

Abstract
Arenaviruses are rodent-borne emerging human pathogens. Diseases caused by these viruses, e.g., Lassa fever (LF) in West Africa and South American hemorrhagic fevers (HFs), are serious public health problems in endemic areas. We have employed replication-competent and replication-deficient strategies to design vaccine candidates potentially targeting different groups “at risk”. Our leader LF vaccine candidate, the live reassortant vaccine ML29, is safe and efficacious in all tested animal models including non-human primates. In this study we showed that treatment of fatally infected animals with ML29 two days after Lassa virus (LASV) challenge protected 80% of the treated animals. In endemic areas, where most of the target population is poor and many live far from health care facilities, a single-dose vaccination with ML29 would be ideal solution. Once there is an outbreak, a fast-acting vaccine or post-exposure prophylaxis would be best. The 2nd vaccine technology is based on Yellow Fever (YF) 17D vaccine. We designed YF17D-based recombinant viruses expressing LASV glycoproteins (GP) and showed protective efficacy of these recombinants. In the current study we developed a novel technology to clone LASV nucleocapsid within YF17D C gene. Low immunogenicity and stability of foreign inserts must be addressed to design successful LASV/YFV bivalent vaccines to control LF and YF in overlapping endemic areas of West Africa. The 3rd platform is based on the new generation of alphavirus replicon virus-like-particle vectors (VLPV). Using this technology we designed VLPV expressing LASV GP with enhanced immunogenicity and bivalent VLPV expressing cross-reactive GP of Junin virus (JUNV) and Machupo virus (MACV), causative agents of Argentinian and Bolivian HF, respectively. A prime-boost regimen required for VLPV immunization might be practical for medical providers, military, lab personnel, and visitors in endemic areas.
Keywords: Lassa, Junin, Macupo, Arenaviruses, Vaccines, YF17D vector, VEEV TC-83-based replicons
Introduction
Arenaviruses represent a fast-growing group of rodent-borne viruses which are an example of how environmental changes disrupt the natural animal virus-host balance and result in unexpected diseases. Amongst more than two dozen arenaviruses discovered so far, six viruses cause diseases, mostly HFs, in humans: LASV, the Old World arenavirus, causes LF in West Africa; JUNV, MACV, GUAV (Guanarito), and SABV (Sabia) viruses from clade B of the New World arenavirus group cause South American HFs [1]. Lymphocytic choriomeningitis virus (LCMV), the prototype arenavirus, is distributed worldwide and can cause neurological disease in adults, brain malformation in newborns, and a LF-like fatal disease in immunocompromized individuals [2,3]. The recently discovered the NW arenavirus, Chapare virus (CHAV), was associated with fatal HF in Bolivia [4] and Lujo virus (LUJV), a new member of the OW group, caused an extremely severe HF outbreak in Southern Africa [5,6].
Several new viruses recently isolated in Africa rapidly expended the OW group which until recently has included non-pathogenic viruses Mopeia (MOPV), Morogo (MORV), Mobala (MOBV), Ippy (IPPYV) [1]. In addition, Kodoko virus (KODV), which is related to but distinct from LCMV was isolated in Guinea [7]. Screenings of M. natalensis trapped in Zimbabwe, where the first patient infected with LUJV was identified, resulted in the isolation of Luna virus (LUNV) genetically related to MOBV [8]. Novel tentative African arenaviruses also include Merino Walk virus (MWV) isolated from a rodent, Myotomys unisulcatus, collected at Eastern Cape, South Africa [9] and Menekre (MENV) and Gbagroube (GBAV) viruses, those sequences were detected in Hylomyscus sp. and Mus (Nannomys) setulosus, respectively [10]. Notably, GBAV sequence was closely related to LASV, while MENV sequence clustered with IPPYV-MOBV-MOPV. Detection of LASV-like sequences in Mus setulosus suggests that co-evolution of African arenaviruses and their hosts can potentially include host-switching events predicting isolation of novel arenavirus species in the future. Interestingly, a group of novel viruses distantly related to arenaviruses but also to filoviruses, was isolated from snakes with fatal IBD (inclusion body disease) [11]. Isolation of arenaviruses from non-mammal hosts indicates that these viruses can infect very broad range of species with unpredictable pathogenic potential for humans.
Highly pathogenic arenaviruses are dominant viral species among NIAID/CDC Category A Priority Pathogens. With the exception of LCMV, each individual arenavirus species is found in a localized area in Africa or in North/South America. However, these viruses pose a serious biological threat to public health in non-endemic areas as well, and the development of effective prevention strategies against infections caused by highly pathogenic arenaviruses is the NIH/NIAID priority. This manuscript is dealing with the most prevalent African arenavirus, LASV, which infects several hundred thousand individuals and kills thousands of them annually, and with two of the most prevalent South American arenaviruses, JUNV and MACV, causative agents of Argentine and Bolivian HF, respectively. In efforts to design experimental vaccines to effectively control these viruses we are adopting three vaccine platfors: (i) reassortant technology [12–14]; (ii) yellow fever YF17D-vectored vaccines [15,16]; and (iii) alphavirus-replicon technology [17]. In this paper, we have provided evidences of post-exposure activity of our leader vaccine candidate against LF, the reassortant ML29; we extended application of YFV17D-based strategy to clone LASV nucleocapsid (NP) in this vector; and we designed alphavirus replicon particles expressing genetically modified LASV GPC with cross-presenting potential and bivalent JUNV/MACV vaccine using advanced VEEV TC-83-based replicon strategy.
Materials and Methods
Post exposure activity of reassortant ML29 in guinea pigs
Josiah/SL strain of LASV was obtained from Centers for Disease Control and prevention (Atlanta, GA). A MOP/LAS reassortant (clone ML29) was previously described [12,18,19]. All work with infectious samples was performed within the maximum bio-containment (BSL-4) laboratory at the Texas Biomedical Research Institute, San Antonio, Texas. The viruses were grown in Vero E6 cells cultured in Dulbecco’s modified minimum Eagle’s medium (DMEM, GIBCO-BRL) with 2% fetal calf serum (FCS, GIBCO-BRL), 1% penicillin-streptomycin, and L-glutamine (2 mM) at 37°C in 5% CO2 [12]. Strain 13 guinea pigs (300–500 g, female) were purchased from USAMRIID (Fort Detrick, Frederick, MD). Animals from two experimental groups (5 animals per group) were infected with LASV, 1×103 PFU (1LD50=0.3 PFU), subcutaneously (s.c.) and treated with ML29 (1×103 PFU, s.c.) on day 2 and day 4 after LASV infection. Two control groups included: (i) animals vaccinated with clone ML29, 1×103 PFU, s.c., and challenged on day 30 with 1×103 PFU of LASV (vaccine control group); and (ii) animals inoculated with PBS and challenged with LASV (challenge control group). All animals were observed twice daily for clinical manifestations according to an approved scoring sheet (decreased activity, ruffled fur, loss of weight, labored breathing, hunched posture). Death or survival past 21 days was defined as an endpoint [17,20]. Albumin and AST levels in plasma were measured using the Premiere Plus photometer system (Stanbio Laboratory, Inc., San Antonio, TX). Viremia and viral load in tissues were measured by quantitative RT-PCR (qRT-PCR) as previously described [14].
Construction of YF17D/LASV recombinant viruses
Cloning of LASV glycoproteins in YF17D was previously described [15,16]. In order to simplify the construction of YFV17D/LASV-NP recombinants, a new vector was designed (Figure 3). This vector contains several adaptations to allow proper expression of the heterologous insert, while avoiding nucleotide sequence duplication to minimize recombination. The authentic translation initiation codon has been inactivated, resulting in the use of the second AUG codon that is in the same reading frame as the original AUG. This deletion has no significant effect on YF17D replication and virus production (P. Bredenbeek and C. Rice; unpublished). The second AUG codon and two nucleotides from Val codon are located at the 3′ end of a sequence called 5′-CS which is involved in a long range RNA interaction with CS1 at 3′ UTR of YFV. This interaction is critical for replication and was therefore left unchanged. Immediately downstream of the Met and Val codons, a short multiple cloning site (MCS) was designed for the inframe insertion of LASV NP fragments using a shuttle plasmid. The 3X FLAG epitope was added downstream of the MCS to facilitate detection of expressed proteins. The 3X FLAG epitope sequence was fused with the FMDV 2A protein to secure cleavage between the heterologous insert and the YFV17D C protein. As a result, the expressed YFV17D C contained an additional NH2-terminal proline required for the FMDV 2A protein “cleavage”. In vitro RNA transcription, electroporation, preparation of virus stocks, plaque assay, immunofluorescence, and western blot were previously described [15,16].
Figure 3. Cloning strategy of LASV NP in YFV17D vector.
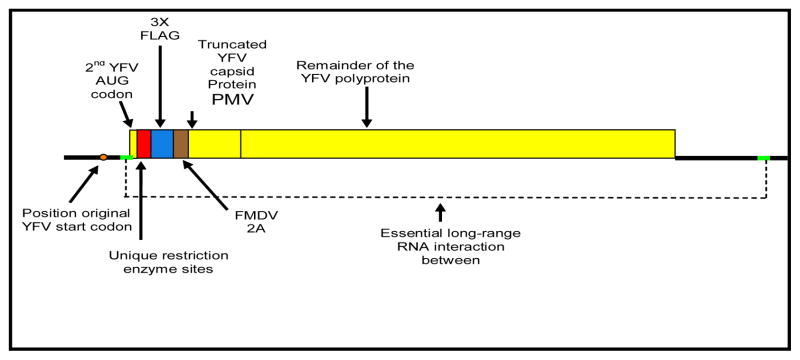
LASV NP fragments were inserted using the Acc65I and Sal I sites in the polylinker sequence (red box). The original YF17D translation initiation codon has been inactivated. Translation of the recombinant polyprotein starts at the second AUG codon. The NH2-terminus of the YF17D C protein starts with Pro-Met-Val (PMV). Proteins encoding DNA fragments that are cloned in the correct ORF relative to the remainder of the YF17D polyprotein will be fused to the 3X Flag epitope (blue box) to allow easy identification. Brown box represents the so-called FMDV 2A cleavage site.
VEEV TC-83-based replicon vaccines
To make TC-83-based VLPV expressing metabolically stable LASV GP (Josiah, lineage IV, acc. no. AY628203) SKI-1/S1P cleavage site was disrupted by introducing mutation R256A, transmembrane and cytoplasmic domains were deleted, and truncated GP was fused at position P427 with the phage T4-derived fibritin trimeric motif GYIPEAPRDGQAYVRKDGEWVLLSTFL [21]. An eight-glycine linker was introduced between truncated GP and the fibritin motif to provide structural flexibility. The final sequence, named ΔLGPfib, was assembled and human codon optimized. The LASV Josiah GP wildtype (LGPwt) and ΔLGPfibc DNAs were cloned into HindIII-BamHI sites within a ClaI-flanked polylinker in the shuttle vector, and sub-cloned as ClaI fragments into the ClaI site of the VEEV TC-83 replicon clones. For production of VLPV [17,22], RNA mixture containing 1 μg of each RNA (ΔLGPfib vector, E1 & E2-helper, and C-helper) were co-transfected into CHO-K1 cells. Transfected cells were transferred into 75 cm2 flasks and incubated at 37°C for VLPV harvesting.
In attempts to design bivalent JUNV/MACV VLPV the synthetic GPC genes of JUNV Candid #1 (Genbank: AY746353) and MACV Carvallo (GenBank: AY619643.1) were assembled from synthetic oligonucleotides and/or PCR products and fragments were cloned into pGA4 using KpnI and SacI restriction sites or into pcDNA3.1 using HindIII and XhoI restriction sites, respectively. The codon usage was adapted to the codon bias of Homo sapiens genes. In addition, regions of very high (>80%) or very low (<30%) GC content and undesirable cis-acting sequence motifs (e.g., internal TATA-boxes, chi-sites and ribosomal entry sites, AT-rich or GC-rich sequence stretches, and etc.) have been avoided where possible. The final constructs were verified by sequencing and cloned at the ClaI site of the VEEV TC-83 replicon. Immunofluorescence and Western blot were performed as previously described [15,16].
Results and Discussion
There are several reasons to justify a replication-competent, “live” vaccine as an attractive approach to control LF: (i) cell-mediated immunity (CMI) plays the major role in LF patient recovery and in protection; (ii) a live vaccine provides the most effective natural pathway to process and present protective antigens to MHC molecules; (iii) epidemiological observations in LF endemic areas of West Africa provide evidence that a single (survived) exposure will induce long-term protection against disease [23]; (iv) a vaccine candidate formulated to contain both LASV NP and GP antigens will induce a broad cross-reactivity and a large pool of CD4+ memory T cells against all phylogenetic groups of LASV [12,24,25]; (v) a single-shot immunization approach is a crucial for population of remote rural areas of West Africa, which has a very limited medical infrastructure and where implementation of prime-boosting immunization is not practical [26–28]. We are currently developing two live-attenuated platforms. One of them based on reassortant technology and the second one employs genetic backbone of live-attenuated vaccine YF17D, the most efficacious and safe vaccine ever developed [29].
Post-exposure application of live reassortant vaccine ML29 protects experimental animals against fatal Lassa fever
Reassortant technology is currently used to manufacture licensed live attenuated influenza (FluMist®, MedImmune, Inc.) and rotavirus(Rotarix®, GSK Biologicals; Rotateq®, Merck & Co., Inc.) vaccines. The bi-segmented feature of the arenavirus RNA genome [30] allows to produce reassortants during co-replication in vitro. The MOP/LAS reassortant, clone ML29 [30,19], was designed to keep the attenuated profile of MOPV, but to induce a robust CMI responses against LASV from different phylogenetic groups. Clone ML29 was selected from a library of MOPV/LASV reassortants and encodes the major antigens (NP and GPC) of LASV, and the L protein (RdRp, RNA-dependent RNA polymerase) and zinc-binding (Z) protein of MOPV [18]. Since clone ML29 has 70% of MOPV genome, it has the same classification as the vector virus, MOPV. MOPV is considered risk group 2 by the EU biosafety regulation and risk group 3 by the CDC. Eighteen mutations distinguish the ML29 genome from the parental arenaviruses and likely additionally contributed to the attenuated phenotype [12,19]. In all available animal models, including NHPs (rhesus macaques and marmosets), ML29 was safe, highly immunogenic, and induced cross-reactive sterilizing CMI responses fully protected animals against fatal LF [12–14].
Several examples of natural reassortment between genetically-related bunyaviruses raised the concern that vaccination with an attenuated reassortant vaccine in endemic areas will affect the natural pattern of the disease and enhance pathogenicity. To address this concern, challenge and vaccine application was performed simultaneously on day 0. We showed [14] that all animals vaccinated with ML29 survived after simultaneous challenge with high doses of LASV-Josiah. Importantly, the vaccination also protected 60% animals from challenge with high dose of the distantly-related strain, LASV-803213/NIG. These observations clearly indicate that co-replication in vivo of wild-type LASV and ML29 results in attenuated infection. These observations also justified experiments to test post-exposure activity of the ML29.
Two groups of five strain 13 guinea pigs were challenged with LASV-Josiah (>3,000 LD50/animal) on day 0 and then treated with ML29 reassortant vaccine 2 days and 4 days post infection. Two control groups included a non-vaccinated challenge control and a positive vaccination control (vaccination with ML29 and challenge on day 30). In non-vaccinated group all animals died with typical signs of a fatal LF disease (Figure 1). All LASV-infected guinea pigs that were treated with ML29 four days after challenge also became febrile, developed clinical (fever, weight loss) and biochemical (low levels of albumin and high levels of ALT in plasma) signs of fatal LF disease and were euthanized. In experimental group treated 2 days after LASV exposure, animals showed some signs of illness but 4 from 5 treated animals recovered and survived to project end, 35 days after challenge. The one animal that succumbed to disease in this group had a greater than 7 day delayed time to death compared to the non-vaccinated control.
Figure 1. Post-challenge ML29 vaccination.

Two groups of strain 13 guinea pigs (5 animals/group) were challenged with LASV-Josiah (1000 PFU) and 2 and 4 days after the challenge animals received ML29 (1,000 PFU). Control groups received PBS (challenge control). Survival (A), weight loss (B), temperature (C), plasma levels of albumin (D) and ALT (insertion in panel D) were monitored. Survived animals were necropsied at the end of the experiment, on day 36 after LASV challenge.
Detection of viral load by qRT/PCR showed that in the non-vaccinated group and in animals treated with ML29 four days after LASV challenge, viral loads in all tested tissues and in plasma (not shown) were more than 10,000 copies/μg and especially high in target organs, spleen, LN, lungs (Figure 2). In good confirmation with our previous results [14], a preventive ML29 vaccination resulted in sterilizing immunity effectively control viral loads in tissues (Figure 2B). Treatment of LASV-exposed animals with ML29 two days after exposure significantly reduced levels of LASV replication, lower than crucial death/survival threshold, 10,000 copies/μg [31].
Figure 2. Viral RNA load in tissues.
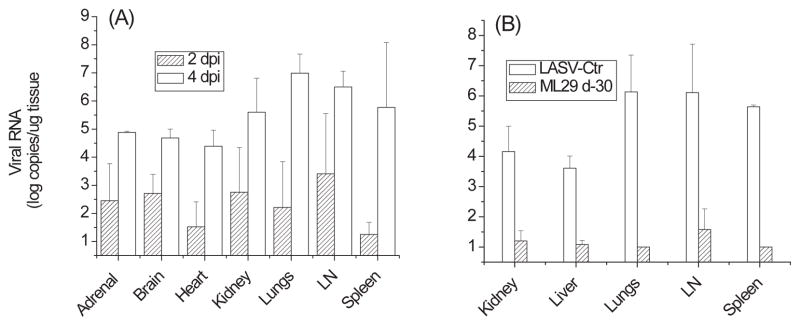
Tissue samples from animal groups described in figure 1 and from a vaccine control group (immunization with ML29 and challenge on day 30 with LASV, see Materials and Methods) were homogenized using the TissueLyser (Qiagen) RNA was extracted following the Trizol Reagent protocol (Invitrogen). Real-time qRT-PCR was performed using an ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA) and RNA Ultrasense one-step real-time qRT-PCR system (Invitrogen) according to manufacturer’s recommendations. The primer/probe set for LASV-Josiah RNA targeted a GPC region of the S segment using LASV 36E2 and 80F2 primers [66]. Standards used in qRT-PCR were generated from serial 10-fold dilutions of RNA isolated from LASV-Josiah stock (10−1 PFU/ml – 10−7 PFU/ml) that were enumerated in triplicate by conventional plaque assay as previously described [12]. Sensitivity of qRT-PCR was 100 PFU-equivalent per ml of blood or per g of tissue.
Currently, at least two hypotheses are under evaluation to explain post-exposure activity of ML29. The ML29 L RNA has more stable predicted “pan-handle” structure [30,19] than the LASV L RNA. As a result, a more stable promoter and higher affinity to RdRp will effectively out-compete the LASV during co-replication in target cells (DCs, macrophages). It has been shown that replication of MOPV, in contrast to LASV, results in immune activation of target cells and stronger T-cell responses [32]. Another possibility is that the ML29 treatment will rescue fatally infected animals by activation of ML29-specific innate immune responses overcoming LASV infection. Perhaps both proposed mechanisms are contributed to the survival. The hypothesis that co-replication of both viruses induces a potent innate immune responses driven by ML29 is currently testing in rhesus macaques, the most appropriate model of human LF.
Replication-competent rVSV-based vaccines expressing filovirus glycoproteins (MARV or EBOV) demonstrated post-exposure activity in NHP models [33–35]. However, this treatment was effective only in a very narrow time-window between exposure to the virus (EBOV or MARV) and treatment, 20–30 min. Still this post-exposure treatment strategy can be useful in case of lab accidents in BSL-4 facilities [36] or for treatment of medical personnel exposed to highly pathogenic viruses, LASV, MARV, EBOV, during case patient management.
Yellow fever 17D-based vectors expressing LASV nucleocappsid antigen
There are several arguments in support of the statement: “For the long term, a yellow fever/Lassa fever chimera vaccine for use in EPI in West Africa is a very attractive solution” [37,38]. The YF17D is one of the most efficacious and safe vaccines ever developed with a highly favorable benefit-risk profile and excellent cost-effectiveness. Since 1937 more than 540 million doses have been delivered world-wide and no reversion to wild-type YF virus has been reported. Rare observed adverse effects are associated with host factors and are not due to virus reversion [39–41]. During the past 25 years the absence of effective public health policy, social-ecological factors and vaccine shortage resulted in a resurgence of YF (~ 200,000 cases and 30,000 deaths annually) in South America and Africa. Currently, YF and LF are two the most prevalent viral hemorrhagic fevers circulating in overlapping endemic areas of West Africa. In South America, some arenaviral HFs are also circulated in YF endemic areas.
Based on outstanding YF17D safety records and recent success in molecular biology of flaviviruses, the genetic backbone of YF17D has been used for construction of chimeric YF17D-based viruses expressing prM and E proteins of closely-related flaviviruses, Japanese encephalitis (JE), Dengue, and West Nile virus. ChimeriVax™-based vaccines against these flaviviruses are currently undergoing Phase II–III clinical testing (reviewed by [42]) and a chimeric JE vaccine with the brand name IMOJEV® is the first YF17D-vectored vaccine licensed for adults and pediatric population in Australia and Thailand [43].
To generate recombinant YF17D viruses expressing LASV genes we developed technology to clone and express LASV glycoproteins between YF17D E and NS1 genes [15].
The YF17D-based recombinants expressing truncated LASV ΔGPC (w/out a SSP, a stable signal peptide) or LASV GP1 and GP2 were replication-competent, deeply attenuated, induced immune responses against both pathogens, YF virus and LASV, and protected 80% of guinea pigs against fatal LF in proof-of-concept challenge experiments [38,15]. Cloning of shorter inserts, LASV GP1 and GP2, between YFV17D E and NS1 genes enhanced genetic stability of recombinant viruses [15,16,38].
LASV-infected individuals in endemic areas of West Africa have very strong memory CD4+ T-cell responses against the LASV NP, which are partially cross-reactive with other LASV strains [25]. This finding has an important implication for vaccine design against LF. Our hypothesis is that CD8+ cytotoxic T-cells directed against NP-derived epitopes which are presented shortly after infection are required to restrict the initial viral replication as much as possible. Notably, mice that were tolerated against all NP-derived T-cell epitopes were severely compromised in their ability to control LCMV at early stage of the infection [44]. During the later stages of the infection, these CTLs become exhausted and CTLs against epitopes derived from the late viral proteins, GP1 and GP2, will be responsible for effective LASV control. Currently there is a consensus that anti-NP immunity is involved in virus control at early stage of the infection and also contributed to longterm cross-reactive immunity [25,45]. Simultaneous immunization with a cocktail of recombinant YF17D-based viruses that express LASV GP1, GP2 and NP can potentially induce CD8+ CTLs to effectively control LASV [13,44,46,47]. To achieve this goal, in the current study we have cloned LASV NP in YF17D vector.
The LASV GP cloning strategy cannot be applied for LASV NP because these proteins have different biochemical features, expression profiles, and maturation pathways. Initial YF17D/LASV-NP recombinants were engineered by cloning LASV NP using a novel insertion site, the capsid gene, C. A fusion-PCR-based strategy was designed to clone the LASV NP gene immediately downstream of the cleavage site for the YF17D NS3 protease at the 3′ end of the C-gene, while maintaining the requirements for efficient cleavage at this site. This strategy would result in the addition of 3 YF17D- derived amino acids (Arg, Ser, His) at the NH2-terminus of the LASV NP. At C-terminus, the LASV NP was fused with the signal sequence of the YF17D prM via the foot and mouth disease virus (FMDV) 2A protein to obtain the required cleavage between the LASV NP protein and the signal peptide of the YFV17D prM gene. This would release the prM signal peptide and allow proper translocation of the YFV newly synthesized YFV membrane proteins to the lumen of the endoplasmic reticulum (ER).
Although the intended plasmid was successfully constructed using the low copy number plasmid pACNR-FLYF17D as a backbone [15], it turned out to be extremely unstable. In order to circumvent the instability problem, an alternative strategy was designed that aimed at constructing a limited number of YF17D recombinants expressing various fragments of the LASV NP gene (Figure 3). Four overlapping fragments of the LASV NP were obtained by PCR and cloned into a shuttle plasmid that contained the YF17D 5′ end and most of the viral structural genes of the YF17D vector. After sequencing to verify the nucleotide sequence of the LASV NP inserts, the recombinant YF17D-LASV gene fragment was cloned into the full-length YF17D clone to create the full-length viral cDNA for the intended YF17D/LASV-NP recombinants. Four YF17D/LASV NP recombinants were constructed: (i) YF17D/LASV-NP(1-876), contains the N-terminal fragment, 293 aa; (ii) YF17D/LASV-NP(870-1707), contains the C-terminal part, 236 aa; (iii) YF17D/LASV-NP(456-1296), contains the middle part of the LASV NP, 287 aa; and (iv) YF17D/LASV-NP(1-1296), contains the first 431 aa residues. The plasmids were linearized using the unique Afl II site at the 3′ end and used for in vitro RNA transcription to obtain full-length recombinant RNA that was subsequently used to transfect BHK-21J cells by electroporation.
The transfected cells were used for analysis of protein expression by IFA, Western blotting, and for detection of infectious virus by CPE. Positive staining for YF17D NS3 protein has been considered as a marker for viral replication; and positive staining using an α-FLAG antibody as a marker of LASV NP expression. As seen in figure 4, YF17D NS3 positive cells could be detected in the BHK cells transfected with either YF17D/LASV-NP (1-876) or –NP (870-1707). These cells also reacted with the α-FLAG antibody, suggesting proper expression of the LASV NP fragments. This antibody did not react with cells transfected with YF17D RNA that did not contain an insert (Figure 4, panel B). Similar results were obtained for two other YF17D-based recombinants expressing LASV NP (435-1296) and NP (1-1296).
Figure 4. Immunofluorescence of BHK-21J cells transfected with RNA transcripts obtained from YF17D.

panel A, B and C; pACNR-FLYF/LASV/NP (1-876), panel D, E and F; and pACNR-FLYF/LASVNP(870-1707), panel G, H and I. Cells were stained with an antiserum against YFV NS3 (top panels A, D, and G) or an anti-FLAG epitope antibody (panel B, E and H). Bottom panel shows the results of merging the anti-YF17D NS3 and the anti-FLAG staining. The cells were fixed at 30 hr post electroporation and processed for IFA using standard protocols [45].
Western blot analysis revealed that staining with the α-FLAG antibody detected LASV NP-specific proteins with expected size (Table 1). We also observed a clear cytopathic effect (CPE) in the BHK cells that were transfected with RNA transcripts synthesized from recombinant DNAs. The CPE formation started at 48 hr post electroporation (p.e.) and the medium was harvested at 60 hr p.e. when nearly all cells had died. The medium was cleared of cell debris and used for plaque assays to determine the viral titers which varied from 1×105 to 5×106 PFU/ml (data not shown). Two recombinant constructs expressing overlapping LASV NP fragments, 1-876 and 870-1707, had infectious titers around 1–5×106 PFU/ml and have been selected for further in vivo experiments.
Table 1.
The predicted molecular weight of the LASV-NP-FLAG proteins and the estimated size of these proteins as detected by Western blot analysis (staining with α-FLAG antibody).
| Recombinant YF17D/LASV-NP viruses | Expected size (kDa) | Estimated observed size (kDa) |
|---|---|---|
| YF17D/LASV-VNP(1-876) | 40 | 39 |
| YF17D/LASV-NP(435-1296) | 36 | 36 |
| YF17D/LAS-NP(870-1707) | 36 | 36 |
| YF17D/LASV-NP(1-1296) | 53 | 53 |
| YF17D…3XFLAG-FMDV2A (empty vector) | 5 | 5.6 |
In summary, we have developed two strategies to clone and express inserts of moderate size(200–300 aa residues) between the YF17D genes encoding the viral proteins E and NS1 (for LASV GP1 and GP2) and within YF17D C gene (for LASV NP) resulted in genetically stable recombinant viruses during 10 passages in tissue culture. Ten passages of recombinant YF17D-based viruses will be sufficient to manufacture non-GMP lots for toxicology and pre-clinical studies and for making Phase I lots during vaccine development [42]. We confirmed that insertion of foreign genes into YFV17D backbone resulted in additional attenuation of recombinant viruses (reviewed by [42]). While safety is not concern, low immunogenicity of YFV17D-based recombinants in pre-clinical evaluation in NHPs can be a potential problem (Lukashevich, unpublished). A deletion of the NP C-terminal domain involved in suppression of IFN-I induction can be a helpful solution [48]. In addition, immunogenicity of YF17D/LAS-GP recombinants can be also enhanced by expression of genetically modified metabolically stable glycoproteins, for example GP1, for cross-priming CD8+ T cell responses [49,50] (Figure 5). If successful, YF17D-based recombinant technology can be applied not only to control LF and YF in West Africa, but also YF and arenaviral HFs in co-endemic areas of South America.
Figure 5. Generation of LASV GP VLPV.

(A) ΔLGPfib, C-truncated non-cleavable GP fused with T4 fibritin (FIB); LGPwt, “wild”-type GP: SSP, the stable signal peptide (aa 1–58); GP1 (59-259) and GP2 (260-491) subunits; CS, cleavage site for the SKI-1/S1P. The fusion peptide and transmembrane domain of the GP2 are shown in black boxes; CD, cytoplasmic domain. (B) GP genes were cloned into alphavirus vector and packed in VLPV (see text). (C) Western blot of cell extracts from Vero cells treated with VLPV (>2 VLPV/cell). 24 hours after treatment (exposure), cells lysates were prepared for SDS/PAGE. Samples were not boiled and run on 8% gel under non-reducing conditions. Proteins were blotted onto nitrocellulose membranes, treated with mAbs against LASV GP1 and virus-specific proteins were detected as previously described [45]. The GP1 ran off the gel under these gel electrophoresis conditions; 75 and 250kDa are proteins markers.
Advanced alphavirus replicon-based arenaviral vaccines
LASV replicon particles with a cross-presenting potential and enhanced immunogenicity
Alphavirus replicon particles are replication-defective virions which are not able to spread beyond the initially infected cells, predominantly DCs and macrophages [51]. These particles are produced by removing the structural genes from the replicon RNA vector and replacing them with foreign genes of interest. Recombinant RNA replicons are encapsidated using helper genes encoding alphavirus capsid and glycoprotein provided in trans. Alphavirus replicon technology offers attractive features in terms of safety, immunogenicity, and efficacy and currently numerous vaccine candidates are in pre-clinical development [51]. Alphavirus replicon vectors (or virus-like particle vectors, VLPV) were developed based on attenuated strain of VEEV [22]. It was shown that transfection of cells with VEEV-based VLPV expressing LASV NP resulted in a highly effective production of a foreign gene. Nearly 20% of total cells proteins were identified as LASV NP antigen [22]. In addition, the split helper system into two separate molecules encoding capsid and glycoprotein significantly reduced the occurrence of replication-competent virus (RCV). The original VEEV-based replicon technology requires BSL-3 containment due to possibility of RCV generation. In this study to further improve safety of alphavirus replicon-based vaccines, the genetic backbone of VEEV TC-83 vaccine was used for construction of bicistronic vectors.
One of the unique features of the VLPV technology is an ability to target professional antigen-presenting cells and to promote CD8+ T cells responses to co-delivered antigens [52], the feature which is especially attractive for LASV vaccine design. In addition, alphavirus VLPV stimulate innate immune responses, posses intrinsic systemic and mucosal adjuvant activities, and practically have no anti-vector immunity concerns. The ability of rationally designed VLPV to express several genes from the same pathogen (e.g., LASV) or from two different pathogens (e.g., LASV and EBOV) was already successfully tested and proved in a guinea pigs challenge model [17]. Notably, co-expression of two major antigens of the same pathogen (e.g., EAV, equine arteritis virus) induced higher protection and had more favorable safety profile than replicons expressing individual EAV envelope proteins [53]. Currently we are using bivalent approach to make vaccines against pathogenic arenaviruses: (i) against LASV, by co-expression of LASV GPC from distantly-related phylogenetic groups, I and IV [54]; and (ii) against JUNV and MACV, by cloning and co-expression of GPC of both viruses.
In spite of the attractive features mentioned above, VLPV platform requires a prime-boost immunization strategy to achieve desirable levels of immune responses and protection. In practical terms, this strategy would be applicable for “organized” target groups (e.g., first responders, personnel of local hospitals in endemic areas, international travelers visiting endemic areas, military personnel, and staff of BSL4 labs). In attempts to enhance immunogenicity of experimental VLPV, we designed an experimental vaccine expressing genetically modified metabolically stable LASV GPC to enhance cell-mediated immune responses through a cross-priming pathway. Effective vaccines for cross-priming CD8+ T cell responses should express metabolically stable antigens [49,50]. Recent study showed that long-lived stable antigens better improve vaccine immunogenicity compared with antigens subjected to accelerated proteosomal degradation [55].
Naturally, LASV GPC is not cross-presented antigen. Strategy to make metabolically stable LASV GP is depicted in Figure 5. In brief, cleavage site was disrupted by mutagenesis, transmembrane and cytoplasmic domains were deleted, and truncated GP was fused with the phage T4-derived fibritin trimerization motif [21]. The final sequence, named ΔLGPfib, was human codon optimized and and cloned within the VEEV TC-83 replicons. For production of VLPV, RNA transcripts encoding ΔLGPfib vector and and two TC-83 helpers, E1 & E2 and C, were co-transfected into CHO-K1 cells and incubated for VLPV harvesting. As a control, unmodified LASV GPC sequence was used (named LGPwt) for making VLPV expressing original LASV GPC sequence.
Cells expressing LGPwt and ΔLGPfib were positively stained in IFA with a cocktail of LASV GP-specific mAbs (L52-121-22-BA02, L52-2121-22-BA02, and L52-135-17A, provided by USAMRIID) [17] or with GP2-specific rabbit polyclonal antibodies [15] indicating that GP1 and truncated GP2 subunits were correctly folded and genetic modification did not affect antigenic identity of ΔLGPfib. Western blot analysis of Vero cells exposed to VLPV showed that the LASV GP-specific protein from ΔLGPfib-exposed cells migrated as a heterogeneous band between 200–250 kDa (Figure 5C). In LGPwt-exposed cells un-cleaved portion of LGPwt had an expected mol. weight, ~72 kDa [15]. Notably, in cells exposed to ΔLGPfib VLPV, the trimer-stabilized LASV GP was expressed as a cell-associated protein, co-stained with the polyclonal α-calnexin antibodies, and only a small portion (<10%) of the ΔLGPfib was recovered from culture medium (not shown). Similarly modified non-cleavable variant of LCMV-GP with a fibritin tail also accumulated as stable, soluble GP trimers in the ER of transfected cells [56]. More importantly, this modification converted GP into a potent immunogen for CD8+ CTL cross-priming [56]. In line with these results, VLPV expressing ΔLGPfib protected mice against LASV challenge in our CBA/J-ML29 model [57] and induced stronger CMI responses than those induced by VLPV expressed GPwt (Figure 6). Our hypothesis is that ΔLGPfib VLPV augment cross-presentation of LASV GP that normally does not enter the cross-presentation pathway. We are going to optimize vaccine formulation by testing 4 bivalent VLPVs expressing LGPwt and ΔLGPfib derived from distantly-related LASV isolates from phylogenetic groups I and IV.
Figure 6.
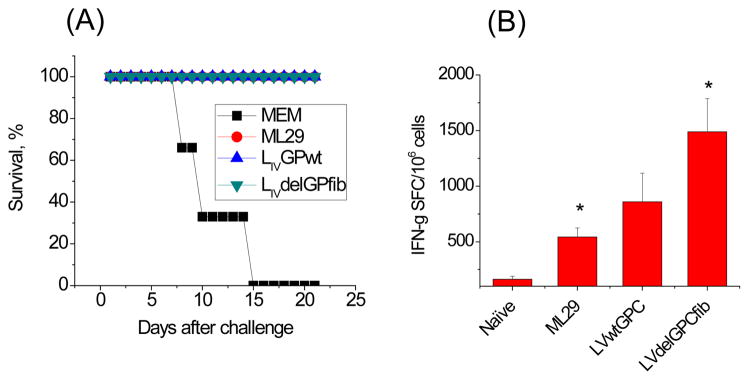
Efficacy and immunogenicity of TC-83-based VLPV in a CBA/J-ML29 mouse model [57]. (A) Mice immunized with VLPV and ML29 (vaccine control) were fully protected against fatal challenge. (B) Mice immunized with ΔLGPfib VLPV induced stronger cell-mediated immunity in comparison with ML29- and LGPwt-immunized mice.
Design of bivalent JUNV/MACV VLPV
Argentine and Bolivian HF are the most prevalent infectious diseases caused by pathogenic arenaviruses in South America. Before introduction of Candid #1 [58,59], up to 800 case of AHF were diagnosed each year. MACV has caused human disease outbreaks with at least 1,200 cases and resulted in 200 deaths. The disease recently re-emerged in Bolivia with fatality rate ~20% [60,61] is clinically similar to AHF. While the Candid #1 vaccine has proven to be safe and efficacious in adults, this vaccine is not recommended for pregnant women and children under 15 years old [58,59]. This vaccine has an IND status in the US.
Monocistronic TC-83 replicon vectored JUNV GPC has already proven to be efficacious in a guinea pigs challenge model [62]. For bi-cistronic VLPV, codon-optimized JUNV GPC and MACV GPC genes were cloned into TC-83 vector downstream from the TC-83 subgenomic 26S promoters as shown in Figure 7A. Replicon and two helper RNA species were synthesized by invitro transcription from plasmid DNA constructs. Preparation of VLPV was carried out in BHK-21 cells co-transfected with bi-cistronic vector and two helper RNAs as previously described [17].
Figure 7. Expression of JUNV GPC from bi-cistronic vector in BHK-21 cells, by indirect immunofluorescence.

(A) construction of bivalent JUNV/MACV VLPV. (B) detection of JUNV GPC my monoclonal antibodies in IFA. BHK-21 cells were incubated with VLPV containing bi-cistronicTC-83 vector expressing JUNV GPC and MACV GPC. VLPVs were diluted 100-fold and incubated with BHK-21 cell monolayer for 16 hr at 37C in 8-well chamber slides. Then, cells were fixed with cold acetone, and indirect IFA was performed using JUNV-specific monoclonal antibody, GB03-BE08. (C) Western blot analysis using GB03-BE08 and rabbit polyclonal against MACV GPC.
Cells exposed to bi-cistronic VLPV were positively stained with JUNV monoclonal antibodies specific to JUNV [63] (Figure 7B) or with rabbit antibody against MACV GPC (not shown). Monoclonal GB03-BE08 [63,64] (BeiResources, NR-2564) specifically interacted with JUNV and did-not cross react with MACV GPC. Western Blot analysis confirmed expression of both GPCs in cells exposed to bivalent JUNV/MACV VLPV (Figure 7C). These results indicate that bicistronic VEEV TC-83 vector can be successfully used for cloning and expression of GPC genes from JUNV and MACV.
In summary, in this paper we have presented three vaccine platforms to design vaccines against pathogenic arenaviruses. Our the most advanced vaccine candidate, the reassortant ML29, is safe, highly immunogenic and proved to be protective not only as preventive vaccine against LF but also as a post-exposure treatment. A single-dose injection induces cross-protective sterilizing immunity against distantly-related LASV isolates. This experimental vaccine is well positioned to control LASV in general population of West Africa. Unfortunately, the current status of ML29 as a risk group 3 pathogen (based on MOPV risk group assignment) is the major obstacle for further development of ML29 in the US.
The recent success of YF17D-based chimeric technology justifies YF17D-vectored bivalent vaccines to control both infections, LF and YF, in West Africa and South American arenaviral HFs and YF in South America. Excellent safety profile of YF17D indicates that safety will not be a problem for these experimental vaccines. Insertion of foreign genes results in additional attenuation and, probably, is connected with low immunogenicity in NHPs. In spite of successful cloning and expression of LASV genes in YF17D vector, genetic stability and low immunogenicity must be addressed to make efficacious YF17D-vectored arenaviral vaccines. If successful, YFV/LASV vaccine is well positioned to be included in immunization program in West Africa [37].
Safety profile of VEEV TC-83, lack of anti-vector immunity, intrinsic systemic, mucosal adjuvant activities, and ability to express several genes from the same or different pathogens, make TC-83-based replicon technology as an attractive solution to design bivalent vaccines against pathogenic arenaviruses. While these vaccines are not applicable for population of remote areas with poor medical infrastructure, prime-boost immunization might be practical for medical providers, military and lab personnel, visitors in endemic areas.
Acknowledgments
The authors thank Dr. Sina Bavari (USAMRIID, Frederick, MD) for providing rabbit antibodies against MACV. This work was supported by grants R01AI052367 (I.S.L.), R01AI068961 (I.S.L.), R01AI093450 (I.S.L./P.P.), and R43AI094700 (P.P.) from the National Institutes of Health.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Salvato MS, Clegg JCS, Buchmeier MJ, Charrel RN, Gonzalez JP, et al. Ninth Report of the International Committee on Taxonomy of Viruses. Academic Press, Elsevier Inc; 2012. Family arenaviridae, virus taxonomy, classification and nomenclature of viruses; pp. 715–723. [Google Scholar]
- 2.Bonthius DJ. Lymphocytic choriomeningitis virus: an underrecognized cause of neurologic disease in the fetus, child, and adult. Semin Pediatr Neurol. 2012;19:89–95. doi: 10.1016/j.spen.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacNeil A, Ströher U, Farnon E, Campbell S, Cannon D, et al. Solid organ transplant-associated lymphocytic choriomeningitis, United States, 2011. Emerg Infect Dis. 2012;18:1256–1262. doi: 10.3201/eid1808.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson BR, Delgado S, Aguda R, et al. A newly discovered arenavirus associated with a fatal hemorrhagic fever case in Bolivia. XIII Intern. Confer. on Negative Strand Viruses; 2006. p. Abstracts, 212. [Google Scholar]
- 5.Paweska J, Sewlall NH, Ksiazek TG, Blumberg LH, Hale MJ, et al. Nosocomial outbreak of novel arenavirus infection, southern Africa. Emerg Infect Dis. 2009;15:1598–1602. doi: 10.3201/eid1510.090211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briese T, Paweska JT, McMullan LK, Hutchison SK, Street C, et al. Genetic detection and characterization of lujo virus, a new hemorrhagic fever-associated arenavirus from southern africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lecompte E, Meulen JT, Emonet S, Daffis S, Charrel RN. Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology. 2007;364:178–183. doi: 10.1016/j.virol.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Ishii A, Thomas Y, Moonga L, Nakamura I, Ohnuma A, et al. Novel arenavirus, Zambia. Emerg Infect Dis. 2011;17:1921–1924. doi: 10.3201/eid1710.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palacios G, Savji N, Hui J, Travassos da Rossa, Popov V, et al. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J Gen Virol. 2010;91:1315–1324. doi: 10.1099/vir.0.017798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulibaly-N’Golo D, Allali B, Kouassi SK, Fichet-Calvet E, Becker-Ziaja B, et al. Novel Arenavirus Sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Cóte d’Ivoire: Implications for Evolution of Arenaviruses in Africa. PLoS One. 2011;6:e20893. doi: 10.1371/journal.pone.0020893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, et al. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. M Bio. 2012;3:e00180–12. doi: 10.1128/mBio.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukashevich IS, Patterson J, Carrion R, Moshkoff D, Ticer A, et al. A live attenuated vaccine for Lassa fever made by reassortment of Lassa and Mopeia viruses. J Virol. 2005;79:13934–13942. doi: 10.1128/JVI.79.22.13934-13942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukashevich IS, Carrion R, Jr, Salvato MS, Mansfield K, Brasky K, et al. Safety, immunogenicity, and efficacy of the ML29 vaccine for Lassa fever in small non-human primates. Vaccine. 2008;26:5246–5254. doi: 10.1016/j.vaccine.2008.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrion R, Jr, Patterson JL, Johnson C, Gonzales M, Moreira CR, et al. A ML29 reassortant virus protects guinea pigs against a distantly-related Nigerian strain of Lassa virus and can provide sterilizing immunity. Vaccine. 2007;25:4093–4102. doi: 10.1016/j.vaccine.2007.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bredenbeek P, Molenkamp R, Spaan WJ, Deubel V, Marianneau P, et al. A recombinant Yellow Fever 17D vaccine expressing Lassa virus glycoproteins. Virology. 2006;345:299–304. doi: 10.1016/j.virol.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Dalebout TJ, Bredenbeek PJ, Carrion R, Jr, Brasky K, et al. Yellow fever 17D-vectored vaccines expressing Lassa virus GP1 and GP2 glycoproteins provide protection against fatal disease in guinea pigs. Vaccine. 2011;29:1248–1257. doi: 10.1016/j.vaccine.2010.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pushko P, Geisbert J, Parker M, Jahrling P, Smith J. Individual and bivalent vaccines based on alphavirus replicons protect guinea pigs against infection with Lassa and Ebola viruses. J Virol. 2001;75:11677–11685. doi: 10.1128/JVI.75.23.11677-11685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukashevich IS. Generation of reassortants between African arenaviruses. Virology. 1992;188:600–605. doi: 10.1016/0042-6822(92)90514-p. [DOI] [PubMed] [Google Scholar]
- 19.Moshkoff D, Salvato MS, Lukashevich IS. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes. 2006;34:169–176. doi: 10.1007/s11262-006-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters CJ, Jahrling PB, Liu CT, Kenyon RH, McKee KT, Jr, et al. Experimental studies of arenaviral hemorrhagic fevers. Curr Top Microbiol Immunol. 1987;134:5–68. doi: 10.1007/978-3-642-71726-0_2. [DOI] [PubMed] [Google Scholar]
- 21.Boudko SP, Londer YY, Letarov AV, Sernova NV, Engel J, et al. Domain organization, folding and stability of bacteriophage T4 fibritin, a segmented coiled-coil protein. Eur J Biochem. 2002;269:833–841. doi: 10.1046/j.1432-1033.2002.02734.x. [DOI] [PubMed] [Google Scholar]
- 22.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, et al. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 23.Fisher-Hoch SP, McCormick JB. Lassa fever vaccine. Expert Rev Vaccines. 2004;3:103–111. doi: 10.1586/14760584.3.2.189. [DOI] [PubMed] [Google Scholar]
- 24.Meulen J, Badusche M, Satoguina J, Strecker T, Lenz O, et al. Old and New World arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by Lassa virus-specific human CD4+ T-cell clones. Virology. 2004;321:134–143. doi: 10.1016/j.virol.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Ter Meulen J, Badusche M, Kuhnt K, Doetze A, Satoguina J, et al. Characterization of human CD4+ T-cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J Virol. 2000;74:2186–2192. doi: 10.1128/jvi.74.5.2186-2192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukashevich IS. A live reassortant vaccine for Lassa Fever. In: Vela EM, editor. Molecular Pathogenesis of Hemorrhagic Fever Viruses Transw Res Net. Kerala, India: 2010. pp. 143–170. [Google Scholar]
- 27.Salvato MS, Lukashevich IS. Vaccines against Lassa fever: New Generation Vaccines. 4. Marcel Dekker Inc; 2009. pp. 895–904. [Google Scholar]
- 28.Fisher-Hoch SP, McCormick JB. Lassa fever vaccine. Expert Rev Vaccines. 2004;3:189– 197. doi: 10.1586/14760584.3.2.189. [DOI] [PubMed] [Google Scholar]
- 29.Monath TP. Yellow fever vaccine. Expert Review of Vaccines. 2005;4:553–574. doi: 10.1586/14760584.4.4.553. [DOI] [PubMed] [Google Scholar]
- 30.Lukashevich IS, Salvato MS. Lassa Virus Genome. Current Genomics. 2006;7:351–379. [Google Scholar]
- 31.McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, et al. Lassa fever. Effective therapy with ribavirin. N Engl J Med. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- 32.Pannetier D, Reynard S, Russier M, Journeaux A, Tordo N, et al. Human Dendritic Cells Infected with the Nonpathogenic Mopeia Virus Induce Stronger T-Cell Responses than Those Infected with Lassa Virus. J Virol. 2011;85:8293–8306. doi: 10.1128/JVI.02120-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daddario-DiCaprio KM, Geisbert TW, Ströher U, Geisbert JB, Grolla A, et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet. 2006;367:1399– 1404. doi: 10.1016/S0140-6736(06)68546-2. [DOI] [PubMed] [Google Scholar]
- 34.Geisbert TW, Daddario-DiCaprio KM, Williams KJ, Geisbert JB, Leung A, et al. Recombinant Vesicular Stomatitis Virus Vector Mediates Postexposure Protection against Sudan Ebola Hemorrhagic Fever in Nonhuman Primates. J Virol. 2008;82:5664–5668. doi: 10.1128/JVI.00456-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feldmann H, Jones SM, Daddario-DiCaprio KM, Geisbert JB, Ströher U, et al. Effective Post-Exposure Treatment of Ebola Infection. PLoS Pathog. 2007;3:e2. doi: 10.1371/journal.ppat.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Günther S, Feldmann H, Geisbert TW, Hensley LE, Rollin PE, et al. Management of accidental exposure to Ebola virus in the biosafety level 4 laboratory, Hamburg, Germany. J Infect Dis. 2011;204:S785–S790. doi: 10.1093/infdis/jir298. [DOI] [PubMed] [Google Scholar]
- 37.Fisher-Hoch S, McCormick J. Towards a human Lassa fever vaccine. Rev Med Virol. 2001;11:331–341. doi: 10.1002/rmv.329. [DOI] [PubMed] [Google Scholar]
- 38.Editorial. YF17D vector for Lassa fever vaccine. International Innovation. :77–79. [Google Scholar]
- 39.Hayes EB. Acute viscerotropic disease following vaccination against yellow fever. Trans R Soc Trop Med Hyg. 2007;101:967–971. doi: 10.1016/j.trstmh.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 40.Lindsey NP, Schroeder BA, Miller ER, Braun MM, Hinckley AF, et al. Adverse event reports following yellow fever vaccination. Vaccine. 2008;26:6077–6082. doi: 10.1016/j.vaccine.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Silva ML, Espírito-Santo LR, Martins MA, Silveira-Lemos D, Peruhype-Magalhães V, et al. Clinical and immunological insights on severe, adverse neurotropic and viscerotropic disease following 17D yellow fever vaccination. Clin Vaccine Immunol. 2010;17:118–126. doi: 10.1128/CVI.00369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guy B, Guirakhoo F, Barban V, Higgs S, Monath TP, et al. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine. 2010;28:632–649. doi: 10.1016/j.vaccine.2009.09.098. [DOI] [PubMed] [Google Scholar]
- 43.Appaiahgari MB, Vrati S. Clinical development of IMOJEV®—a recombinant Japanese encephalitis chimeric vaccine (JE-CV) Expert Opin Biol Ther. 2012;12:1251–1263. doi: 10.1517/14712598.2012.704908. [DOI] [PubMed] [Google Scholar]
- 44.Schildknecht A, Welti S, Geuking MB, Hangartner L, Van den Broek M. Absence of CTL Responses to Early Viral Antigens Facilitates Viral Persistence. J Immunol. 2008;180:3113–3121. doi: 10.4049/jimmunol.180.5.3113. [DOI] [PubMed] [Google Scholar]
- 45.Russier M, Pannetier D, Baize S. Immune Responses and Lassa Virus Infection. Viruses. 2012;4 doi: 10.3390/v3340x3000x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shedlock DJ, Talbott KT, Cress C, Ferraro B, Tuyishme S, et al. A highly optimized DNA vaccine confers complete protective immunity against high-dose lethal lymphocytic choriomeningitis virus challenge. Vaccine. 2011;29:6755–6762. doi: 10.1016/j.vaccine.2010.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baize S, Marianneau P, Loth P, Reynard S, Journeaux A, et al. Early and Strong Immune Responses Are Associated with Control of Viral Replication and Recovery in Lassa Virus-Infected Cynomolgus Monkeys. J Virol. 2009;83:5890–5903. doi: 10.1128/JVI.01948-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carnec X, Baize S, Reynard S, Diancourt L, Caro V, et al. Lassa virus nucleoprotein mutants generated by reverse genetics induce robust type I IFN response in human dendritic cells and macrophages. J Virol. 2011;85:12093–12097. doi: 10.1128/JVI.00429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. doi: 10.1126/science.1096378. [DOI] [PubMed] [Google Scholar]
- 50.Huckriede A, Bungener L, Holtrop M, de Vries J, Waarts BL, et al. Induction of cytotoxic T lymphocyte activity by immunization with recombinant Semliki Forest virus: indications for cross-priming. Vaccine. 2004;22:1104–1113. doi: 10.1016/j.vaccine.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Vander Veen R, Harris DL, Kamrud KI. Alphavirus replicon vaccines. Anim Health Res Rev. 2012;13:1–9. doi: 10.1017/S1466252312000011. [DOI] [PubMed] [Google Scholar]
- 52.Thompson JM, Whitmore AC, Staats HF, Johnston RE. Alphavirus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine. 2008;26:4267–4275. doi: 10.1016/j.vaccine.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balasuriya U, Heidner HW, Davis NL, Wagner HM, Hullinger PJ, et al. Alphavirus replicon particles expressing the two major envelope proteins of equine arteritis virus induce high level protection against challenge with virulent virus in vaccinated horses. Vaccine. 2002;20:1609–1617. doi: 10.1016/s0264-410x(01)00485-6. [DOI] [PubMed] [Google Scholar]
- 54.Bowen MD, Rollin PE, Ksiazek TG, Hustad HL, Bausch DG, et al. Genetic diversity among Lassa virus strains. J Virol. 2000;74:6992–7004. doi: 10.1128/jvi.74.15.6992-7004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schliehe C, Bitzer A, van den Broek M, Groettrup M. Stable antigen is most effective for eliciting CD8+ T-cell responses after DNA vaccination and infection with recombinant vaccinia virus in vivo. J Virol. 2012;86:9782–9793. doi: 10.1128/JVI.00694-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freigang S, Eschli B, Harris N, Geuking M, Quirin K, et al. A lymphocytic choriomeningitis virus glycoprotein variant that is retained in the endoplasmic reticulum efficiently cross-primes CD8(+) T cell responses. Proc Natl Acad Sci U S A. 2007;104:13426–13431. doi: 10.1073/pnas.0704423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goicocheaa MA, Zapata JC, Bryant J, Davis H, Salvato MS, et al. Evaluation of Lassa virus vaccine immunogenicity in a CBA/J-ML29 mouse model. Vaccine. 2012;30:1445–1452. doi: 10.1016/j.vaccine.2011.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Enria DA, Barbera Oro JG. Junin Virus Vaccines. Curr Top Microbiol Immunol. 2002;263:239–261. doi: 10.1007/978-3-642-56055-2_12. [DOI] [PubMed] [Google Scholar]
- 59.McKee KTJ, Oro JG, AL Kuehne JA, Spisso Mahlandt BG. Safety and immunogenicity of a live-attenuated Junin (Argentine hemorrhagic fever) vaccine in rhesus macaques. Am J Trop Med Hyg. 1993;48:403–411. doi: 10.4269/ajtmh.1993.48.403. [DOI] [PubMed] [Google Scholar]
- 60.Aguilar PV, Camargo W, Vargas J, Guevara C, Roca Y, et al. Reemergence of Bolivian Hemorrhagic Fever, 2007–2008. Emerg Infect Dis. 2009;15:1526–1528. doi: 10.3201/eid1509.090017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Radoshitzky SR, Kuhn JH, de Kok-Mercado F, Jahrling PB, Bavari S. Drug discovery technologies and strategies for Machupo virus and other New World arenaviruses. Expert Opin Drug Discov. 2012;7:613–632. doi: 10.1517/17460441.2012.687719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seregin AV, Yun NE, Poussard AL, Peng BH, Smith JK, et al. TC83 replicon vectored vaccine provides protection against Junin virus in guinea pigs. Vaccine. 2010;28:4713–4718. doi: 10.1016/j.vaccine.2010.04.077. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez A, Pifat DY, Kenyon RH, Peters CJ, McCormick JB, et al. Junin virus monoclonal antibodies: characterization and cross-reactivity with other arenaviruses. J Gen Virol. 1989;70:1125–1132. doi: 10.1099/0022-1317-70-5-1125. [DOI] [PubMed] [Google Scholar]
- 64.Demby AH, Chamberlain J, Brown DW, Clegg CS. Early diagnosis of Lassa fever by reverse transcription-PCR. J Clin Microbiol. 1994;32:2898–2903. doi: 10.1128/jcm.32.12.2898-2903.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]