

Abstract
CONTEXT:
Idiopathic pulmonary fibrosis (IPF) and chronic hypersensitivity pneumonitis (HP) are diffuse parenchymal lung diseases characterized by a mixture of inflammation and fibrosis, leading to lung destruction and finally death.
AIMS:
The aim of this study was to compare different pathophysiological mechanisms, such as angiogenesis, coagulation, fibrosis, tissue repair, inflammation, epithelial damage, oxidative stress, and matrix remodeling, in both disorders using bronchoalveolar lavage (BAL).
METHODS:
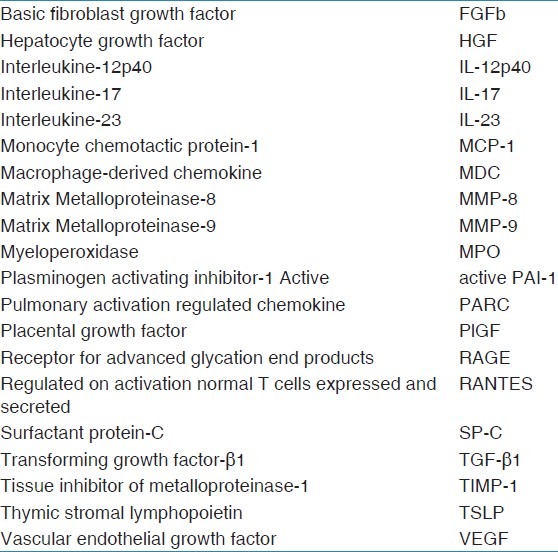
At diagnosis, patients underwent bronchoscopy with BAL and were divided into three groups: Control (n = 10), HP (n = 11), and IPF (n = 11), based on multidisciplinary approach (clinical examination, radiology, and histology): Multiplex searchlight technology was used to analyze 25 proteins representative for different pathophysiological processes: Eotaxin, basic fibroblast growth factor (FGFb), fibronectin, hepatocyte growth factor (HGF), interleukine (IL)-8, IL-12p40, IL-17, IL-23, monocyte chemotactic protein (MCP-1), macrophage-derived chemokine (MDC), myeloperoxidase (MPO), matrix metalloproteinase (MMP)-8, MMP-9, active plasminogen activating inhibitor 1 (PAI-1), pulmonary activation regulated chemokine (PARC), placental growth factor (PlGF), protein-C, receptor for advanced glycation end products (RAGE), regulated on activation normal T cells expressed and secreted (RANTES), surfactant protein-C (SP-C), transforming growth factor-β1 (TGF-β1), tissue inhibitor of metalloproteinase-1 (TIMP-1), tissue factor, thymic stromal lymphopoietin (TSLP), and vascular endothelial growth factor (VEGF).
RESULTS:
All patients suffered from decreased pulmonary function and abnormal BAL cell differential compared with control. Protein levels were increased in both IPF and HP for MMP-8 (P = 0.022), MMP-9 (P = 0.0020), MCP-1 (P = 0.0006), MDC (P = 0.0048), IL-8 (P = 0.013), MPO (P = 0.019), and protein-C (P = 0.0087), whereas VEGF was decreased (P = 0.0003) compared with control. HGF was upregulated in HP (P = 0.0089) and active PAI-1 was upregulated (P = 0.019) in IPF compared with control. Differences in expression between IPF and HP were observed for IL-12p40 (P = 0.0093) and TGF-β1 (P = 0.0045).
CONCLUSIONS:
Using BAL, we demonstrated not only expected similarities but also important differences in both disorders, many related to the innate immunity. These findings provide new clues for further research in both disorders.
Keywords: Bronchoalveolar lavage, enzyme-linked immunosorbent assay, hypersensitivity pneumonitis, interstitial lung disease, idiopathic pulmonary fibrosis
Diffuse parenchymal lung diseases (DPLDs) are a group of more than 200 diverse chronic pulmonary disorders characterized by progressive fibrosis leading to destruction of lung architecture resulting in respiratory failure and finally death. Being mostly non-specific, distinguishing between different DPLDs is difficult. The classification is often based on differences in etiology with IPF being the most common of DPLDs of unknown origin and chronic HP is an important representative for DPLDs of known cause.[1]
Both IPF and chronic HP lead to end-stage pulmonary fibrosis with excessive collagen deposition, extracellular matrix deposition, and destruction of lung architecture, leading to respiratory failure.[2] Although they share similarities in clinical presentation, prognosis and treatment are different, indicating the importance to differentiate between these disorders for which BAL may be used.[3,4]
The pathophysiological mechanism of HP is accepted to be a hypersensitivity reaction, induced by a T-cell-mediated reaction after sensitization with antigens such as organic dusts, bacteria, and molds,[5] but the exact mechanisms are unresolved. Despite intensive research, the pathophysiological mechanisms of IPF remain enigmatic. Complicating factors are (1) the majority of current knowledge is based on the mouse model of bleomycin-induced fibrosis, which is an useful model but not specific for IPF, (2) the late diagnosis of IPF precludes the ability to study early mechanisms and etiology, (3) the discrepancy between clinical, histological, and radiological findings for diagnosis, and (4) the scarcity of the disease.
In the pathogenesis of pulmonary fibrosis, many contributing mechanisms have been described, including angiogenesis, coagulation, fibrogenesis, tissue repair, inflammation, epithelial damage, matrix remodeling, and oxidative stress. As both disorders progress toward end-stage parenchymal lung fibrosis, an important question is whether similar mechanisms drive this fibrosis. BAL represents the closest sampling tool of the lung besides biopsies and the earliest time point possible to research these disorders. Therefore, our aim was to study the differences and similarities in mechanistic pathways involved in IPF and chronic HP using BAL fluid. For this study, we focused on 25 proteins related to different mechanisms using multiplex SearchLight® Assay System. Its use helps to provide new information to unravel mechanisms triggering pulmonary fibrosis.[3]
Methods
Study design
This study was approved by the Ethics Committee (S51293) and Biosafety (MS20101568) of the University Hospital UZ Leuven, Belgium. At the time of diagnosis, patients underwent spirometry tests and bronchoscopy with BAL as part of the diagnostic work-up. The total lung capacity (TLC), forced vital capacity (FVC), forced expiratory volume in 1 s (FEV1), and diffusing capacity (DLCO) were measured according to international guidelines.[6] During bronchoscopic evaluation, BAL was performed following standard guidelines.[7,8] BAL consisted of four aliquots of sterile saline (50 ml) instilled in the right middle lobe or lingula; returned fraction 1 was used for microbiological evaluation, fractions 2, 3 and 4 were pooled and used to assess cellular differentiation, BAL return and protein analysis.[9]
After multidisciplinary consultation, patients with IPF (n = 11) and HP (n = 11) were selected and compared with a control group (n = 10) without evidence for a pulmonary disease. BAL samples were analyzed by means of multiplex searchlight technology to unravel different pathological mechanisms.
Patient selection
Patients were diagnosed within a multidisciplinary consensus as suggested by international guidelines. IPF patient diagnosis was based on clinical, radiological, and/or pathological data following international guidelines.[10] Patients were diagnosed with chronic HP based on their clinical course and insidious onset over a period of months, history of antigen exposure, radiological, and/or histopathological data.[11,12] Both disorders showed the presence of fibrosis in radiological data. For the control group, history was recorded and BAL, biopsy, and radiological imaging were performed to exclude pulmonary illness/fibrosis (e.g., esophageal cancer, connective tissue disease without pulmonary affection). Medical history provided information regarding age, gender, and smoking history (pack years and amount of current-/ex-/non-smokers). Plasma C-reactive protein (CRP) levels were measured to assess systemic inflammation.[13] The selection was refined by excluding patients with lack of clinical data, no full pulmonary function data, lack of sufficient BAL sample, and highly increased CRP levels (>20 mg/l). Finally, 10 controls, 11 IPF, and 11 HP patients were included.
Bronchoalveolar lavage protein measurement
Protein expression was analyzed in undiluted or 1:10 diluted BAL supernatant using the custom multiplex SearchLight® Assay System (Aushon, Billerica, MA, USA). Twenty-five proteins based on the different pathophysiological mechanisms were selected: Eotaxin, basic fibroblast growth factor (FGFb), fibronectin, HGF, IL-8, IL-12p40, IL-17, IL-23, MCP-1, MDC, MPO, MMP-8, MMP-9, active PAI-1, PARC, PlGF, Protein-C, RAGE, RANTES, SP-C, TGF-β1, TIMP-1, tissue factor, TSLP, and VEGF (for abbreviations, see Table 1). If concentration was below detection limit, a value of 50% of the detection limit was attributed (detection limits: FGFb = 2.0 pg/ml; IL-12p40 = 0.6 pg/ml; IL-17 = 0.8 pg/ml; IL-23 = 19.5 pg/ml; RANTES = 0.4 pg/ml; SP-C = 65.4 pg/ml; TGF-β1 = 19.5 pg/ml; and TSLP = 2.4 pg/ml).
Table 1.
Abbreviations for factors analyzed in bronchoalveolar lavage fluid
Analysis
Results are expressed as median (interquartile range [IQR]). Significances among the groups were tested by Kruskal–Wallis one-way analysis of variance (ANOVA) in combination with Mann–Whitney U test. Correlation analysis using the Spearman rank test was performed on both HP and control and on both the IPF and control. Statistical analysis was performed using Prism 4.1 software (GraphPad, San Diego, CA, USA). Results were considered significant if P < 0.05.
Results
Patient characteristics
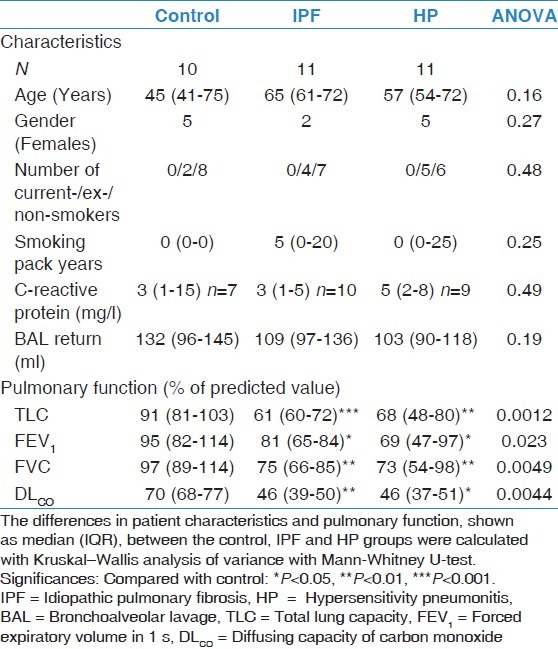
Patient characteristics are summarized in Table 2. No significant differences were observed for age, sex, smoking history, plasma CRP level, or BAL return fraction between the control, IPF, and HP groups.
Table 2.
Patient characteristics
Pulmonary function and bronchoalveolar lavage cell differentiation
Pulmonary function measurements are described in Table 2 and BAL cell differentiation in Table 3. Both IPF and HP patients showed significant decrease in pulmonary function parameters (TLC, FVC, FEV1, and DLCO) compared with the control group. No differences were found between IPF and HP patients.
Table 3.
Cellular differentiation of bronchoalveolar lavage fluid
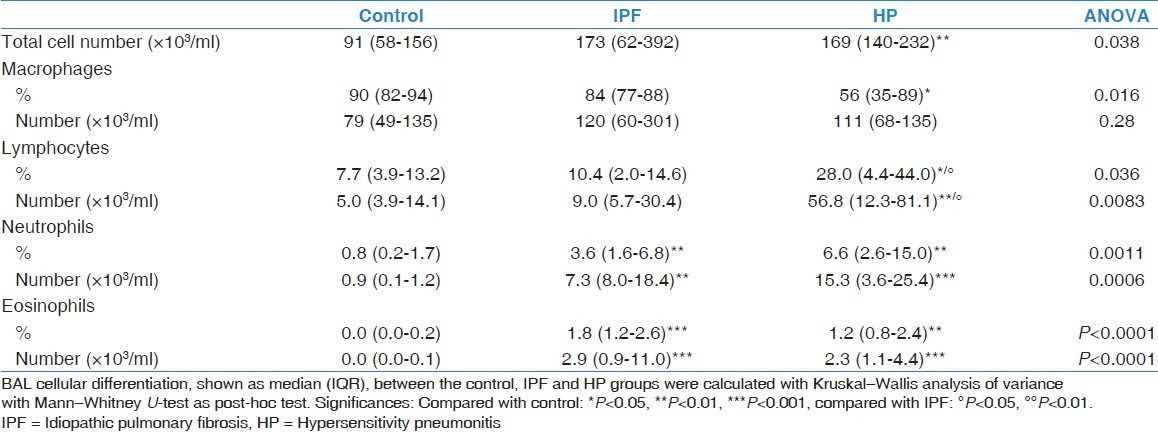
In IPF, an increase in percentage and total number of neutrophils (P = 0.0025; P = 0.0017) and eosinophils (P = 0.0002; P = 0.0002) was observed compared with the control group.
In HP, total white blood cell number was significantly increased compared with the control group (P = 0.0067). The number of macrophages did not change but a decrease in their percentage (P = 0.0167) was observed. An increase in percentage and total number of lymphocytes (P = 0.0412; P = 0.0067), neutrophils (P = 0.0017; P = 0.0008), and eosinophils (P = 0.0002; P = 0.0002) was observed compared with control.
When comparing IPF with HP, an increase in percentage of lymphocytes (P = 0.00216) was observed in HP.
Bronchoalveolar lavage protein measurement
Protein measurements are summarized in Table 4. ANOVA statistics were not significant for RAGE, SP-C, TIMP-1, fibronectin, eotaxin, IL-17A, IL-23, PARC, RANTES, TSLP, PlGF, FGFb, and tissue factor and will therefore not be further discussed.
Table 4.
Protein profile in bronchoalveolar lavage fluid
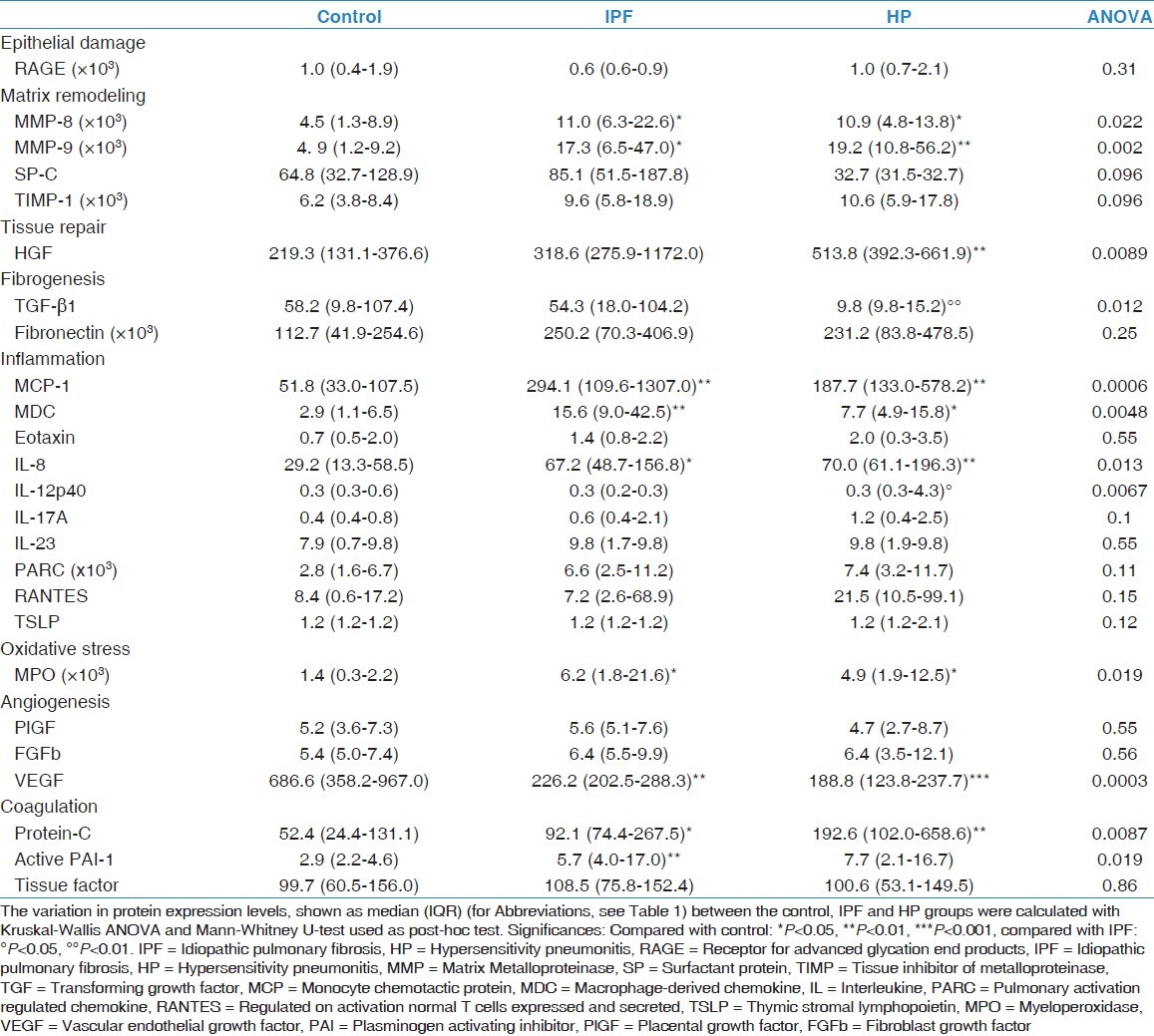
When compared with the control group, an increase in MMP-8 (P = 0.038; P = 0.0083), MMP-9 (P = 0.010; P = 0.0006), MCP-1 (P = 0.0011; P = 0.0014), MDC (P = 0.0044; P = 0.018), IL-8 (P = 0.018; P = 0.0083), MPO (P = 0.015; P = 0.022), and Protein-C (P = 0.045; P = 0.0054) was observed, whereas VEGF was decreased (P = 0.0014; P = 0.0005) in both disorders.
In IPF, active PAI-1 was increased (P = 0.0022), a trend toward increase for HGF (P = 0.053) and a decrease for IL-12p40 (P = 0.072) was found compared with control.
In HP, compared with control, HGF was significantly upregulated (P = 0.0022) and a trend toward increase for active PAI-1 (P = 0.091) and decrease for TGF-β1 (P = 0.057) was found.
In HP, compared with IPF, TGF-β1 (P = 0.0045) was decreased and IL-12p40 (P = 0.072) was increased.
Correlation analysis
Correlation results are summarized in Table 5. Regarding IPF, there was a significant correlation between DLCO and BAL protein expression of HGF (R = –0.49; P = 0.023), MCP-1 (R = –0.47; P = 0.030), MDC (R = –0.58; P = 0.0064), IL-8 (R = –0.45; P = 0.040), IL-12p40 (R = 0.50; P = 0.020), VEGF (R = 0.60; P = 0.0039), Protein-C (R = –0.50; P = 0.022), and active PAI-1 (R = –0.49; P = 0.024). In addition, a correlation was found between FVC and MDC (R = –0.48; P = 0.029), IL-8 (R = –0.47; P = 0.031), VEGF (R = 0.56; P = 0.0087), and Protein-C (R = –0.67; P = 0.0008).
Table 5.
Correlation analysis of bronchoalveolar lavage protein expression with pulmonary function
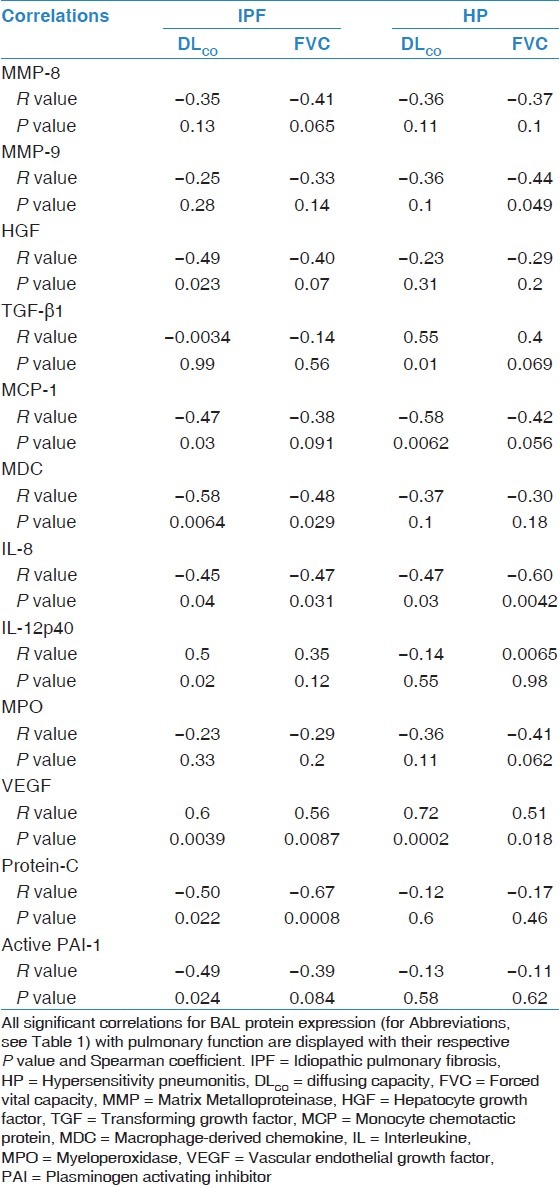
In patients with HP, there was a significant correlation between DLCO and TGF-β1 (R = 0.55; P = 0.010), MCP-1 (R = –0.58; P = 0.0062), IL-8 (R = –0.47; P = 0.030), and VEGF (R = 0.72; P = 0.0002). Additionally, there was a significant correlation between FVC and MMP-9 (R = –0.44; P = 0.049), IL-8 (R = –0.60; P = 0.0042), and VEGF (R = 0.51; P = 0.018).
Discussion
In this study, we examined the involvement of different pathophysiological mechanisms in BAL in IPF and HP, using multiplex protein analysis of 25 different proteins. In both IPF and HP, we observed differences in key factors of angiogenesis (VEGF), coagulation (protein-C and active PAI-1), fibrogenesis (TGF-β1), tissue repair (HGF), inflammation (MCP-1, MDC, and IL-8), matrix remodeling (MMP-8 and MMP-9), and oxidative stress (MPO). Patients with IPF or HP demonstrated decreased pulmonary function combined with altered differential cell counts of BAL. Although IPF and HP differ in onset and clinical presentation, only a few significant differences between them were found.
The first observation in the well-characterized groups was that BAL cellular pattern in both disorders was different, as patients with HP suffer from lymphocytic inflammation compared with IPF and control. This might suggest a discriminative role for BAL in diagnosis of IPF and HP.
The role of innate immunity in IPF and HP has been shown by the upregulation of MCP-1 and MDC. MCP-1, involved in macrophage recruitment, was negatively correlated with DLCO for IPF and HP.[14,15] Suga et al. investigated MCP-1 in HP and found no increase in its expression. This difference might be due to the high lymphocytosis (average > 70%) found in their patients.[13] Alternatively, this can be explained by a higher number of patients with acute HP in comparison with our study. In seasonal summer-type HP, an increase in MCP-1 was interpreted as a possible causative mechanism following repeated stimulations.[16] MDC was negatively correlated with DLCO and FVC in IPF and negatively correlated with DLCO in HP. MDC contributes to pulmonary fibrosis by recruiting macrophages and triggering T-helper cell 2 inflammation, further inducing fibrogenic proteins by stimulating macrophages.[17]
IL-8 functions as a major chemoattractant for neutrophils and has a role in angiogenesis.[18] In both IPF and HP, IL-8 was significantly upregulated and correlated negatively with DLCO and FVC. Neutrophil numbers and percentages were increased in both disorders, indicating an important role for IL-8 as neutrophil attractant. IL-8 has been proposed as an early phase marker for IPF and an indicator of continuous exposure to provoking agents for HP.[19]
MPO expression, a marker for oxidative stress produced by neutrophils, was increased in IPF and HP, corroborating with literature.[20] A shift in the redox balance of IPF has been demonstrated to favor oxidants involving an IL-8-dependent mechanism.[21] In addition, increased levels of MMP-8 and MMP-9, expressed by neutrophils, were found without a compensatory increase in TIMP-1 in both disorders, corroborating the findings of Henry et al.[22,23] This imbalance enhances the activity of MMPs influencing remodeling and development of fibrosis.
Despite current issues on BAL and neutrophils, we observed an important role for immunological mechanisms including neutrophilia and associated oxidative stress and matrix remodeling, in perpetuating both disorders. The activation of the innate arm of the immune system can be linked with reflux,[24] Pseudomonas colonization,[25] and air pollution.[26] Although there is no direct proof for the involvement of innate activation and its different activators, our results might point to a certain role in the process. We recently demonstrated in lung transplant patients that traffic-related air pollution can cause substantial airway immunological reactions with an increase in mortality. Yet very importantly, it was observed that macrolide therapy was protective for this immunological reaction and related development of chronic rejection and mortality. With the studied disorders, HP and IPF, showing similar mechanisms regarding innate immunity in chronic rejection after lung transplantation, one should consider decreasing the innate immune response. In this regard, as in chronic rejection, a potential agent might be the macrolide azithromycin, which was recently studied in the bleomycin mouse model where it modulated both innate and adaptive immunity and consequently reduced parenchymal fibrosis.[27]
Previous studies have shown an increased ratio of angiogenic and angiostatic chemokines in BAL of IPF and HP patients, indicating upregulation of angiogenesis.[28,29] In this study, we observed a downregulation of VEGF in IPF and HP. It has been suggested that VEGF is increased in non-fibrotic, more inflammatory regions and decreased in severe fibrosis.[30,31] This decrease of VEGF at the site of fibrosis may be mediated by proteolytic degradation or may result from epithelial cell apoptosis or injury.[32] Thickening of the barrier between the epithelial surface and the intravascular space due to fibrosis may alter the secretion of VEGF. This also suggests the reason for positive correlation of VEGF with DLCO and FVC in both disorders.[33]
Protein-C was upregulated in both disorders and when activated, it has shown a potential anti-fibrotic effect by diminishing inflammation and coagulation.[34,35] This increased expression was remarkably higher in patients with HP compared with IPF. This is in agreement with literature where protein-C has shown to be increased in IPF, but its activated form was decreased.[36] Enhanced plasminogen activity provides an environment supporting tissue repair in the lung by increasing levels of active HGF. We are the first to report an upregulation of HGF in HP. HGF stimulates the proliferation of alveolar type II epithelial cells and inhibits epithelial to myofibroblast transition.[37,38] It has been shown that production and activation of HGF are reduced in fibroblasts in IPF secondary to a defect in prostaglandin E2 secretion.[39] The expression of HGF may also be restrained by the anti-fibrinolytic effect of PAI-1, which was upregulated and correlated with DLCO in patients with IPF, as shown in literature.[40] These data show that the role of the coagulation pathway may be different between IPF and chronic HP. The enhanced plasminogen activity may provide an environment explaining a slower progression of pulmonary fibrosis in chronic HP when compared to IPF.
Apart from similarities, significant differences between IPF and HP were also found. IL-12p40 was upregulated in HP compared with IPF. IL-12p40 directs the immune response toward the appropriate location and correct pathogen.[41] This points toward the role of the hypersensitivity reaction present in patients with HP.[42] TGF-β1, known as the fibrotic molecule in IPF and experimental fibrosis, was not significantly different in both disorders when compared with control,[43] due to large variance in samples. A significant decrease in patients with HP compared with IPF was observed. This difference between IPF and HP has been documented previously by Hagimoto et al.[44] The increase in TGF-β1 is thought to be produced by alveolar macrophages, in an early inflammatory phase and by epithelial cells during the later fibrotic phase.[45] This could explain the lower concentration of TGF-β1 in HP as they are in a chronic phase of the disorder, with less fibrosis than in IPF.
VEGF (angiogenesis), IL-8, MCP-1, MDC, IL-12p40 (inflammation), HGF (tissue repair), TGF-β1 (fibrogenesis), MMP-9 (matrix remodeling), protein-C, and active PAI-1 (coagulation) were correlated with DLCO and/or FVC for IPF and HP, reflecting the degree of severity. This indeed suggests that pulmonary fibrosis is the result of a combination of different pathological mechanisms.
The major limitation of this study is the low number of samples that were used. Both IPF and HP are orphan diseases and to establish homogenous groups well-documented cases are needed. We are convinced that with higher number of patients, the results would be more pronounced and might have less variability. BAL sampling was performed at the time of diagnosis, potentially reflecting different stages of pulmonary fibrosis, which might explain the broad range of protein expression in each group [Figure 1]. However, time of diagnosis presents the earliest possible phase for BAL sampling in the clinic. This study confirms the usefulness of BAL for research purposes uncovering similarities and differences between these disorders.
Figure 1.
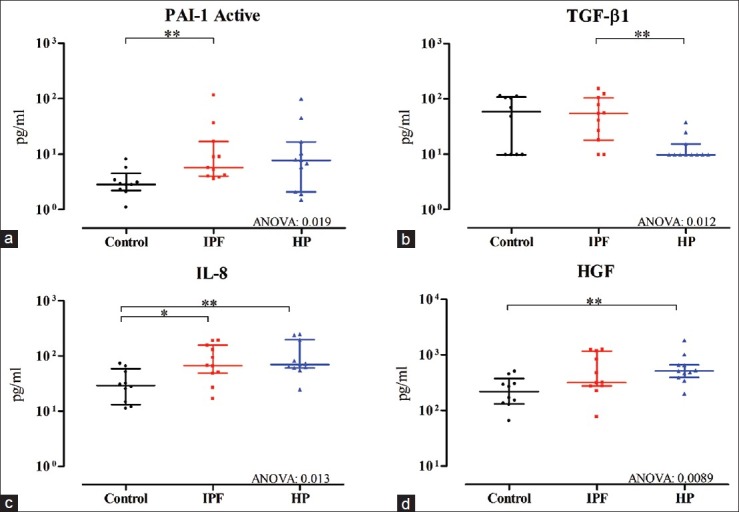
The expression of (a) active PAI-1, (b) TGF-β1, (c) IL-8, and (d) HGF shown as median with IQR (for Abbreviations, see Table 1) in BAL fluid of 10 controls, 11 IPF, and 11 HP patients. Significances: *P<0.05, **P<0.01 remove:, and ***P<0.001
This research provides a better understanding of the involvement of underlying mechanisms and differences between these diseases are compared with the use of animal models. This study demonstrates that though common mechanisms appear, some mechanistic discrepancies were discovered, providing new clues for further research in both disorders.
Footnotes
Source of Support: Wim Wuyts is a Senior Clinical Investigator of the Research Foundation – Flanders: 1.8.325.12N. Stijn Willems is research fellow of the Agency for Innovation by Science and Technology in Flanders (IWT) and Bart Vanaudenaerde is a senior research fellow of the FWO. Geert M Verleden is holder of the GSK (Belgium) chair in respiratory pharmacology at the Katholieke Universiteit Leuven (KUL), and is supported by grants from the KUL (OT/050/10) and the FWO.Flanders: G.0643.08 and G.0723.10.
Conflict of Interest: None declared.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayakawa H, Shirai M, Sato A, Yoshizawa Y, Todate A, Imokawa S, et al. Clinicopathological features of chronic hypersensitivity pneumonitis. Respirology. 2002;7:359–64. doi: 10.1046/j.1440-1843.2002.00406.x. [DOI] [PubMed] [Google Scholar]
- 3.Wells AU. The clinical utility of bronchoalveolar lavage in diffuse parenchymal lung disease. Eur Respir Rev. 2010;19:237–41. doi: 10.1183/09059180.00005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capelozzi VL, Faludi EP, Balthazar AB, Fernezlian SD, Filho JV, Parra ER. Bronchoalveolar lavage improves diagnostic accuracy in patients with diffuse lung disease. Diagn Cytopathol. 2011 doi: 10.1002/dc.21743. [DOI] [PubMed] [Google Scholar]
- 5.McSharry C, Anderson K, Bourke SJ, Boyd G. Takes your breath away – The immunology of allergic alveolitis. Clin Exp Immunol. 2002;128:3–9. doi: 10.1046/j.1365-2249.2002.01849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner RM, Hankinson JL. Standardization of spirometry – 1987 ATS update (American Thoracic Society) J Occup Med. 1988;30:272–3. [PubMed] [Google Scholar]
- 7.Technical recommendations and guidelines for bronchoalveolar lavage (BAL). Report of the European Society of Pneumology Task Group. Eur Respir J. 1989;2:561–85. [PubMed] [Google Scholar]
- 8.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–14. doi: 10.1164/rccm.201202-0320ST. [DOI] [PubMed] [Google Scholar]
- 9.Haslam PL, Baughman RP. Report of ERS Task Force: Guidelines for measurement of acellular components and standardization of BAL. Eur Respir J. 1999;14:245–8. doi: 10.1034/j.1399-3003.1999.14b01.x. [DOI] [PubMed] [Google Scholar]
- 10.American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 11.Schuyler M, Cormier Y. The diagnosis of hypersensitivity pneumonitis. Chest. 1997;111:534–6. doi: 10.1378/chest.111.3.534. [DOI] [PubMed] [Google Scholar]
- 12.Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, et al. Interstitial lung disease guideline: The British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63:v1–58. doi: 10.1136/thx.2008.101691. [DOI] [PubMed] [Google Scholar]
- 13.Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Willems-Widyastuti A, Scheers H, Van Raemdonck DE, et al. Circulating and intrapulmonary C-reactive protein: A predictor of bronchiolitis obliterans syndrome and pulmonary allograft outcome. J Heart Lung Transplant. 2009;28:799–807. doi: 10.1016/j.healun.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J. 1999;14:376–82. doi: 10.1034/j.1399-3003.1999.14b23.x. [DOI] [PubMed] [Google Scholar]
- 15.Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med. 1994;149:655–9. doi: 10.1164/ajrccm.149.3.8118632. [DOI] [PubMed] [Google Scholar]
- 16.Sugiyama Y, Kasahara T, Mukaida N, Matsushima K, Kitamura S. Chemokines in bronchoalveolar lavage fluid in summer-type hypersensitivity pneumonitis. Eur Respir J. 1995;8:1084–90. doi: 10.1183/09031936.95.08071084. [DOI] [PubMed] [Google Scholar]
- 17.Inoue T, Fujishima S, Ikeda E, Yoshie O, Tsukamoto N, Aiso S, et al. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur Respir J. 2004;24:49–56. doi: 10.1183/09031936.04.00110203. [DOI] [PubMed] [Google Scholar]
- 18.Goodman RB, Strieter RM, Frevert CW, Cummings CJ, Tekamp-Olson P, Kunkel SL, et al. Quantitative comparison of C-X-C chemokines produced by endotoxin-stimulated human alveolar macrophages. Am J Physiol. 1998;275:L87–95. doi: 10.1152/ajplung.1998.275.1.L87. [DOI] [PubMed] [Google Scholar]
- 19.Martina S, Martina V, Monika M, Jan P, Libor K, Ilja S. Angiostatic versus angiogenic chemokines in IPF and EAA. Respir Med. 2009;103:1651–6. doi: 10.1016/j.rmed.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Cantin AM, North SL, Fells GA, Hubbard RC, Crystal RG. Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. J Clin Invest. 1987;79:1665–73. doi: 10.1172/JCI113005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenz AG, Hinze-Heyn H, Schneider A, Behr J, Häussinger K, Heindi S, et al. Influence of inflammatory mechanisms on the redox balance in interstitial lung diseases. Respir Med. 2004;98:737–45. doi: 10.1016/j.rmed.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Henry MT, McMahon K, Mackarel AJ, Prikk K, Sorsa T, Maisi P, et al. Matrix metalloproteinases and tissue inhibitor of metalloproteinase-1 in sarcoidosis and IPF. Eur Respir J. 2002;20:1220–7. doi: 10.1183/09031936.02.00022302. [DOI] [PubMed] [Google Scholar]
- 23.Suga M, Iyonaga K, Okamoto T, Gushima Y, Miyakawa H, Akaike T, et al. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2000;162:1949–56. doi: 10.1164/ajrccm.162.5.9906096. [DOI] [PubMed] [Google Scholar]
- 24.Vos R, Blondeau K, Vanaudenaerde BM, Mertens V, Van Raemdonck DE, Sifrim D, et al. Airway colonization and gastric aspiration after lung transplantation: Do birds of a feather flock together? J Heart Lung Transplant. 2008;27:843–9. doi: 10.1016/j.healun.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 25.Vos R, Vanaudenaerde BM, Geudens N, Dupont LJ, Van Raemdonck DE, Verleden GM. Pseudomonal airway colonisation: Risk factor for bronchiolitis obliterans syndrome after lung transplantation? Eur Respir J. 2008;31:1037–45. doi: 10.1183/09031936.00128607. [DOI] [PubMed] [Google Scholar]
- 26.Nawrot TS, Vos R, Jacobs L, Verleden SE, Wauters S, Mertens V, et al. The impact of traffic air pollution on bronchiolitis obliterans syndrome and mortality after lung transplantation. Thorax. 2011;66:748–54. doi: 10.1136/thx.2010.155192. [DOI] [PubMed] [Google Scholar]
- 27.Wuyts WA, Willems S, Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Rinaldi M, et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res. 2010;36:602–14. doi: 10.3109/01902148.2010.492895. [DOI] [PubMed] [Google Scholar]
- 28.Antoniou KM, Tzouvelekis A, Alexandrakis MG, Sfiridaki K, Tsiligianni I, Rachiotis G, et al. Different angiogenic activity in pulmonary sarcoidosis and idiopathic pulmonary fibrosis. Chest. 2006;130:982–8. doi: 10.1378/chest.130.4.982. [DOI] [PubMed] [Google Scholar]
- 29.Cui A, Anhenn O, Theegarten D, Ohshimo S, Bonella F, Sixt SU, et al. Angiogenic and angiostatic chemokines in idiopathic pulmonary fibrosis and granulomatous lung disease. Respiration. 2010;80:372–8. doi: 10.1159/000245332. [DOI] [PubMed] [Google Scholar]
- 30.Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, et al. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am J Respir Crit Care Med. 2004;170:242–51. doi: 10.1164/rccm.200308-1151OC. [DOI] [PubMed] [Google Scholar]
- 31.Navarro C, Ruiz V, Gaxiola M, Carrillo G, Selman M. Angiogenesis in hypersensitivity pneumonitis. Arch Physiol Biochem. 2003;111:365–8. doi: 10.3109/13813450312331337612. [DOI] [PubMed] [Google Scholar]
- 32.Ai S, Cheng XW, Inoue A, Nakamura K, Okumura K, Iguchi A, et al. Angiogenic activity of bFGF and VEGF suppressed by proteolytic cleavage by neutrophil elastase. Biochem Biophys Res Commun. 2007;364:395–401. doi: 10.1016/j.bbrc.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 33.Meyer KC, Cardoni A, Xiang ZZ. Vascular endothelial growth factor in bronchoalveolar lavage from normal subjects and patients with diffuse parenchymal lung disease. J Lab Clin Med. 2000;135:332–8. doi: 10.1067/mlc.2000.105618. [DOI] [PubMed] [Google Scholar]
- 34.Pekovich SR, Bock PE, Hoover RL. Thrombin-thrombomodulin activation of protein C facilitates the activation of progelatinase A. FEBS Lett. 2001;494:129–32. doi: 10.1016/s0014-5793(01)02296-7. [DOI] [PubMed] [Google Scholar]
- 35.Yasui H, Gabazza EC, Taguchi O, Risteli J, Risteli L, Wada H, et al. Decreased protein C activation is associated with abnormal collagen turnover in the intraalveolar space of patients with interstitial lung disease. Clin Appl Thromb Hemost. 2000;6:202–5. doi: 10.1177/107602960000600404. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi H, Gabazza EC, Taguchi O, Wada H, Takeya H, Nishioka J, et al. Protein C anticoagulant system in patients with interstitial lung disease. Am J Respir Crit Care Med. 1998;157:1850–4. doi: 10.1164/ajrccm.157.6.9709078. [DOI] [PubMed] [Google Scholar]
- 37.Hattori N, Mizuno S, Yoshida Y, Chin K, Mishima M, Sisson TH, et al. The plasminogen activation system reduces fibrosis in the lung by a hepatocyte growth factor-dependent mechanism. Am J Pathol. 2004;164:1091–8. doi: 10.1016/S0002-9440(10)63196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shukla MN, Rose JL, Ray R, Lathrop KL, Ray A, Ray P. Hepatocyte growth factor inhibits epithelial to myofibroblast transition in lung cells via Smad7. Am J Respir Cell Mol Biol. 2009;40:643–53. doi: 10.1165/rcmb.2008-0217OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchand-Adam S, Fabre A, Mailleux AA, Marchal J, Quesnel C, Kataoka H, et al. Defect of pro-hepatocyte growth factor activation by fibroblasts in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:58–66. doi: 10.1164/rccm.200507-1074OC. [DOI] [PubMed] [Google Scholar]
- 40.Kotani I, Sato A, Hayakawa H, Urano T, Takada Y, Takada A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb Res. 1995;77:493–504. doi: 10.1016/0049-3848(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 41.Abdi K. IL-12: The role of p40 versus p75. Scand J Immunol. 2002;56:1–11. doi: 10.1046/j.1365-3083.2002.01101.x. [DOI] [PubMed] [Google Scholar]
- 42.Schuyler M, Gott K, Cherne A. Mediators of hypersensitivity pneumonitis. J Lab Clin Med. 2000;136:29–38. doi: 10.1067/mlc.2000.107694. [DOI] [PubMed] [Google Scholar]
- 43.Khalil N, O’Connor RN, Flanders KC, Unruh H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: An immunohistochemical study. Am J Respir Cell Mol Biol. 1996;14:131–8. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- 44.Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, et al. TGF-beta 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol. 2002;168:6470–8. doi: 10.4049/jimmunol.168.12.6470. [DOI] [PubMed] [Google Scholar]
- 45.Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. 1989;170:727–37. doi: 10.1084/jem.170.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]