

Abstract
Targeted delivery of cancer therapeutics and imaging agents aims to enhance the accumulation of these molecules in a solid tumor while avoiding uptake in healthy tissues. Tumor-specific accumulation has been pursued with passive targeting by the enhanced permeability and retention effect, as well as with active targeting strategies. Active targeting is achieved by functionalization of carriers to allow specific interactions between the carrier and the tumor environment. Functionalization of carriers with ligands that specifically interact with overexpressed receptors on cancer cells represents a classic approach to active tumor targeting. Cell-penetrating peptides (CPPs) provide a non-specific and receptor-independent mechanism to enhance cellular uptake that offers an exciting alternative to traditional active targeting approaches. While the non-specificity of CPP-mediated internalization has the intriguing potential to make this approach applicable to a wide range of tumor types, their promiscuity is, however, a significant barrier to their clinical utility for systemically administered applications. Many approaches have been investigated to selectively turn on the function of systemically delivered CPP-functionalized carriers specifically in tumors to achieve targeted delivery of cancer therapeutics and imaging agents.
Introduction
Targeted delivery of anti-cancer therapeutics to solid tumors aims to enhance accumulation of the drug within the tumor while minimizing accumulation in healthy tissues in order to maximize therapeutic efficacy and to minimize off-target side effects. Targeting delivery to the disease site is particularly critical for many current cancer therapeutics, which generally exert their therapeutic action by targeting features that are not exclusive to cancerous cells, such as rapid cancer cell division1 or the propensity of cancer cells toward apoptosis,2 so that off-target delivery to a sub-set of healthy cells that share these features also results in significant systemic toxicity and undesirable side-effects. Targeted delivery thus remains an active area of investigation to improve the therapeutic efficacy of anti-cancer drugs while reducing their undesirable side effects in healthy organs.
Two major approaches have been investigated to target drugs and imaging agents to tumors: passive and active targeting. Passive targeting of macromolecules and nanoparticle carriers to solid tumors is possible because of the enhanced permeability and retention (EPR) effect that is a consequence of aberrant physiological features of the tumor environment including a leaky tumor vasculature and a lack of draining lymphatic vessels.3 The pores in the disorganized neovasculature of many solid tumors, resulting from anomalous angiogenesis, permit the diffusion of molecules from the vascular to the extravascular space. Molecules with a prolonged systemic circulation –such as macromolecules and nanoparticles– can take advantage of the leakiness in the tumor vasculature by diffusing into the extravascular compartment of tumors and accumulating, over time, in the tumor tissue. The lack of an organized lymphatic system in the tumor reduces the clearance of macromolecules and nanoparticles, which further prolongs their residence in the tumor.
The second approach –active targeting– attempts to enhance the accumulation of a drug or imaging agent in solid tumors by the specific interaction of a carrier with targets that are overexpressed by tumors as compared to healthy tissue. Antibody-antigen and ligand-receptor interactions are two examples of highly specific biomolecular interactions that can be exploited to target carriers to tumors. Although active targeting based on ligand-receptor or antibody-antigen interactions have shown enhanced tumor accumulation and improved therapeutic effect by targeting functionalized carriers to tumors,4 the application of these approaches are limited by the inherent heterogeneity of cancer classes. First, the heterogeneity of cancer types limits active targeting approaches exploiting upregulated receptors or overexpressed cell-surface antigens to only those types of cancer that overexpress that target compared to healthy tissues, as the expression of these targets can vary widely across tumor classes.5 Second, these targets are furthermore heterogeneous in their expression between patients with a specific type of cancer, such that only a subset of those patients with the appropriate level of target expression can be expected to benefit from a specific active targeting approach.6 Finally, the spatial distribution of the target can also be heterogeneous within a single tumor,7–9 such that the carrier may accumulate unevenly throughout the tumor tissue. Because of these limitations of traditional active targeting, it is clear that new alternatives are needed for the creation of carriers that can provide targeted delivery of drugs and imaging agents in a variety of cancers and for a large subset of cancer patients. Cell-penetrating peptides are a potential class of molecules that can be exploited to achieve these goals due to their non-specific mechanism of cellular uptake that is applicable to a variety of cell types and tumor classes. Their non-specificity, however, presents a challenge in their use in systemically administered applications for targeted delivery. This review summarizes the many approaches to spatially control the function of cell-penetrating peptides that aim to harness the power of these molecules to create improved carriers capable of providing targeted delivery of cancer drugs and imaging agents to a variety of tumor types.
CELL-PENETRATING PEPTIDES
Cell-penetrating peptides (CPPs) are a family of peptides that show efficient receptor-independent cellular uptake.10, 11 Over two decades ago, short peptides from HIV’s trans-activator of transcription (TAT) protein were first discovered to penetrate cell membranes and efficiently internalize into cells.12 The discovery of TAT was quickly followed by the identification of other peptides that exhibited similar behavior, such as Antennapedia, a transcription factor from drosophila (penetratin),10 and anti-microbial peptides derived from bovine neutrophils (bactenecin),13 resulting in the definition of CPPs as a class of peptides that achieve cellular uptake in a variety of cell types. Today this class has grown to include CPPs such as arginine-oligomers (e.g., arginine8)14–16 as well as synthetic derivatives.17–21 The list of CPPs will continue to grow with novel techniques now being utilized to predict22, 23 and screen24 for new CPP sequences.
A subset of CPPs is cationic and rich in basic residues; for such CPPs, arginines are believed to play a particularly important role in cellular internalization. While the exact mechanism of arginine-rich CPP cellular uptake remains unclear, it is believed that ionic attractions and bidentate hydrogen bonding of arginine’s guanidine head group with anionic components of the lipid membrane and cell surface proteoglycans initiate interactions that lead to their internalization.25, 26 Events that are potentially implicated in CPP internalization include pore formation,27, 28 inverted micelle formation,29 membrane potential-driven translocation,30, 31 and endocytosis,32, 33 which provide potential explanations for the function of both charged CPPs and alternative classes of CPPs that may be amphiphilic or hydrophobic in nature.
The non-specific cellular uptake of CPPs can be conferred to larger cargo by covalent conjugation, as CPPs have been shown to facilitate the internalization of associated peptides and proteins,34, 35 DNA,36 siRNA,37, 38 quantum dots,39 metal complexes,40, 41 liposomes,42 and micelles.43, 44 The mechanism of cellular uptake, however, varies greatly between short CPP sequences alone and CPP-cargo conjugates (Box 1).45 Short CPP sequences appear to easily traverse the cell membrane barrier, and are diffusely localized throughout the cytosol, while CPPs conjugated to larger cargo tend to display a punctate accumulation within intracellular vesicles.
Box 1. Effects of cargo on the uptake mechanism of CPP-cargo conjugates.
Differences in the mechanism of cellular uptake are seen between short CPP sequences and CPP-cargo conjugates. CPPs alone appear to traverse the cell membrane in an endocytosis-independent manner, whereas conjugation of CPPs to cargo ranging in size from small peptides to large proteins exhibit uptake by an endocytic mechanism.32, 45, 46 This shift in the mechanism of cellular uptake was demonstrated in human osteosarcoma cells where fluorescently labelled CPP arginine7 (R7) or R7 conjugated to a single additional amino acid (R7W) directly translocated across the cell membrane, as seen by their diffuse cytosolic fluorescence.45 In contrast, the same R7 CPP when appended with 9 to 11 additional amino acids exhibited uptake indicative of endocytic mechanisms as suggested by the appearance of punctate intracellular fluorescence. The exact endocytic pathways exploited by any CPP-cargo conjugate is likely to be dependent on the type and density of the CPP as well as the size and composition of the associated cargo. For example, arginine8-functionalized liposomes have been shown to exploit distinct pathways of uptake dependent on the surface density of the arginine8 moiety.46 Liposomes with a low density of CPP functionalization were taken up by clathrin-mediated endocytosis and shuttled to lysosomes, while those with a high density of CPP functionalization were internalized by macropinocytosis and were less likely to be routed to degradative lysosomes. In this example, a high density of arginine8 thus provided a potential advantage in internalization by exploiting a less degradative pathway of cellular uptake. Effects of CPP functionalization on the mechanism of uptake should therefore be considered in the design and characterization of carriers for CPP-mediated drug and imaging agent delivery.
Despite the observation that the exact mechanism of cellular uptake of CPPs appears to be quite sensitive to the type of attached cargo, the ability of CPPs to facilitate non-specific and receptor-independent cellular uptake of a variety of carriers provides the opportunity to target diverse cell types. Because of the promiscuous cellular internalization of CPPs, initial success has largely been realized with their local delivery. Examples include Penetratin™ 1 Peptide from Q-Biogene47 and Chariot™ Protein Delivery Reagent from Active Motif,48 which have been developed as in vitro laboratory reagents for intracellular delivery. In vivo applications of CPPs are now in clinical trials for use in local transdermal delivery for treatment of scarring,49 wrinkles,50 and psoriasis.51 Systemic delivery of CPP-cargo conjugates is also being investigated for treatment of myocardial infarction52 and stroke.53
CPPs are particularly exciting in their use for cancer therapy and detection, as they hold the potential to create a single drug carrier that is capable of targeting a diverse range of tumors across a heterogeneous patient population, overcoming the limitations of many active targeting approaches. Additionally, the potential of CPPs to direct cellular internalization to less degradative pathways than receptor-mediated endocytosis could minimize the breakdown of labile therapeutic cargo. CPPs are limited, however, by their promiscuous cellular uptake, which greatly hinders their utility to target specific sites in vivo. Systemic administration of CPPs for in vivo applications has revealed that their non-specificity creates a significant challenge, as their systemic injection leads to uptake in a variety of tissues, thus posing the risk of increased toxicity and off-target side effects. This off-target accumulation has been demonstrated in mice by the retention of arginine9 CPPs at the site of tail vein injection and the accumulation of arginine9 CPPs in the liver.54 The challenge of using CPPs systemically is also compounded by charge-mediated blood clearance that can decrease circulation time, a parameter that is important to maximize to passively target solid tumors by the EPR effect. It is clear that suboptimal biodistribution and pharmacokinetics make the direct use of CPPs impractical for applications requiring systemic administration.
Despite the challenges in employing CPPs for in vivo applications requiring systemic administration, investigation of their use for the delivery of drugs and imaging agents has actively continued because of their robust uptake and potentially less degradative pathways of cellular entry. These advantages make them an interesting alternative to traditional receptor-dependent methods and provide an exciting approach –if their problems can be resolved– to potentially overcome the limitations of other active targeting methods. To ultimately exploit the advantages of CPPs for targeted delivery to tumors by systemic administration novel approaches are clearly necessary –and are actively being pursued by many groups– to trigger the presentation of CPPs in situ only at the disease site to achieve targeted tumor accumulation. These approaches are summarized in the remainder of this review.
CONTROL OF CELL-PENETRATING PEPTIDES WITH ACTIVATABLE CARRIERS
The use of CPPs for targeted systemic delivery requires precise control of CPP presentation only at a target site in vivo. To achieve this control over CPP presentation researchers have focused on exploiting stimulus-responsive materials to trigger the selective display of CPPs within a site of disease, such as a solid tumor. These triggers for CPP activation may be intrinsic to the pathological environment of a tumor, such as low pH caused by lactic acid build up55 or overexpression of ECM remodeling proteases,56 or may be extrinsic to the body such as the focused application of heat or light to a disease site. Within this paradigm to control CPP presentation five approaches have been investigated: 1) controlled display of CPPs by removal of “stealth” polymers; 2) triggered display of CPPs by actuation of molecular tethers; 3) controlled presentation of CPPs by dissociation from ionic inhibitors; 4) manipulation of CPP charge by ionizable residues; and 5) modulation of the interfacial density of critical CPP residues by temperature-triggered micelle assembly. These approaches are summarized in Table 1.
Table 1.
Summary of carriers for controlled activation of CPPs
| Mechanism of Activation | System Description | Cell-Penetrating Peptide | Activation Trigger | Cargo | Ref |
|---|---|---|---|---|---|
Controlled display of CPPs by removal of “stealth” polymers
|
TAT-functionalized liposomes with cleavable PEG coating | YGRKKRRQRRR | pH sensitive cleavage of hydrazone linker for PEG release | Rhodamine as model cargo | 59,60 |
| GFP plasmid as model gene cargo | 61 | ||||
| Chemotherapeutic doxorubicin | 62 | ||||
| Human transcriptional factor Hph-1-functionalized quantum dots with cleavable PEG coating | YARVRRRGPRR | MMP-2 cleavage of peptide linker for PEG release | Quantum dot for fluorescent imaging | 63 | |
| Arginine-rich CPP-functionalized dextran-coated iron oxide particles with cleavable PEG coating | RRRRGRRRRKG | MMP-2 cleavage of peptide linker for PEG release | Iron oxide particle for MR imaging | 65 | |
| TAT-functionalized liposomes with cleavable PEG coating | YGRKKRRQRRR | MMP-2 cleavage of peptide linker for PEG release | Rhodamine as model cargo | 66 | |
| TAT-functionalized micelle with dissociable PEG coating | YGRKKRRQRRR | pH sensitive dissociation of polysulfonamide-PEG for PEG release | FITC as model cargo | 67 | |
| Chemotherapeutic doxorubicin | 68 | ||||
| TAT-functionalized liposome with cleavable PEG coating | AYGRKKRRQRRR | Cysteine-mediated cleavage of disulfide linker for PEG release | Calcein as model cargo | 69 | |
| DiR and DiD as model cargo | 70 | ||||
Controlled exposure of CPPs by actuation of molecular tethers
|
TAT-functionalized micelles with ionizable linkers for triggered CPP display | YGRKKRRQRRR | pH sensitive extension of linker beyond PEG coating for TAT presentation | Chemotherapeutic doxorubicin | 72 |
Controlled presentation of CPPs by dissociation from ionic inhibitors
|
Arginine8 with cleavable anionic inhibitory domain | RRRRRRRR | Prostate-specific antigen cleavage of peptide linker for dissociation of anionic inhibitor domain | FITC as model cargo | 74 |
| MMP-14 cleavage of peptide linker for dissociation of anionic inhibitor domain | Technetium-99m for SPECT imaging | 78 | |||
| Arginine9 with cleavable anionic inhibitory domain | RRRRRRRRR | MMP-2 cleavage of peptide linker for dissociation of anionic inhibitor domain | Fluorescein and Cy5 as model cargo | 54, 73, 75 | |
| MMP-2/9 cleavage of peptide linker for dissociation of anionic inhibitor domain | 177Lu and 126I for PET/SPECT imaging | 79 | |||
| Arginine9-functionalized dendrimeric nanoparticle with cleavable anionic inhibitor domains | RRRRRRRRR | MMP-2 cleavage of peptide linker for dissociation of anionic inhibitor domain | Cy5 and gadolinium for fluorescent and MR imaging | 76,77 | |
| Arginine5 with cleavable anionic inhibitory domains | RRRRR | MMP-2 cleavage of peptide linker for dissociation of anionic inhibitor domain | BQH3 and Alexa750 for photoacoustic imaging | 80 | |
| TAT-functionalized asparaginase with ionic-associated heparin inhibitor | YGRKKRRQRRR | Competitive binding of protamine for dissociation of heparin inhibitor | Anti-tumor asparaginase enzyme | 81,83 | |
| Penetratin-functionalized HPMA polymer with cleavable inhibitory neutralizing groups | RRMKWKK | UV light cleavage of linker for dissociation of neutralizing groups | (KLAKLAK)2 proapoptotic peptide | 84 | |
|
Controlled charge of CPPs by ionizable residues |
Modified transportan with lysine→histidine substitutions | AGYLLGHINLHHL-AHLHHIL | Ionization of histidine residues below pKa of 6.5 | Chemotherapeutic camptothecin | 85 |
Controlled density of CPPs by temperature-triggered micelle assembly
|
Arginine5-functionalized elastin-like polypeptide diblock copolymers | RRRRR | Hyperthermia-triggered micelle assembly for modulation of arginine density | Alexa488 as model cargo | 89 |
Controlled display of CPPs by removal of “stealth” polymers
A simple method to control the presentation of CPPs is to shield them with “stealth” polymers that can be selectively shed in the target tissue. Hydrophilic polymers, such as polyethylene glycol (PEG) are ideal for providing a protective corona over CPP-functionalized nanoparticle carriers because PEG prolongs the circulation time57 and limits cellular uptake58 of PEGylated nanoparticles compared to bare nanoparticles. In one implementation of this concept, rhodamine-labeled TAT-functionalized liposomes were also functionalized with PEG through pH-sensitive hydrazone linkers, wherein the PEG created a steric barrier covering the TAT peptide.59, 60 Incubation at low pH resulted in cleavage of the PEG from the liposome and loss of the PEG chains from the interface, revealing TAT on the liposome surface and restoring the cellular uptake of these vehicles. This approach has been used for local gene delivery to tumors with PEG-coated TAT-liposomes loaded with a GFP-encoding plasmid.61 Liposomes with PEG chains attached via a pH-labile linker showed 3-fold greater transfection as evident by GFP expression in tumor cells in comparison to TAT-liposomes with stable pH-insensitive PEG chains. This technique was extended to multifunctional liposomes, which were additionally functionalized with cancer-specific antibodies to increase tumor accumulation of nanoparticles by active targeting.62 Doxorubicin-loaded TAT-functionalized liposomes modified with antibody and pH-cleavable PEG demonstrated increased cytotoxicity in breast cancer MCF-7, melanoma B16-F10, and cervical cancer HeLa cells after pre-incubation of liposomes at pH 5, which removed the PEG and exposed the underlying TAT, as compared to multifunctional liposomes at pH 7.4. It is anticipated that the additional component of cancer-specific antibodies will improve the local accumulation and controlled activation of liposomes only at the targeted tumor site when administered systemically in vivo.
In an alternative implementation of this approach, the PEG shield can be detached with cleavage by overexpressed enzymes in tumors, such as matrix metalloproteinases (MMPs) that play a role in the remodeling of the tumor extracellular matrix. This approach has been demonstrated for the controlled uptake of quantum dots for tumor imaging.63 Quantum dots functionalized with a CPP derived from human transcriptional factor Hph-164 were shielded with PEG chains attached to the quantum dot through MMP-2 cleavable peptide linkers. Removal of the PEG upon incubation with MMP-2 was confirmed by the increased intensity of quantum dot emission due to the loss of FRET quenching between the donor quantum dot and an acceptor Cy5 fluorophore conjugated to the PEG. The attachment of PEG to the CPP-modified quantum dots decreased their cellular uptake in GFP-expressing human melanoma cells (Figure 1). After incubation with MMP-2, causing cleavage of the PEG linker, cellular uptake was restored. Similar results have been obtained with CPP-modified dextran-coated iron oxide particles shielded with MMP-2 cleavable PEG.65 Most recently, this approach has been extended to the controlled cellular uptake of liposomes functionalized with MMP-2 cleavable PEG terminated with cancer-specific antibodies.66 The addition of antibodies to this multifunctional system provides the opportunity for active tumor targeting of the liposomes prior to the enzymatic removal of PEG in the tumor environment and subsequent cellular uptake mediated by the now exposed TAT decorating the liposome surface. These studies suggest the potential of this approach to achieve localized uptake of cargo in tumors that have high protease expression.
Figure 1.
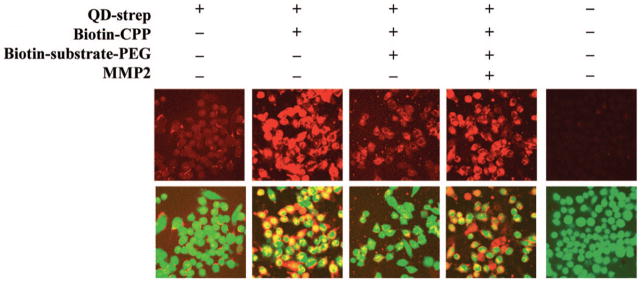
MMP-2 cleavage of PEG controlled cellular uptake of quantum dots. Streptavidin-coated quantum dots (QD-strep) (red – top row) achieved little internalization by GFP-expressing MDA-MB-435 melanoma cells (green – bottom row, merged with top row). Functionalization of the quantum dot surface with biotin-CPP increased cellular uptake, while addition of removable PEG (biotin-substrate-PEG) decreased the internalization by blocking interactions of the CPP with the cell surface. Cellular uptake was recovered in the presence of MMP-2 with cleavage of the substrate-PEG linker, as the CPPs could interact with the cell surface following the removal of PEG. Reprinted with permission from Ref 63 Copyright 2009 American Chemical Society.
A third variation of this approach involves the noncovalent attachment of PEG to a carrier by ionic interactions between cationic CPPs and a diblock copolymer composed of an anionic block and a PEG block. The anionic block can be designed such that its pKa is only slightly below physiological pH, causing the charge of this block to be neutralized in the acidic conditions of the tumor environment. This pH-triggered transition of the anionic block from negative to neutral charge causes dissociation of this block from the CPP, releasing the PEG, and permitting cellular uptake of the CPP-modified carrier. This approach has been demonstrated with micelles of PEG-poly(L-lactic acid) (PEG- PLLA) functionalized with TAT on the micelle corona.67 Mixing these micelles with poly(L-cystine bisamide-g-sulfadiazine)-PEG led to ionic complexation of the positively charged TAT and negatively charged sulfonamide, resulting in the assembly of a protective PEG shell extending beyond the TAT decorated corona of the PEG-PLLA micelle.68 When the pH was lowered to 7.0, the polysulfonamide became neutrally charged, causing its dissociation from TAT and allowing cellular uptake of the micelles. When loaded with the chemotherapeutic doxorubicin, cell death could be prevented at pH 7.4, a pH at which the PEG coating presumably remained intact in cell culture (Figure 2). However, at the depressed pH of 7.0 the cytotoxicity was similar to free drug as the polysulfonamide-PEG chains disassociated from the micelles and TAT-mediated cellular uptake delivered the cytotoxic payload.
Figure 2.
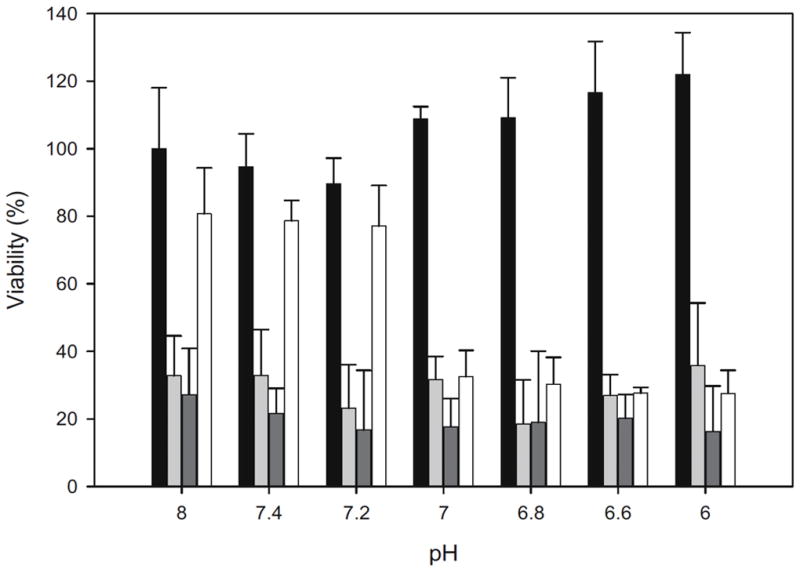
Cytotoxicty was controlled by pH-mediated removal of PEG. Protective PEG was added to TAT-functionalized micelles by ionic interactions between the cationic TAT and PEG-conjugated anionic polysulfonamide. Neutralization of polysulfonamide charge in acidic conditions caused the dissociation of PEG that allowed interaction of TAT with the cell surface. Doxorubicin-loaded PEG-shielded TAT-micelles (white) demonstrated selective cytotoxicity, with increased cell death occurring only at pH 7.0 and below. No pH-dependence in cytotoxicity was shown with controls including TAT-micelles (dark grey), free doxorubicin (light grey), or untreated cells (black). Reprinted from Ref 68 with kind permission from Springer Science and Business Media.
Finally, this method can also be implemented with a mechanism of PEG cleavage that is not endogenous to the disease site. One example of this approach is the modification of TAT-functionalized DOPE liposomes by the attachment of PEG through disulfide bonds.69 The stealth PEG coating can thus be removed in the presence of a reducing agent such as cysteine. Inclusion of 8% cleavable PEG in the TAT-functionalized liposome provided optimal reduction of cellular uptake in HepG2 liver carcinoma cells when the PEG coating was intact. Upon incubation with 20 mM cysteine, the cellular uptake of liposomes increased 4-fold by removal of the PEG and interaction of TAT with the cell surface. This method has been extended to TAT-functionalized DSPE liposomes modified with PEG through disulfide bonds.70 Upon systemic injection the protective PEG coating permitted accumulation of fluorophore-loaded liposomes in C26 colon cancer tumors by the EPR effect. The PEGylated liposomes significantly decreased off-target accumulation in the liver, a side effect that was observed with TAT-functionalized liposomes lacking the PEG coating. After 24 hours, systemic administration of cysteine removed the PEG from the locally accumulated liposomes and increased the uptake in tumors by 56% as compared to control animals that received PBS instead of the cysteine injection (Figure 3). This controlled CPP exposure led to a 130% increase in cellular uptake compared to PEGylated liposomes, TAT-functionalized liposomes, and non-cleavable PEGylated TAT-functionalized liposome controls. While this approach requires external administration of the cleavage agent, it allows activation of a CPP to be temporally controlled, which can provide advantageous synergy with the EPR effect.
Figure 3.
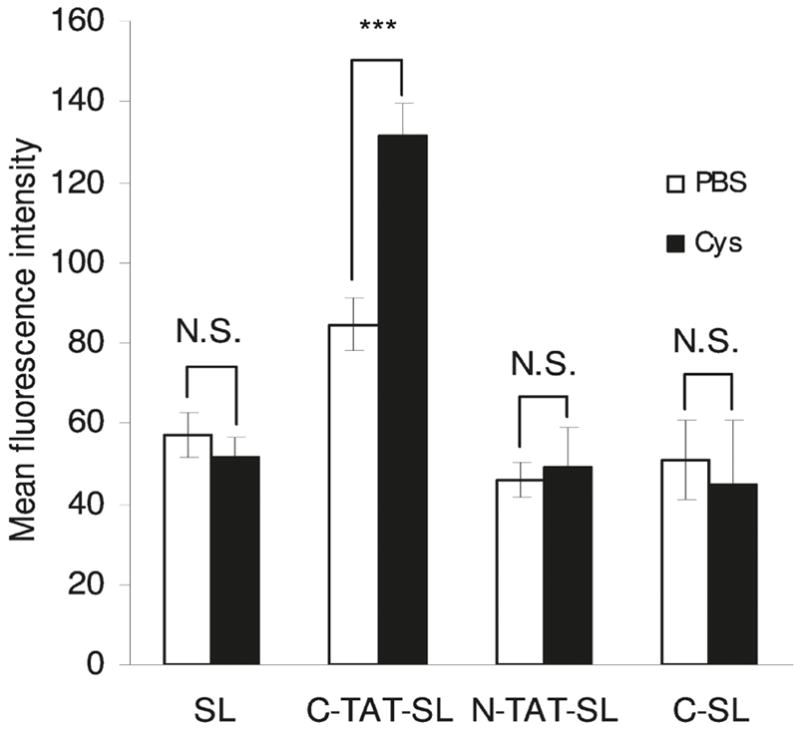
Tumor accumulation was enhanced with removal of PEG from tumor-localized stealth liposomes (SL). TAT-functionalized liposomes with cleavable disulfide-linked PEG (C-TAT-SL) accumulated in C26 colon cancer tumors via the EPR effect prior to the intravenous administration of thiol-reactive cysteine (Cys). Local cleavage and release of the PEG increased the cellular uptake in the tumor by 56% in cysteine administered animals, compared to the PBS administered controls, as measured by flow cytometry of excised tumor cells. This tumor uptake was 130% greater than that achieved with controls including non-functionalized stealth liposomes (SL), non-cleavable PEG coated TAT-liposomes (N-TAT-SL), and cleavable PEG coated non-functionalized liposomes (C-SL). Reprinted with permission from Ref 70. Copyright 2011 American Chemical Society.
Triggered display of CPPs by actuation of molecular tethers
“Stealth” polymers can be used to selectively shield CPPs without the need for cleavage of the protective polymer by using environmentally responsive linkers to trigger the presentation of a CPP.71 This approach has the advantage that the protective characteristics of PEG can be realized without the need to shed the PEG coating, which can lead to off-target uptake if the activated CPP carrier escapes the target tissue. This approach has been investigated with micelles composed of two block copolymers: poly(histidine)-PEG and poly(lactic acid)-PEG-poly(histidine)-TAT.72 Self-assembly of a mixture of these block copolymers resulted in micelles whose hydrophobic core was composed of poly(histidine) and poly(lactic acid) and whose corona was composed of PEG. The short poly(histidine) linker within the poly(lactic acid)-PEG-poly(histidine)-TAT copolymer acted as an actuator that controlled the location of the TAT peptide within the micelle structure. At pH 7.4 the neutral poly(histidine) linker was relatively hydrophobic, so that the attached TAT peptide remained buried near the interface between the hydrophilic corona and the hydrophobic core. A decrease in the pH to 7.0 caused ionization of the poly(histidine) segment, leading to an increase in its hydrophilicity that led to its extension within the PEG shell and display of the appended TAT peptide beyond the micelles’ corona. TAT presentation by pH-triggered actuation led to a 30-fold increase in uptake of micelles in MCF-7 human breast cancer cells at pH 7.0 compared to the uptake achieved at pH 7.4. Further decrease to a pH of 6.8 enhanced the cellular uptake to 70-fold greater than at physiologic pH. Doxorubicin was encapsulated in the micelle core to demonstrate the controlled cytotoxicity of these pH-responsive carriers. Drug loaded micelles demonstrated cytotoxicity only upon lowering the pH to 7.0 and exhibited greater cytotoxicity than free doxorubicin in drug-resistant NCI/ADR-RES ovarian cancer cells at this pH. The cytotoxicity of these pH-sensitive carriers was attributed both to the controlled cellular uptake of drug-loaded micelles and doxorubicin release by the pH-triggered destabilization of the micelle caused by the ionization of the histidine component that resided in the micelle core. Efficacy of doxorubicin-loaded micelles was also demonstrated in vivo by regression of A2780/AD drug resistant ovarian cancer xenografts in nude mice (Figure 4). Three intravenous doses of pH-activatable micelles achieved prolonged tumor regression that was superior to controls, including a pH sensitive micelle without TAT functionalization, a micelle that displayed TAT at all pH, and free doxorubicin.72 This therapeutic effect was demonstrated in several tumor lines to confirm the broad applicability of this approach to tumors with an acidic microenvironment.
Figure 4.

Triggered display of TAT in response to the acidic tumor environment improved tumor regression in drug resistant ovarian tumor xenografts. Three intravenous doses of doxorubicin-loaded pH-sensitive activatable TAT-functionalized micelles (PHSMpop-upTAT - circle) achieved superior tumor regression in comparison to controls including a non-functionalized pH-sensitive micelle (PHSM - square), a pH-sensitive micelle that displayed TAT at all pH (PHSMTAT - triangle), and free doxorubicin (DOX – inverted triangle). The in vitro cytotoxicity of these constructs at physiological (7.4) and acidic (6.8) pH demonstrated the controlled cytotoxicity of the activatable PHSMpop-upTAT (inset graph). Reprinted from Ref 72. Copyright 2008 with permission from Elsevier.
Controlled presentation of CPPs by dissociation from an ionic inhibitor
Due to the cationic character of many CPPs, and the importance of ionic interactions in initiating cellular uptake, complexing positively charged CPPs with negatively charged groups serves as an alternative means to inhibit the function of CPPs by preventing their interaction with the cell surface. In the simplest approach, the cationic residues of the CPP can be directly coupled to anionic residues in a size-matched peptide. Conjugating these oppositely charged components through a flexible peptide linker encourages their association. This peptide linker can itself be a protease substrate whose cleavage enables the dissociation of the CPP from its inhibitory peptide. Protease-triggered cleavage of the linker thus provides a controlled material response that will initiate cellular uptake that is otherwise prevented when the CPP and inhibitor peptide are complexed together.
In one implementation of this concept a CPP and an anionic peptide were connected by a peptide linker that included a MMP-2 cleavage site.73 Increased tumor specificity has been proposed by the use of linkers cleavable by cell membrane-bound enzymes as opposed to soluble proteases such as MMPs, which may exhibit off-target expression, thereby resulting in premature or off-target presentation of the CPP. Choosing a linker sequence specifically cleaved by prostate-specific antigen (PSA) was the first attempt to trigger activation of a CPP in response to this extracellular cell-surface protease that is selective to and overexpressed by some prostate cancers.74
The most thoroughly characterized system exploiting this approach was utilized for tumor targeted delivery of imaging agents. A Cy5-labeled arginine9 oligomer and an inhibitory glutamate8 oligomer were connected through an MMP-2 cleavable PLGLAG peptide linker and were shown to form hairpin structures due to intramolecular ionic interactions within a single peptide chain.73 Incubation of cleaved constructs with human fibrosarcoma cells resulted in a 17-fold increase in uptake compared to uncleaved controls, as measured by flow cytometry. For in vivo applications a similar activatable construct was conjugated to PEG to increase the typically short plasma half-lives of small peptides. Systemic delivery of these enzyme-activatable PEGylated constructs showed at least a 3-fold increase in tumor accumulation in comparison to non-cleavable construct with a scrambled peptide linker sequence. This approach has been tested in a variety of MMP-2 overexpressing tumors. Accumulation in subcutaneous fibrosarcoma, melanoma, cervical, prostate, colon, and breast cancer tumors ranged from approximately 2 to 6-fold higher than the accumulation of a negative control construct whose D-amino acid linker sequence prevented its recognition and cleavage by tumor enzymes.75
Investigation of this approach for the controlled uptake of larger cargo has been explored with the conjugation of ionically-inhibited CPPs to dendrimers.75, 76 Attachment of the CPP terminus of the cationic CPP-anionic peptide complex to a dendrimeric nanoparticle allowed retention of the CPP on the particle surface following MMP-2 cleavage of the linker that released the inhibitory anionic peptide. A polyamidoamine dendrimer provided a substrate for both the attachment of Cy5 fluorophore and the magnetic resonance (MR) contrast agent gadolinium to enable tumor imaging by two orthogonal modalities. Systemically injected dendrimers preferentially accumulated in MMP-2 overexpressing tumors and provided detectable signals over 48 hours, which enabled surgical resection planning by MR imaging (MRI) (Figure 5). Fluorescence of the Cy5 label was utilized during surgery to enable intra-operative imaging of tumor margins. Due to the high retention of these particles by tumor cells, a single systemically administered dose provided sufficient signal to also permit post-surgical evaluation of resection with MRI. This approach improved tumor resection by ensuring complete removal of tumor margins that could potentially be left behind in conventional resection protocols.77 This imaging-assisted approach was shown to prolong tumor free survival and is a promising means of improving clinical success of tumor resections to achieve cancer-free outcomes.
Figure 5.
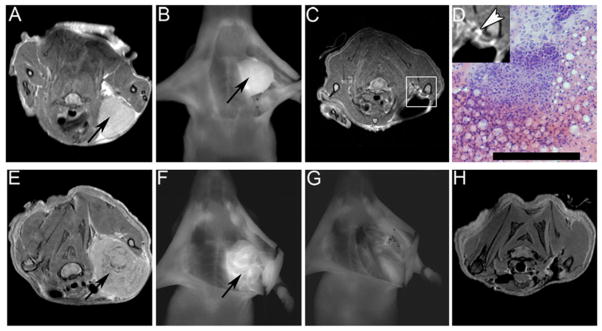
Dendrimeric nanoparticles functionalized with MMP-2 activatable CPPs permitted the targeted tumor delivery of Cy5 fluorophore and MR contrast agent, gadolinium. The dual imaging properties of these particles permitted surgical planning with MRI, real-time fluorescence imaging for intraoperative assistance of tumor resection, and post-operative evaluation with MRI. Accumulation of nanoparticles provided MR contrast to identify a fibrosarcoma xenograft tumor (arrow - A). Intraoperative visualization of the tumor was achieved with fluorescence imaging prior to resection (B). Post-operative MRI revealed residual tumor tissue (box – C) whose cancerous character was confirmed with histology following removal with a second surgery (D). Similar surgical assistance was provided for melanoma xenografts. The tumor was identified with MRI (arrow – E) and fluorescence provided intraoperative visualization (F). Surgical resection was performed such that no visible fluorescence remained at the surgical site (G) and post-operative MRI confirmed the successful removal of all cancer cells (H). Tumor resection assistance with guidance by activatable CPP-nanoparticles accumulated in tumor tissue lead to prolonged tumor-free survival after removal of murine melanoma and mammary adenocarcinoma tumors. Reprinted with permission from Ref 77.
This approach has also been extended to delivery of imaging agents suitable for PET and SPECT. In one such approach an MMP-14 cleavable construct was evaluated for the delivery of a technetium radionuclide chelated to the terminus of the construct’s CPP component.78 Alternatively, MMP-2/9 cleavable constructs have been directly labeled with radionuclides. These radiolabeled MMP-2/9 activatable constructs achieved similar tumor accumulation to those comparable systems described above, with a 6-fold increase in tumor accumulation of activatable constructs, compared to non-activatable controls with scrambled MMP cleavage sequences.79 However, this system also exhibited increased accumulation of activatable constructs in nearly all tissues when compared to non-activatable controls. Dual labeling of the activatable constructs, with 125I conjugated to the attenuating anionic domain and 177Lu conjugated to the cationic polyarginine CPP domain, revealed high ratios of 177Lu/125I signals in the tumor as well as the muscle, heart, lung, and spleen, indicating that accumulation of activated constructs was not specific to the tumor tissue. These results suggest that activation may not be occurring only in the tumor, but that systemic activation in the circulation and accumulation by the EPR effect may contribute to the enhanced tumor accumulation achieved by this method.
Dual labeling of similar activatable CPP carriers has also been developed for use in photoacoustic imaging. In this approach, an MMP-2 cleavable construct was synthesized with a BHQ3 quencher (abs 672 nm) conjugated to an arginine5 CPP domain and an Alexa750 fluorophore (ex/em 749/775 nm) conjugated to the glutamic acid4 anionic inhibitory domain.80 In the intact, unactivated state, both of the attached chromophores contributed to a spectral signal that was a nonlinear combination of their respective properties. Photoacoustic images acquired at 675 and 750 nm showed strong signals from absorption of both chromophores in the intact construct. Upon activation by MMP-2 cleavage of the linker between the domains, allowing release of the anionic inhibitor peptide, the BHQ3-labeled CPP component alone showed high cellular uptake. By subtracting the photoacoustic image acquired at 750 nm from that obtained at 675 nm, the contribution from the non-activated construct was eliminated and the residual signal was attributed to the activated BHQ3-CPP component. This technique has the unique potential to identify specific localization of an activated probe within a mixed population of carriers.
Removal of an inhibitory anionic domain from a cationic CPP can also be achieved by an exogenous factor. For instance, a cationic CPP that is electrostatically bound to an anionic inhibitor can be replaced by a more strongly interacting species that is delivered to the desired site of action.81, 82 This approach has been investigated with the ionic complexation of negatively charged heparin to TAT-functionalized asparaginase, which is an enzyme used to treat acute lymphoblastic leukemia.83 The cellular uptake of asparaginase, measured by flow cytometry, was significantly increased by its conjugation to TAT via a disulfide linker. When complexed with heparin the ionic interactions between this anionic molecule and the cationic TAT prevented cellular uptake in MOLT-4 leukemia cells. Cellular uptake was recovered with the addition of protamine sulfate, a heparin antidote, which binds more strongly to the heparin, displacing it from the TAT and thereby enabling the TAT-asparaginase fusion to interact with the cell surface. Once internalized, the enzyme was liberated from the TAT peptide by cleavage of the disulfide linker in the reductive cytosolic environment within the cell.
Finally, extrinsic triggers can also be used to dissociate an ionic inhibitor from a cationic CPP. This approach has been investigated with UV light as an external trigger to cleave the linker between a CPP and ionic inhibitor. N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer functionalized with a penetratin derivative was rendered inactive by masking the CPP lysine residues with charge neutralizing protective groups conjugated via a photo-labile linker.84 Cellular uptake was thus prevented in this protected state. Exposure to UV light for ten minutes was sufficient to cleave the photo-sensitive linker and release the protecting groups, allowing significant increase in cellular uptake in this activated form (Figure 6). Photo-controlled cytotoxicity was demonstrated by conjugation of the proapoptotic peptide (KLAKLAK)2 to the HPMA copolymer backbone. UV-activated constructs decreased cell viability to 10–20% in four cancer cell lines, in contrast to unactivated constructs that maintained cell viability at 90–100%. The use of an extrinsic trigger presents the opportunity for such a method to be applicable to a wide range of tumor types, though use of endoscopic illumination or near-IR activated constructs may be necessary to ensure access of the photo-trigger to a variety of tumor locations within the body.
Figure 6.
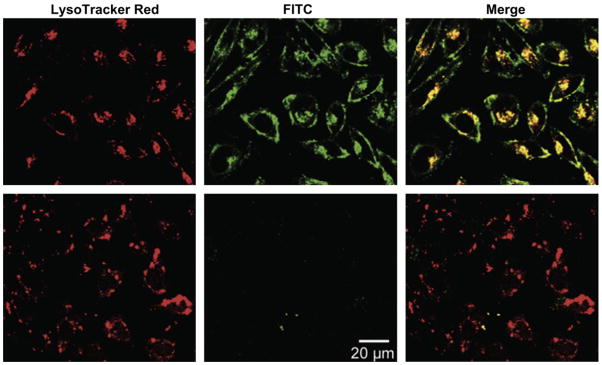
Cellular uptake of CPP-functionalized HPMA was controlled by the dissociation of neutralizing inhibitory domains in response to UV illumination. Charge-neutralizing groups conjugated to the CPP by UV-labile linkers prevented internalization of FITC labeled polymer (green) in PC-3 prostate cancer cells kept in the dark (bottom). Exposure to UV light for 10 minutes was sufficient to remove the neutralizing groups and allow significant increase in cellular uptake (top). Lysosomes were labeled with LysoTracker Red and colocalized, in part, with the internalized HPMA. The delivery to lysosomes provided the acidic conditions necessary to allow intracellular drug release from the carrier using a pH-sensitive hydrazine linker. Reprinted from Ref 84. Copyright 2011 with permission from Elsevier.
Manipulation of CPP charge by ionizable residues
Due to the importance of charge in the initial interactions that lead to cellular internalization of CPPs, an alternative approach to controlling CPP function involves the modulation of CPP ionization from an inactive neutral state to an active cationic state. This approach has been investigated with CPPs that incorporate histidine residues into the CPP to toggle the CPP’s charge. Manipulation of the CPP charge is made possible by the histidine residues, whose pKa of approximately 6.5 makes them neutral at physiologic pH of 7.4 and positively charged below pH 6.5, a pH that may be observed in the extracellular space of tumors.
This approach has been utilized for the selective cellular uptake of a transportan-derived CPP.85 All lysine residues were replaced with histidine residues in this lysine-rich CPP. This histidine-modified CPP showed low cellular uptake at pH 7.4 as the histidine residues remained neutral at this pH, thereby minimizing ionic interactions with the cell surface that are believed to initiate CPP internalization. At pH 6.0, however, the histidine residues became positively charged, promoting the cellular uptake of the CPP (Figure 7). This is in contrast to the original lysine-containing CPP, which demonstrated pH-independent cellular uptake. Conjugation of camptothecin, a cancer chemotherapeutic, to this histidine-modified CPP through a disulfide linker permitted pH-controlled cytotoxicity with intracellular drug release in the reducing environment of the cytosol. The histidine-modified CPP showed increased in vitro cytotoxicity at pH 6.0, in comparison to pH 7.4, due to the increased cellular uptake and intracellular drug delivery achieved when the CPP was positively charged at low pH.
Figure 7.
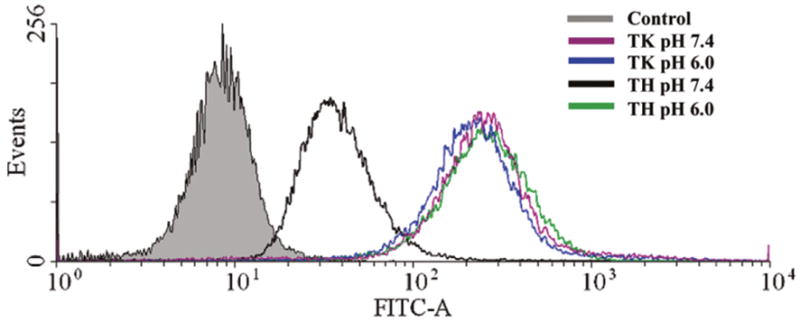
Cellular uptake of a transportan-derived CPP was controlled by the pH-triggered ionization of histidine residues. Peptides whose cationic lysine residues were replaced with histidine residues (TH) exhibited low levels of cellular uptake, as measured by flow cytometry, in HeLa cervical cancer cells at pH 7.4 due to the neutral charge of the histidines at this pH. At pH 6.0 the ionization of the histidine residues conferred positive charge to the CPP and increased cellular uptake. This pH-triggered cellular uptake was in contrast to the original lysine-containing peptide (TK), which demonstrated equivalent cellular uptake at both pH 7.4 and 6.0. Reprinted with permission from Ref 85. Copyright 2011 American Chemical Society.
Modulation of the interfacial density of critical CPP residues by temperature-triggered micelle assembly
Arginine residues have been shown to be critical to the function of many cationic CPPs. For arginine oligomers the CPP function is controlled by the number of sequential arginine residues. A threshold of six residues is believed to be sufficient to achieve cellular uptake, while less than six residues is inadequate in achieving internalization.17 The simple sequence of arginine oligomers is thus a special case in which the CPP’s function can be manipulated by the length of the CPP, and thus the number of presented arginine residues. A variation to this approach stems from the hypothesis that CPP function is not, per se, controlled by the absolute number of sequential arginine residues, but by the local arginine residue density. This hypothesis then suggests that control of the local arginine density may allow one to build a digital “off-on” switch of CPP function by toggling the local density of arginine residues in a nanoscale construct. One means of controlling the local density of arginine residues is by temperature-triggered micelle assembly from thermally responsive block copolymers. This has been investigated with elastin-like polypeptide diblock copolymers (ELPBCs), composed of hydrophobic and hydrophilic ELP blocks.86 Raising the solution above the critical micelle temperature (CMT) selectively desolvates the hydrophobic block and triggers self-assembly of the ELPBC into spherical micelles, wherein the hydrophobic block forms the micelle core and the hydrophilic block forms the micelle corona. Thermally controlled self-assembly can thus serve as a trigger to increase local arginine density when the hydrophilic terminus of the ELPBC is functionalized with arginine residues. When a limited number of arginines, below the threshold necessary for cellular uptake, is appended to the hydrophilic domain, cellular uptake can be prevented in this “off state” wherein the diblock ELPBCs exist as soluble unimers below the CMT. Micelle assembly above the CMT increases the local density of arginines on the micelle corona, exceeding the threshold necessary for cellular uptake and thus triggering internalization at this micelle “on state”.
Modulation of arginine density by temperature-triggered micelle assembly has been explored with ELPBCs whose CMT is between body temperature (37 °C) and the temperature achieved with mild clinical hyperthermia (approximately 42 °C).87, 88 ELPBCs could thus be injected systemically and circulate as soluble unimers in their “off state”, while only in the locally heated tumor tissue could micelle assembly occur and achieve cellular uptake in their “on state”. This approach has been explored in vitro with ELPBCs functionalized with five arginine residues (Arg5-ELPBC).89 At 37 °C HeLa cervical cancer cells exhibited minimal cellular uptake of Arg5-ELPBC in their unimer state, comparable to non-functionalized control ELPBC. It was only at 42 °C, when micelle assembly increased the local arginine density on the micelle corona, that significant internalization was observed (Figure 8). An 8-fold increase in cellular uptake, as measured by flow cytometry, was achieved with Arg5-ELPBC micelle assembly, while non-functionalized ELPBC and temperature-insensitive arginine-functionalized soluble unimer (Arg5-ELP) controls demonstrated no significant change in cellular uptake between 37 and 42 °C.
Figure 8.

Temperature-triggered micelle assembly controlled cellular uptake by modulation of arginine density. Arginine5-functionalized elastin-like polypeptide diblock copolymers (Arg5-ELPBC) achieved cellular uptake in HeLa cervical cancer cells, as quantified by flow cytometry, comparable to controls at 37 °C in their soluble unimer state, with arginine content below the threshold required to achieve significant cellular internalization. At 42 °C the micelle assembly of Arg5-ELPBC enhanced the local arginine density on the micelle corona, which increased cellular uptake 8-fold over that achieved at 37 °C. Non-functionalized diblock copolymers capable of temperature-triggered micelle assembly (ELPBC) and soluble temperature-insensitive arginine-functionalized unimers (Arg5-ELP) demonstrated negligible change in cellular uptake between 37 and 42 °C. Data adapted with permission from Ref 89 Copyright 2012 American Chemical Society.
This approach presents a particularly exciting means of activating CPP function because of its clinically relevant extrinsic trigger. Due to its independence from intrinsic features that may vary in the tumor population, this method could be applicable to a variety of tumor types and disease sites, provided they can be accurately heated. Additionally, this approach provides a means of reversible activation that is not provided by many of the alternative techniques discussed above. When the micelle leaks out of a heated tumor, the cell penetration function turns off as the micelle disassembles into a unimer that presents only five arginine residues. This ability to turn on CPP function in heated tumors and to keep it turned off in unheated healthy tissues may prove to be an important factor in reducing systemic toxicity caused by off-target accumulation, though in vivo experiments are needed – and are in progress– to prove this hypothesis. Should this be the case, evaluation of the in vivo delivery of chemotherapeutics with this approach will further elucidate its potential for targeted accumulation by manipulation of local arginine density.
Conclusion
The non-specific cellular uptake conferred by CPPs make them an interesting class of molecules that can be exploited for the intracellular delivery of therapeutics and imaging agents. The properties of CPPs present an interesting trade-off; while their non-specific and receptor-independent mechanism of cellular uptake provides a valuable means of intracellular delivery that serves as an alternative to approaches of active targeting such as ligand-receptor interactions, their non-specificity creates a significant challenge in targeting delivery by systemic administration to a focal site of disease. To solve this problem, a number of approaches are being actively investigated to trigger CPP function only at a local site of disease following systemic delivery. Harnessing the power of CPPs for tumor targeted delivery has been pursued by a variety of mechanisms, relying on stimulus responsive materials which provide triggered changes in material properties that allow spatially focused presentation of CPPs in response to intrinsic disease characteristics or locally applied extrinsic cues. A number of approaches have shown successful control of CPPs in vitro and some have shown encouraging results in preclinical animal models, suggesting that spatially targeted CPPs for systemic delivery of drugs and imaging agents have exciting potential to impact a variety of solid tumors. Mastering the control of CPPs in systemically administered carriers for targeted delivery may bring to fruition innovative drug and imaging agent vehicles applicable to a wide range of cancer types and a large subset of cancer patients.
Acknowledgments
This work was supported by the NIH through grant R01EB007205 (A.C.) and by a graduate fellowship from the NSF’s Research Triangle MRSEC (NSF DMR-1121107) (S.M.).
Contributor Information
Sarah R. MacEwan, Department of Biomedical Engineering, Duke University, Durham, NC, USA
Ashutosh Chilkoti, Email: Chilkoti@duke.edu, Department of Biomedical Engineering, Duke University, Durham, NC, USA, Center for Biologically Inspired Materials and Material Systems, Duke University, Durham, NC, USA.
References
- 1.Skipper HE. Kinetics of mammary tumor cell growth and implications for therapy. Cancer. 1971;28:1479–1499. doi: 10.1002/1097-0142(197112)28:6<1479::aid-cncr2820280622>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, del Moore GV, Deng J, Anderson KC, Richardson P, Tai YT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338:284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 6.Muss HB, Thor AD, Berry DA, Kute T, Liu ET, Koerner F, Cirrincione CT, Budman DR, Wood WC, Barcos M, et al. c-erbB-2 expression and response to adjuvant therapy in women with node-positive early breast cancer. N Engl J Med. 1994;330:1260–1266. doi: 10.1056/NEJM199405053301802. [DOI] [PubMed] [Google Scholar]
- 7.Fink-Retter A, Gschwantler-Kaulich D, Hudelist G, Mueller R, Kubista E, Czerwenka K, Singer CF. Differential spatial expression and activation pattern of EGFR and HER2 in human breast cancer. Oncol Rep. 2007;18:299–304. [PubMed] [Google Scholar]
- 8.Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008;99:929–935. doi: 10.1111/j.1349-7006.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dexter DL, Leith JT. Tumor heterogeneity and drug resistance. J Clin Oncol. 1986;4:244–257. doi: 10.1200/JCO.1986.4.2.244. [DOI] [PubMed] [Google Scholar]
- 10.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 11.Kerkis A, Hayashi MA, Yamane T, Kerkis I. Properties of cell penetrating peptides (CPPs) IUBMB Life. 2006;58:7–13. doi: 10.1080/15216540500494508. [DOI] [PubMed] [Google Scholar]
- 12.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 13.Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry. 2002;41:14150–14157. doi: 10.1021/bi026661l. [DOI] [PubMed] [Google Scholar]
- 14.Khalil IA, Kogure K, Futaki S, Harashima H. Octaarginine-modified liposomes: enhanced cellular uptake and controlled intracellular trafficking. Int J Pharm. 2008;354:39–48. doi: 10.1016/j.ijpharm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura Y, Kogure K, Futaki S, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–367. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Al-Taei S, Penning NA, Simpson JC, Futaki S, Takeuchi T, Nakase I, Jones AT. Intracellular traffic and fate of protein transduction domains HIV-1 TAT peptide and octaarginine. Implications for their utilization as drug delivery vectors. Bioconjug Chem. 2006;17:90–100. doi: 10.1021/bc050274h. [DOI] [PubMed] [Google Scholar]
- 17.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. Oligocarbamate molecular transporters: design, synthesis, and biological evaluation of a new class of transporters for drug delivery. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 19.Wender PA, Kreider E, Pelkey ET, Rothbard J, Vandeusen CL. Dendrimeric molecular transporters: synthesis and evaluation of tunable polyguanidino dendrimers that facilitate cellular uptake. Org Lett. 2005;7:4815–4818. doi: 10.1021/ol051496y. [DOI] [PubMed] [Google Scholar]
- 20.Vezenkov LL, Maynadier M, Hernandez JF, Averlant-Petit MC, Fabre O, Benedetti E, Garcia M, Martinez J, Amblard M. Noncationic dipeptide mimic oligomers as cell penetrating nonpeptides (CPNP) Bioconjug Chem. 2010;21:1850–1854. doi: 10.1021/bc1002086. [DOI] [PubMed] [Google Scholar]
- 21.Som A, Tezgel AO, Gabriel GJ, Tew GN. Self-activation in de novo designed mimics of cell-penetrating peptides. Angew Chem Int Ed Engl. 2011;50:6147–6150. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 22.Hallbrink M, Kilk K, Elmquist A, Lundberg P, Lindgren M, Jiang Y, Pooga M, Soomets U, Langel U. Prediction of Cell-Penetrating Peptides. International Journal of Peptide Research and Therapeutics. 2005;11:249–259. [Google Scholar]
- 23.Jones S, Holm T, Mager I, Langel U, Howl J. Characterization of bioactive cell penetrating peptides from human cytochrome c: protein mimicry and the development of a novel apoptogenic agent. Chem Biol. 2010;17:735–744. doi: 10.1016/j.chembiol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Marks JR, Placone J, Hristova K, Wimley WC. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J Am Chem Soc. 2011;133:8995–9004. doi: 10.1021/ja2017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 26.Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K, Negishi M, Nomizu M, Sugiura Y, Futaki S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2007;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- 27.Dathe M, Wieprecht T. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim Biophys Acta. 1999;1462:71–87. doi: 10.1016/s0005-2736(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 28.Henriques ST, Melo MN, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem J. 2006;399:1–7. doi: 10.1042/BJ20061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawamoto S, Takasu M, Miyakawa T, Morikawa R, Oda T, Futaki S, Nagao H. Inverted micelle formation of cell-penetrating peptide studied by coarse-grained simulation: importance of attractive force between cell-penetrating peptides and lipid head group. J Chem Phys. 2011;134:095103. doi: 10.1063/1.3555531. [DOI] [PubMed] [Google Scholar]
- 30.Henriques ST, Costa J, Castanho MA. Translocation of beta-galactosidase mediated by the cell-penetrating peptide pep-1 into lipid vesicles and human HeLa cells is driven by membrane electrostatic potential. Biochemistry. 2005;44:10189–10198. doi: 10.1021/bi0502644. [DOI] [PubMed] [Google Scholar]
- 31.Herce HD, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci U S A. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 33.Mueller J, Kretzschmar I, Volkmer R, Boisguerin P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug Chem. 2008;19:2363–2374. doi: 10.1021/bc800194e. [DOI] [PubMed] [Google Scholar]
- 34.Wadia JS, Dowdy SF. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv Drug Deliv Rev. 2005;57:579–596. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Massodi I, Bidwell GL, 3rd, Raucher D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J Control Release. 2005;108:396–408. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR, Juliano RL. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: effects on cellular uptake, binding to target sequences, and biologic actions. Pharm Res. 2002;19:744–754. doi: 10.1023/a:1016136328329. [DOI] [PubMed] [Google Scholar]
- 37.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Ifediba MA, Medarova Z, Ng SW, Yang J, Moore A. siRNA delivery to CNS cells using a membrane translocation peptide. Bioconjug Chem. 2010;21:803–806. doi: 10.1021/bc900488e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi Y, Kim K, Hong S, Kim H, Kwon YJ, Song R. Intracellular protein target detection by quantum dots optimized for live cell imaging. Bioconjug Chem. 2011;22:1576–1586. doi: 10.1021/bc200126k. [DOI] [PubMed] [Google Scholar]
- 40.Splith K, Hu W, Schatzschneider U, Gust R, Ott I, Onambele LA, Prokop A, Neundorf I. Protease-activatable organometal-Peptide bioconjugates with enhanced cytotoxicity on cancer cells. Bioconjug Chem. 2010;21:1288–1296. doi: 10.1021/bc100089z. [DOI] [PubMed] [Google Scholar]
- 41.Oh E, Delehanty JB, Sapsford KE, Susumu K, Goswami R, Blanco-Canosa JB, Dawson PE, Granek J, Shoff M, Zhang Q, et al. Cellular uptake and fate of PEGylated gold nanoparticles is dependent on both cell-penetration peptides and particle size. ACS Nano. 2011;5:6434–6448. doi: 10.1021/nn201624c. [DOI] [PubMed] [Google Scholar]
- 42.Qin Y, Chen H, Zhang Q, Wang X, Yuan W, Kuai R, Tang J, Zhang L, Zhang Z, Liu J, et al. Liposome formulated with TAT-modified cholesterol for improving brain delivery and therapeutic efficacy on brain glioma in animals. Int J Pharm. 2011;420:304–312. doi: 10.1016/j.ijpharm.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Sawant RR, Torchilin VP. Enhanced cytotoxicity of TATp-bearing paclitaxel-loaded micelles in vitro and in vivo. Int J Pharm. 2009;374:114–118. doi: 10.1016/j.ijpharm.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanazawa T, Taki H, Tanaka K, Takashima Y, Okada H. Cell-penetrating peptide-modified block copolymer micelles promote direct brain delivery via intranasal administration. Pharm Res. 2011;28:2130–2139. doi: 10.1007/s11095-011-0440-7. [DOI] [PubMed] [Google Scholar]
- 45.Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–172. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 46.Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 47.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 49.Flynn CR, Cheung-Flynn J, Smoke CC, Lowry D, Roberson R, Sheller MR, Brophy CM. Internalization and intracellular trafficking of a PTD-conjugated anti-fibrotic peptide, AZX100, in human dermal keloid fibroblasts. J Pharm Sci. 2010;99:3100–3121. doi: 10.1002/jps.22087. [DOI] [PubMed] [Google Scholar]
- 50.Brandt F, O’Connell C, Cazzaniga A, Waugh JM. Efficacy and safety evaluation of a novel botulinum toxin topical gel for the treatment of moderate to severe lateral canthal lines. Dermatol Surg. 2010;36 (Suppl 4):2111–2118. doi: 10.1111/j.1524-4725.2010.01711.x. [DOI] [PubMed] [Google Scholar]
- 51.Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 52.Miyaji Y, Walter S, Chen L, Kurihara A, Ishizuka T, Saito M, Kawai K, Okazaki O. Distribution of KAI-9803, a novel delta-protein kinase C inhibitor, after intravenous administration to rats. Drug Metab Dispos. 2011;39:1946–1953. doi: 10.1124/dmd.111.040725. [DOI] [PubMed] [Google Scholar]
- 53.Liu JR, Zhao Y, Patzer A, Staak N, Boehm R, Deuschl G, Culman J, Bonny C, Herdegen T, Eschenfelder C. The c-Jun N-terminal kinase (JNK) inhibitor XG-102 enhances the neuroprotection of hyperbaric oxygen after cerebral ischaemia in adult rats. Neuropathol Appl Neurobiol. 2010;36:211–224. doi: 10.1111/j.1365-2990.2009.01047.x. [DOI] [PubMed] [Google Scholar]
- 54.Aguilera TA, Olson ES, Timmers MM, Jiang T, Tsien RY. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol (Camb) 2009;1:371–381. doi: 10.1039/b904878b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 56.Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287–5297. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 58.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–181. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur J Pharm Biopharm. 2009;71:431–444. doi: 10.1016/j.ejpb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kale AA, Torchilin VP. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J Drug Target. 2007;15:538–545. doi: 10.1080/10611860701498203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273. doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mok H, Bae KH, Ahn CH, Park TG. PEGylated and MMP-2 specifically dePEGylated quantum dots: comparative evaluation of cellular uptake. Langmuir. 2009;25:1645–1650. doi: 10.1021/la803542v. [DOI] [PubMed] [Google Scholar]
- 64.Choi JM, Ahn MH, Chae WJ, Jung YG, Park JC, Song HM, Kim YE, Shin JA, Park CS, Park JW, et al. Intranasal delivery of the cytoplasmic domain of CTLA-4 using a novel protein transduction domain prevents allergic inflammation. Nat Med. 2006;12:574–579. doi: 10.1038/nm1385. [DOI] [PubMed] [Google Scholar]
- 65.Harris TJ, von Maltzahn G, Lord ME, Park JH, Agrawal A, Min DH, Sailor MJ, Bhatia SN. Protease-triggered unveiling of bioactive nanoparticles. Small. 2008;4:1307–1312. doi: 10.1002/smll.200701319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6:3491–3498. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sethuraman VA, Bae YH. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release. 2007;118:216–224. doi: 10.1016/j.jconrel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sethuraman VA, Lee MC, Bae YH. A biodegradable pH-sensitive micelle system for targeting acidic solid tumors. Pharm Res. 2008;25:657–666. doi: 10.1007/s11095-007-9480-4. [DOI] [PubMed] [Google Scholar]
- 69.Kuai R, Yuan W, Qin Y, Chen H, Tang J, Yuan M, Zhang Z, He Q. Efficient Delivery of Payload into Tumor Cells in a Controlled Manner by TAT and Thiolytic Cleavable PEG Co-Modified Liposomes. Mol Pharm. 2010 doi: 10.1021/mp100171c. [DOI] [PubMed] [Google Scholar]
- 70.Kuai R, Yuan W, Li W, Qin Y, Tang J, Yuan M, Fu L, Ran R, Zhang Z, He Q. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol Pharm. 2011;8:2151–2161. doi: 10.1021/mp200100f. [DOI] [PubMed] [Google Scholar]
- 71.Lee ES, Na K, Bae YH. Super pH-sensitive multifunctional polymeric micelle. Nano Lett. 2005;5:325–329. doi: 10.1021/nl0479987. [DOI] [PubMed] [Google Scholar]
- 72.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J Control Release. 2008;129:228–236. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BL. Intracellular cargo delivery by an octaarginine transporter adapted to target prostate cancer cells through cell surface protease activation. Bioconjug Chem. 2006;17:787–796. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- 75.Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, Gross LA, Tsien RY. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol (Camb) 2009;1:382–393. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG, Tsien RY. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc Natl Acad Sci U S A. 2010;107:4317–4322. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Watkins GA, Jones EF, Scott Shell M, VanBrocklin HF, Pan MH, Hanrahan SM, Feng JJ, He J, Sounni NE, Dill KA, et al. Development of an optimized activatable MMP-14 targeted SPECT imaging probe. Bioorg Med Chem. 2009;17:653–659. doi: 10.1016/j.bmc.2008.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Duijnhoven SM, Robillard MS, Nicolay K, Grull H. Tumor targeting of MMP-2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J Nucl Med. 2011;52:279–286. doi: 10.2967/jnumed.110.082503. [DOI] [PubMed] [Google Scholar]
- 80.Levi J, Kothapalli SR, Ma TJ, Hartman K, Khuri-Yakub BT, Gambhir SS. Design, synthesis, and imaging of an activatable photoacoustic probe. J Am Chem Soc. 2010;132:11264–11269. doi: 10.1021/ja104000a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YT, Kwon YM, Spangrude GJ, Liang JF, Chung HS, Park YJ, Yang VC. Preliminary in vivo evaluation of the protein transduction domain-modified ATTEMPTS approach in enhancing asparaginase therapy. J Biomed Mater Res A. 2009;91:209–220. doi: 10.1002/jbm.a.32204. [DOI] [PubMed] [Google Scholar]
- 82.Huang Y, Park YS, Wang J, Moon C, Kwon YM, Chung HS, Park YJ, Yang VC. ATTEMPTS system: a macromolecular prodrug strategy for cancer drug delivery. Curr Pharm Des. 2010;16:2369–2376. doi: 10.2174/138161210791920441. [DOI] [PubMed] [Google Scholar]
- 83.Kwon YM, Li YT, Liang JF, Park YJ, Chang LC, Yang VC. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: a proof-of-concept investigation. J Control Release. 2008;130:252–258. doi: 10.1016/j.jconrel.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shamay Y, Adar L, Ashkenasy G, David A. Light induced drug delivery into cancer cells. Biomaterials. 2011;32:1377–1386. doi: 10.1016/j.biomaterials.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 85.Zhang W, Song J, Zhang B, Liu L, Wang K, Wang R. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconjug Chem. 2011;22:1410–1415. doi: 10.1021/bc200138d. [DOI] [PubMed] [Google Scholar]
- 86.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roemer RB. Engineering aspects of hyperthermia therapy. Annu Rev Biomed Eng. 1999;1:347–376. doi: 10.1146/annurev.bioeng.1.1.347. [DOI] [PubMed] [Google Scholar]
- 88.Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia. 2001;17:1–18. doi: 10.1080/02656730150201552. [DOI] [PubMed] [Google Scholar]
- 89.MacEwan SR, Chilkoti A. Digital switching of local arginine density in a genetically encoded self-assembled polypeptide nanoparticle controls cellular uptake. Nano Lett. 2012;12:3322–3328. doi: 10.1021/nl301529p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further Reading
- Bolhassani A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim Biophys Acta. 2011;1816:232–246. doi: 10.1016/j.bbcan.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Lehto T, Kurrikoff K, Langel U. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin Drug Deliv. 2012;9:823–836. doi: 10.1517/17425247.2012.689285. [DOI] [PubMed] [Google Scholar]
- Margus H, Padari K, Pooga M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol Ther. 2012;20:525–533. doi: 10.1038/mt.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensen N, Walton JGA, Bradley M. Peptides for cell-selective drug delivery. Trends Pharmacol Sci. 2012;33:186–192. doi: 10.1016/j.tips.2012.02.002. [DOI] [PubMed] [Google Scholar]