term prognosis by determining whether neurodegeneration underlies the cognitive complaint. Although a patient may have several potential causes for cognitive complaint, the presence of neurodegeneration strongly suggests a prognosis of near-term decline.
Despite the current lack of studies to date on prognostic ability of biomarkers in unselected clinical populations, results from research studies on highly selected MCI patient populations suggest that about half of MCI patients with medial temporal atrophy will develop dementia within approximately 18 months; and more than 80% will develop dementia within 3 years [60]. Thus, presence of medial temporal atrophy in a patient with objective memory impairment suggests a strong risk of developing dementia within 3 years. Similar risk of near-term decline might, in theory, be provided by FDG-PET, but to date, FDG-PET has only proven useful in distinguishing between neurodegenerative disorders, rather than detecting presence or absence of neurodegeneration. This is perhaps due to relative insensitivity to absolute changes in regional signal in the small medial temporal lobe structures where AD pathology first appears [82]. Additionally, head-to-head comparison of FDG-PET and volumetric MRI (vMRI) in detection of early AD favored vMRI [83]. Thus, we would recommend biomarker assessment of neurodegeneration using vMRI; comparing patient volumes of hippocampus, inferior lateral ventricle and lateral ventricle to age, sex and intracranial volume-adjusted normative values. If volumes show greater than expected neurodegeneration for age, the prognosis is one of near-term clinical decline.
Reduced hippocampal volume and ex vacuo expansion of the inferior lateral ventricle, often in the setting of normal lateral ventricle volumes, suggests focused medial temporal lobe atrophy. This would be consistent with AD but also with other neurodegenerative disorders such as frontotemporal dementia, dementia with Lewy bodies and hippocampal sclerosis, each associated with poor near-term prognosis. In such a case, pursuit and management of other possibly treatable causes should certainly continue in a rigorous and judicious manner, but the awareness of likely concurrent neurodegenerative illness serves to inform expectations and balance the risk/benefit ratio of aggressive pursuit of nondegenerative causes. Possibly overly aggressive approaches that might be avoided in this case include actions that may reduce patient comfort, such as changing or withholding potentially confounding medications that are otherwise beneficial. Even with evidence of neurodegeneration, the final diagnosis remains uncertain, just as it always has in clinical practice, although now prognosis and management is better informed.
Once evidence for a neurodegenerative condition has been established, if additional evidence of probable etiology is desired for the tailoring of symptomatic medications or for recommendation for enrollment in AD clinical trials, an amyloid test may be considered. Education of the patient and caregiver remains critical, and the provision of direct information and realistic expectations while acknowledging remaining uncertainty, and without extinguishing hope, continues as a physician art regardless of the availability of biomarkers.
The finding of normal brain volumes for age confers a better near-term prognosis and, while it does not rule out the possibility of future neurodegenerative disease, it can be used to guide clinical management while possibly providing increased hope to the patient, caregiver and physician. In neurodegenerative disorders, evidence suggests that by the time a cognitive complaint becomes clinically apparent, significant neural dystrophy and degeneration has already taken place. The relationship between hippocampal atrophy, as assessed by MRI, and memory has been extensively studied and there is strong support for the idea that by the time a memory complaint becomes clinically apparent in a neurodegenerative disorder, the medial temporal lobe shows evidence of neurodegeneration. Stated another way, if an elderly patient’s presenting complaint is memory impairment and the cause of the complaint is AD, it would be atypical, though not impossible, for that patient’s hippocampus and inferior lateral ventricle to be at the volume expected for healthy aging. The degree to which this supposition holds across individual patients and the level of deviation from the mean that is acceptable to define ‘normal’ for each brain structure remains to be determined, but the quantification nonetheless provides information that can guide management. Although there is always the possibility of a false negative or an atypical presentation of AD that does not involve medial temporal lobe atrophy in the early stages, the physician noting healthy hippocampal and inferioral lateral ventricle volumes in a patient previously suspected of having neurodegenerative disease might redouble efforts to find another cause for the cognitive complaints. Such a search might lead to successful identification of a treatable cause and subsequent patient recovery. The addition of amyloid testing in such a case is irrelevant. If there is no atrophy, neurodegeneration is taken off of the table as a potential cause. A negative amyloid result would not change management, and a positive amyloid result might only lead to misattribution of the complaint to AD.
The entire picture changes, however, should a therapy be shown to alter the course of AD through removal of amyloid. In such a case, the determination of a patient’s amyloid status will become important for risk–benefit assessment. Depending on the severity of the therapy’s adverse effects, amyloid testing might be reserved for those with objective evidence supporting high risk for near-term clinical decline. That is, given that a large proportion of amyloid-positive patients will die of other causes before developing cognitive symptoms, the risk–benefit ratio might favor treating only those shown to be in the neurodegenerative phase of the disease by clinical progression or vMRI (but not by simple existence of a new or stable memory impairment). Alternatively, if the therapy is associated with minimal side-effects, treatment might be instantiated at the first sign of amyloid positivity, similar to treating high cholesterol in the absence of cardiac symptoms. The flowchart depicted in Figure 4 summarizes the above clinical scenarios under the assumption that amyloid removal agents will not prove sufficiently benign to be used in patients without evidence supporting a prognosis of near-term decline. It is important to note that therapies targeted at other factors, such as tau, or lifestyle strategies aimed at preventing AD are also under investigation. Whether the availability of such treatments would alter the recommended evaluation strategy will depend on their specificity for AD.
Figure 4. Recommended decision tree for clinical evaluation of the elderly patient with cognitive complaints.

AD: Alzheimer’s disease; DLB: Dementia with Lewy bodies; FTD: Frontotemporal dementia; HS: Hippocampal sclerosis; Rx: Clinical treatment; vMRI: Volumetric MRI.
Conclusion
With the growing clinical availability of bio-markers for the detection of AD pathology and neural injury, physicians have access to new tests to aid in diagnosis and prognosis in elderly patients presenting with memory complaints. Research has shown that amyloid pathology can be present in older individuals without cognitive problems, and may precede cognitive impairment by a decade or more. In contrast, presence of brain atrophy detectable with vMRI indicates a rapid course of decline. Thus, after confirming the presence of a cognitive disorder, a physician who desires additional prognostic information is advised to order a biomarker test that can indicate the presence of neuronal degeneration (Figure 4). If neurodegeneration is present, the prognosis is probably one of near-term decline to dementia, although the underlying etiology may be uncertain. By contrast, amyloid bio-markers can confirm presence of AD pathology, but cannot indicate when or whether that pathology will lead to clinical decline. Thus, these tests are currently only recommended if evidence of progressive decline or neurodegeneration has been obtained, to aid in specificity of the neurodegenerative diagnosis.
Future perspective
Our knowledge of AD and its long preclinical phase is rapidly evolving. With the looming AD epidemic that threatens the financial security of most societies, a wide array of research efforts are underway to better understand the pathophysiological basis of this disorder and to develop treatments. To date, clinical trials of amyloid-modifying treatments have failed to prevent clinical decline in AD patients even when amyloid levels were successfully reduced [84]. Although some interpret these findings as evidence against the amyloid hypothesis of AD pathogenesis, others argue that such results would be expected if the cascade of AD neurodegeneration, triggered by amyloid, becomes independent of it once initiated [84]. There is much evidence to support the view that the neurodegenerative process may be (or may become) independent of amyloid, and this raises the possibility that treatments that target other factors related to the development of AD may prove useful in preventing or slowing AD progression. The development of therapies that target nonamyloid factors may result in reduced emphasis on amyloid biomarkers in the future. Alternatively, amyloid-modifying agents may be successful at preventing AD if administered early, after the appearance of amyloid but prior to the development of neuronal injury and cognitive decline. If the enormous challenges involved in demonstrating such a beneficial effect can be overcome [85], amyloid biomarkers may be recommended as part of a routine, preventative treatment strategy. It is possible, however, that secondary prevention of AD based on amyloid positivity may also be too late to be effective. This would necessitate the development of even earlier disease biomarkers. Such real concerns highlight the monumental challenges that have thus far stymied all efforts to halt the pathological progression of AD. Finding a cure may well require an ambitious commitment by society at a level unprecedented in recent history, but necessitated by the impact the increasing prevalence of AD will have in coming years. Unfortunately, governmental leadership and funding for such transformative programs in science and medicine has, of late, been missing. In the meantime, an organized and evidence-based approach to incorporating biomarkers in clinical practice should remain focused on the goal of alleviating the suffering conferred by this devastating illness, both by the disease itself and by the dread it strikes in the elderly who note memory decline, using the greater accuracy in near-term prognosis provided by judicious incorporation of biomarkers to optimize treatment and tailor information provided to patients and their families.
Executive summary.
The changing clinical landscape
With the aging of the population, physicians will see a growing number of older patients presenting with memory or other cognitive complaints.
Cognitive problems can arise from many etiologies, including currently incurable neurodegenerative disorders, the most common of which is Alzheimer’s disease (AD), as well as treatable, nondegenerative disorders.
AD clinical expression & neuropathology
Amyloid pathology and neurofibrillary tangles are the two hallmark features of AD.
Amyloid pathology may be present a decade or more prior to the onset of clinical symptoms, whereas neurofibrillary tangle pathology, which leads to neuronal injury and death, is more closely associated with clinical symptoms.
Research results on biomarkers of AD
Biomarkers have been developed that indicate the presence of brain amyloid pathology or neuronal injury.
In individuals with mild cognitive impairment, amyloid and injury biomarkers are each associated with elevated risk of decline to dementia, although injury biomarkers, such as medial temporal atrophy, are associated with a higher risk of rapid decline.
Amyloid biomarkers are positive in a substantial portion of cognitively healthy older adults and it is currently unknown whether such individuals will eventually develop AD or whether some may be resistant to amyloid.
Revised research and clinical criteria for diagnosis of AD & mild cognitive impairment
Recently proposed diagnostic criteria recommend the inclusion of biomarkers to increase certainty that AD pathophysiology underlies the clinical syndrome and to aid in prognosis of individuals with cognitive impairment. However, the authors of the new criteria warn that widespread clinical use is premature given limited research to date in unselected patient populations and the need for further biomarker standardization and validation.
Although not yet recommended for routine clinical use, physicians are likely to face requests for biomarker tests from informed patient populations, particularly now that an amyloid imaging agent has received regulatory approval for clinical use.
Amyloid biomarkers in clinical practice: potential for harm
Because amyloid pathology may be present for a decade or more prior to the onset of clinical symptoms, a positive amyloid test in the absence of an injury biomarker does not inform prognosis and may lead to an erroneous diagnosis of AD, causing physicians to miss potentially treatable causes of cognitive dysfunction.
Structural MRI in clinical practice
Visual rating scales and fully automated volumetric methods applied to structural MRIs can indicate the presence of medial temporal lobe atrophy, which is predictive of a rapid course of clinical decline.
Recommended clinical evaluation strategy for elderly patients with cognitive complaints
Evaluation of patients with cognitive complaints should begin with verification of objective cognitive impairment. In the absence of such impairment, no biomarker tests are recommended.
In the presence of cognitive impairment, MRI measures of brain atrophy are recommended to help determine whether cognitive complaints arise from a neurodegenerative disorder, which would indicate a prognosis of near-term clinical decline to dementia.
Amyloid biomarkers may be useful once presence of a neurodegenerative disorder is established to tailor treatment, particularly if effective amyloid-modifying treatments become available.
Conclusion
Amyloid biomarkers can confirm presence of AD pathology but cannot indicate when or whether that pathology will lead to clinical decline. Thus, these tests should only be performed when evidence suggests progressive impairment or neurodegeneration.
Future perspective
Although there have been enormous advances in knowledge of biomarkers associated with AD, much research is still needed to determine their predictive ability in the preclinical stage of the disease.
There are monumental challenges involved in the development and testing of treatments to prevent or slow the progression of AD; finding a cure is likely to require an ambitious and unprecedented commitment by society.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
Supported by NIA K01AG029218 (LK McEvoy) and NINDS K02 NS067427, NIA U01 AG10483, P50 AG005131, R01 AG034062 (JB Brewer). LK McEvoy’s spouse is president of, and holds stock and stock options in, CorTechs Labs, Inc. JB Brewer is an investigator for, and receives research funds from Janssen Alzheimer Immunotherapy; he also receives research funds from the General Electric Medical Foundation, holds stock options in CorTechs Labs, Inc. and has served on advisory boards for Elan, Avanir and Lilly Biomarker Business Unit. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪ ▪ of considerable interest
- 1▪.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. First study to report imaging of amyloid in the living human brain. [DOI] [PubMed] [Google Scholar]
- 2.Rowe CC, Ackerman U, Browne W, et al. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94–9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008;7(2):129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- 3.Wong DF, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18) J Nucl Med. 2010;51(6):913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herholz K, Ebmeier K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011;10(7):667–670. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- 5.Department of Economic and Social Affairs. World population ageing. United Nations Publications; UN, NY, USA: 2001. Population Division. [Google Scholar]
- 6.Vincent GK, Velkoff VA. The next four decades: the older population in the United States: 2010 to 2050. US Department of Commerce, Economics and Statistics Administration, US Census Bureau; 2010. Current population reports. [Google Scholar]
- 7.Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 8.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 9.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 11.The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(4 Suppl):S1–S2. [PubMed] [Google Scholar]
- 12.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 13.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30(7):1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360(22):2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- 15.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17(9):1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41(1):17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 20.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 22.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 23.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrup K. Reimagining Alzheimer’s disease – an age-based hypothesis. J Neurosci. 2010;30(50):16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-β in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321(3):823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 26.Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans. 2011;39(4):886–890. doi: 10.1042/BST0390886. [DOI] [PubMed] [Google Scholar]
- 27.Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ittner LM, Gotz J. Amyloid-β and tau – a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2010;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 29.Desikan RS, McEvoy LK, Thompson WK, et al. Amyloid-β associated volume loss occurs only in the presence of phospho-tau. Ann Neurol. 2011;70(4):657–661. doi: 10.1002/ana.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitwell JL, Josephs KA, Murray ME, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71(10):743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimers Dement. 2012;8(1 Suppl):S1–S68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klunk WE. Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging. 2011;32(Suppl 1):S20–S36. doi: 10.1016/j.neurobiolaging.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1(8–9):371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimmer T, Riemenschneider M, Forstl H, et al. β amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. EMBO Mol Med. 2009;65(11):927–934. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol. 2006;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 36.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA. 2011;305(3):275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leinonen V, Alafuzoff I, Aalto S, et al. Assessment of β-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh compound B. Arch Neurol. 2008;65(10):1304–1309. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- 38.Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar β-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68(2):232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008;4(1):38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowe CC, Ng S, Ackermann U, et al. Imaging β-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 42.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68(11):1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 43.Resnick SM, Sojkova J, Zhou Y, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-β measured by [11C] PiB. Neurology. 2010;74(10):807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Aβ (42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65(2):176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagust WJ, Bandy D, Chen K, et al. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46▪▪.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31(8):1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. Documented that the proportion of cognitively healthy older adults testing positive for amyloid pathology increases strongly with advancing age. In 177 healthy older adults, 18% of healthy individuals aged 60–69 years showed elevated amyloid deposition with Pittsburgh compound B imaging, whereas 65% of those over 80 years showed elevated amyloid deposition. [DOI] [PubMed] [Google Scholar]
- 47.Fjell AM, Walhovd KB, Fennema-Notestine C, et al. Brain atrophy in healthy aging is related to CSF levels of Aβ1–42. Cereb Cortex. 2010;20(9):2069–2079. doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker JA, Hedden T, Carmasin J, et al. Amyloid-β associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69(6):1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chetelat G, Villemagne VL, Villain N, et al. Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology. 2012;78(7):477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- 50.Desikan RS, McEvoy LK, Thompson WK, et al. Amyloid-b-associated clinical decline occurs only in the presence of elevated P-tau. Arch Neurol. 2012 doi: 10.1001/archneurol. 2011.3354. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodrigue KM, Kennedy KM, Devous MD, Sr, et al. β-amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78(6):387–395. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stern Y. Cognitive reserve. Neuropsychologia. 2009;47(10):2015–2028. doi: 10.1016/j.neuropsychologia.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kramer PL, Xu H, Woltjer RL, et al. Alzheimer disease pathology in cognitively healthy elderly: a genome-wide study. Neurobiol Aging. 2011;32(12):2113–2122. doi: 10.1016/j.neurobiolaging.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66(12):1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okello A, Koivunen J, Edison P, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology. 2009;73(10):754–760. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jack CR, Jr, Wiste HJ, Vemuri P, et al. Brain β-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain. 2010;133(11):3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302(4):385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 59.Vemuri P, Wiste HJ, Weigand SD, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology. 2009;73(4):294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60▪▪.Heister D, Brewer JB, Magda S, Blennow K, McEvoy LK. Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology. 2011;77(17):1619–1628. doi: 10.1212/WNL.0b013e3182343314. This study compared the relative ability of amyloid and injury biomarkers, separately and together, for predicting risk of dementia. Although the presence of any biomarker was associated with increased risk, presence of atrophy predicted a faster rate of decline. Joint presence of atrophy and cerebrospinal fluid biomarkers improved risk prediction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Rossum IA, Visser PJ, Knol DL, et al. Injury markers but not amyloid markers are associated with rapid progression from mild cognitive impairment to dementia in Alzheimer’s disease. J Alzheimers Dis. 2012;29(2):319–327. doi: 10.3233/JAD-2011-111694. [DOI] [PubMed] [Google Scholar]
- 62.Fennema-Notestine C, Hagler DJ, Jr, McEvoy LK, et al. Structural MRI biomarkers for preclinical and mild Alzheimer’s disease. Hum Brain Mapp. 2009;30(10):3238–3253. doi: 10.1002/hbm.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vemuri P, Wiste HJ, Weigand SD, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology. 2009;73(4):287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McEvoy LK, Fennema-Notestine C, Roddey JC, et al. Alzheimer disease: quantitative structural neuroimaging for detection and prediction of clinical and structural changes in mild cognitive impairment. Radiology. 2009;251(1):195–205. doi: 10.1148/radiol.2511080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Westman E, Cavallin L, Muehlboeck JS, et al. Sensitivity and specificity of medial temporal lobe visual ratings and multivariate regional MRI classification in Alzheimer’s disease. PLoS ONE. 2011;6(7):e22506. doi: 10.1371/journal.pone.0022506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McEvoy LK, Holland D, Hagler DJ, Jr, Fennema-Notestine C, Brewer JB, Dale AM. Mild cognitive impairment: baseline and longitudinal structural MR imaging measures improve predictive prognosis. Radiology. 2011;259(3):834–843. doi: 10.1148/radiol.11101975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 68▪▪.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. Influential paper that describes the rationale for revising the diagnostic criteria of mild cognitive impairment and Alzheimer’s disease to incorporate biomarkers. [DOI] [PubMed] [Google Scholar]
- 69▪.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. Describes the revised recommendations on the incorporation of biomarkers into the clinical diagnosis of Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70▪▪.Albert MS, Dekosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging and Alzheimer’s Association workgroup. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. Describes the revised recommendations on the incorporation of biomarkers into the diagnosis of mild cognitive impairment. It provides a nice overview of biomarker studies and identifies several areas for future research to improve understanding of the potential clinical utility of these biomarkers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71▪.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. Proposes objective criteria for identifying individuals in a preclinical stage of Alzheimer’s disease to aid research on the sequence of events that occur during the long asymptomatic phase of the disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeCarli C, Frisoni GB, Clark CM, et al. Qualitative estimates of medial temporal atrophy as a predictor of progression from mild cognitive impairment to dementia. Arch Neurol. 2007;64(1):108–115. doi: 10.1001/archneur.64.1.108. [DOI] [PubMed] [Google Scholar]
- 73.Shen Q, Loewenstein DA, Potter E, et al. Volumetric and visual rating of magnetic resonance imaging scans in the diagnosis of amnestic mild cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 2011;7(4):e101–e108. doi: 10.1016/j.jalz.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jack CR, Jr, Barkhof F, Bernstein MA, et al. Steps to standardization and validation of hippocampal volumetry as a biomarker in clinical trials and diagnostic criterion for Alzheimer’s disease. Alzheimers Dement. 2011;7(4):474–485.e474. doi: 10.1016/j.jalz.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fischl B, Van Der Kouwe A, Destrieux C, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14(1):11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 76.Morra JH, Tu Z, Apostolova LG, et al. Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Hum Brain Mapp. 2009;30(9):2766–2788. doi: 10.1002/hbm.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barnes J, Foster J, Boyes RG, et al. A comparison of methods for the automated calculation of volumes and atrophy rates in the hippocampus. Neuroimage. 2008;40(4):1655–1671. doi: 10.1016/j.neuroimage.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 78.Chupin M, Gerardin E, Cuingnet R, et al. Fully automatic hippocampus segmentation and classification in Alzheimer’s disease and mild cognitive impairment applied on data from ADNI. Hippocampus. 2009;19(6):579–587. doi: 10.1002/hipo.20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brewer JB, Magda S, Airriess C, Smith ME. Fully-automated quantification of regional brain volumes for improved detection of focal atrophy in Alzheimer disease. Am J Neuroradiol. 2009;30(3):578–580. doi: 10.3174/ajnr.A1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brewer JB. Fully-automated volumetric MRI with normative ranges: translation to clinical practice. Behav Neurol. 2009;21(1):21–28. doi: 10.3233/BEN-2009-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 82.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. 2005;32(4):486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 83.Karow DS, McEvoy LK, Fennema-Notestine C, et al. Relative capability of MR Imaging and FDG PET to depict changes associated with prodromal and early Alzheimer disease. Radiology. 2010;256(3):932–942. doi: 10.1148/radiol.10091402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Golde TE, Schneider LS, Koo EH. Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69(2):203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85▪▪.Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011;3(111):111cm133. doi: 10.1126/scitranslmed.3002609. Describes the daunting challenges involved in demonstrating efficacy of potential Alzheimer’s disease treatments in clinical trials. It points to the need for decades-long cohort studies of aging to elucidate the trajectories and relations among various Alzheimer’s disease biomarkers, and the challenges associated with testing potential secondary preventative therapies in asymptomatic adults at risk for Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Strozyk D, Blennow K, White LR, Launer LJ. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60(4):652–656. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- 87.Buerger K, Ewers M, Pirttila T, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129(Pt 11):3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 88.McEvoy LK, Brewer JB. Quantitative structural MRI for early detection of Alzheimer’s disease. Expert Rev Neurother. 2010;10(11):1675–1688. doi: 10.1586/ern.10.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.NeuroQuant®. www.cortechslabs.com.