

Abstract
In this study, we attempt to target the mitogen-activated protein kinase (MAPK) pathway in acute myeloid leukemia (AML) cells using a recombinant anthrax lethal toxin (LeTx). LeTx consists of protective antigen (PrAg) and lethal factor (LF). PrAg binds cells, is cleaved by furin, oligomerizes, binds three to four molecules of LF, and undergoes endocytosis, releasing LF into the cytosol. LF cleaves MAPK kinases, inhibiting the MAPK pathway. We tested potency of LeTx on a panel of 11 human AML cell lines. Seven cell lines showed cytotoxic responses to LeTx. Cytotoxicity of LeTx was mimicked by the specific mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 (MEK1/2) inhibitor U0126, indicating that LeTx-induced cell death is mediated through the MEK1/2-extracellular signal-regulated kinase (ERK1/2) branch of the MAPK pathway. The four LeTx-resistant cell lines were sensitive to the phosphatidylinositol 3-kinase inhibitor LY294002. Co-treatment of AML cells with both LeTx and LY294002 did not lead to increased sensitivity, showing a lack of additive/synergistic effects when both pathways are inhibited. Flow cytometry analysis of MAPK pathway activation revealed the presence of phospho-ERK1/2 only in LeTx-sensitive cells. Staining for Annexin V/propidium iodide and active caspases showed an increase in double-positive cells and the absence of caspase activation following treatment, indicating that LeTx-induced cell death is caspase-independent and nonapoptotic. We have shown that a majority of AML cell lines are sensitive to the LF-mediated inhibition of the MAPK pathway. Furthermore, we have demonstrated that LeTx-induced cytotoxicity in AML cells is nonapoptotic and dependent on phospho-ERK1/2 levels.
Introduction
Acute myeloid leukemia (AML) is one of the most common leukemias in adults with an estimated 13,780 newly diagnosed cases and an estimated 10,200 deaths in the United States in 2012 [1]. Though a high proportion of AML patients enter complete remission following combination induction and consolidation chemotherapy, most patients eventually relapse because of persistence of chemotherapy-resistant blasts in the bone marrow [2]. Hence, alternative approaches employing novel, more selective mechanisms for targeting AML blasts are being sought. One such approach consists of targeting the mitogen-activated protein kinase (MAPK) pathway in AML cells using a recombinant anthrax lethal toxin (LeTx) [3–5].
The MAPK pathway is a highly conserved signaling pathway whose activation leads to cell proliferation and survival [5,6]. Mutations leading to the constitutive activation of the Ras-Raf-mitogenactivated protein/extracellular signal-regulated kinase kinase 1/2 (MEK1/2)-extracellular signal-regulated kinase (ERK1/2) pathway are a hallmark of several tumors such as melanoma and are found in a variety of cancers, including AML, thus constituting an attractive target for novel AML therapies [7–10].
LeTx is a binary toxin produced by the gram positive bacterium Bacillus anthracis and consists of two separate proteins: the cell binding and internalization moiety protective antigen (PrAg) and the catalytic moiety lethal factor (LF) [11,12]. PrAg binds to cells through its ubiquitously expressed cell surface receptors tumor endothelial marker-8 and capillary morphogenesis gene-2 and is subsequently cleaved by cell surface furin-like proteases leading to the release of a 20-kDa fragment and the generation of an active 63-kDa fragment (PrAg63) [5,13]. The latter then forms oligomers, binds three to four molecules of LF, and undergoes endocytosis [14]. On acidification of the endosome, PrAg63 oligomers undergo a conformational change leading to pore formation and allowing LF to translocate into the cytosol [15]. LF is a zinc metalloprotease that cleaves and inactivates MEKs, leading to the inhibition of the MAPK pathway and subsequent growth inhibition and cell death [16–18]. We have previously shown that cytotoxicity of LeTx to human melanoma cell lines carrying the V600E BRAF mutation is dependent on the activation status of the MAPK pathway, particularly MEK1/2, allowing the use of this toxin for the selective targeting of tumors with constitutive MAPK activation [4,5,9,10,18].
We and others have previously demonstrated the potency and selectivity of LeTx to melanoma cell lines in vitro and in an in vivo melanoma model [4,5]. However, unlike melanoma, in which the importance of N-Ras and BRAF mutations (found in up to 95% of cases) is well established, the importance of MAPK pathway mutations in AML has been poorly investigated [19]. Moreover, very few attempts have been made to target the MAPK pathway in AML, with the exception of targeting the receptor tyrosine kinase fms-like tyrosine kinase 3 in AML cells carrying fms-like tyrosine kinase 3 mutations [20,21]. Hence, a deeper investigation of the MAPK pathway in human AML cell lines along with the possibility of selectively targeting AML through the inhibition of the MAPK pathway are warranted.
In this study, we attempt to target the MAPK pathway in AML cell lines using a recombinant LeTx and to characterize the response of AML cells to the LeTx-mediated inhibition of the MAPK pathway.
Materials and Methods
Expression and Purification of LeTx
Recombinant LeTx proteins PrAg and LF, as well as FP59 (fusion of the PrAg binding domain of LF and the catalytic domain of Pseudomonas aeruginosa exotoxin A), were expressed and purified as described previously [22,23]. LY294002 was purchased from Cell Signaling Technology (Danvers, MA).
Cells and Cell Lines
Human AML cell lines HL60, U937, ML1, ML2, Mono-Mac-1, Mono-Mac-6, KG-1, SigM5, TF1-vRaf, TF1-vSrc, and TF1-HaRas were grown as described previously [24].
Proliferation Inhibition Assay (Cytotoxicity)
Sensitivity of AML cell lines to LeTx was determined using a proliferation inhibition assay as described previously [5]. We have also used a recombinant protein, termed FP59, consisting of the PrAg binding domain of LF fused to the catalytic domain of P. aeruginosa exotoxin A. Binding of FP59 to PrAg and its translocation into the cytosol are identical to LF; however, it does not target the MAPK pathway but rather ADP-ribosylates elongation factor 2 leading to inhibition of protein synthesis and cell death. PrAg/FP59 was used as a control for catalytic domain entry into the cytosol of AML cells. Briefly, aliquots of 104 cells/well, in 100 µl of cell culture medium, containing a fixed concentration of 10-9 M LF or FP59, were plated onto a flat-bottom 96-well plate (Corning Inc, Corning, NY). Then, 50 µl of PrAg in media were added to each well to yield concentrations ranging from 10-8 to 10-13 M. When LY294002 or U0126 were used, they were added as described above for PrAg but in concentrations ranging from 10-4 to 10-9 M. Following a 48-hour incubation at 37°C/5% CO2, 50 µl of XTT cell proliferation reagent (Roche, Basel, Switzerland) was added to each well and the plates incubated for another 4 hours. Absorbance was then read at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA). Nominal absorbance and percent maximal absorbance were plotted against the log of concentration, and a nonlinear regression with a variable slope sigmoidal dose-response curve was generated along with inhibitory concentration 50 (IC50) using GraphPad Prism 5 software (GraphPad Software, San Diego, CA). All assays were performed at least twice with an interassay range of 30% or less for IC50.
Cell Cycle Analysis
The impact of LeTx treatment on the cell cycle of AML cells was determined using propidium iodide (PI) staining on flow cytometry. Briefly, cells incubated with three different concentrations of LeTx (10,000, 300, and 4.5 pM) or media alone in flat-bottom 96-well plates (Corning Inc) for 24 and 48 hours at 37°C/5% CO2 were harvested and fixed in 70% ethanol for a minimum of 24 hours, at -20°C. Cells were then incubated in 500 µl of PI staining solution (50 µg/ml) for 40 minutes at 37°C. Samples were then read on a C6 flow cytometer (BD Accuri, Ann Arbor, MI) and total cell DNA content was measured on FL2-A. Percent of cells in G0/G1, S, and G2/M phases was determined in control cells and in cells treated with the three different concentrations of LeTx (10,000, 300, and 4.5 pM) following gating for the cell population on width versus forward scatter.
Inhibition Assays
AML cells were incubated with either the small molecular weight phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (Cell Signaling Technology) alone and in combination with LeTx or with the small molecular weight MEK1/2 inhibitor U0126 (Cell Signaling Technology). Briefly, 104 cells/well were plated onto 100 µl of medium in a flat-bottom, 96-well plate. Then, 100 µl of either medium (control cells) or medium containing LeTx (10-8 MPrAg/10-9 MLF), LY294002 (20 and 50 µM), U0126 (20 and 50 µM), or a combination of the above were added. Cells were then incubated for 48 hours at 37°C/5% CO2 followed by the addition of 50 µl of XTT cell proliferation reagent (Roche). Cells were incubated for another 4 hours and absorbance was read at 450 nm using a 96-well plate reader (Thermo Fisher Scientific). Data were analyzed using GraphPad Prism 5 software (GraphPad Software). The absorbance and the percent absorbance of controls were compared between the different treatment groups.
Intracellular Staining and Flow Cytometry Analysis
Activation of the MEK1/2-ERK1/2 pathway in AML cell lines [untreated and following a 10-hour incubation with LeTx (10-8 M PrAg/10-9 M LF)] was assessed by determining the presence or absence of phospho-ERK1/2 using flow cytometry as described previously [25]. Approximately 3 x 106 cells were fixed in 70% ethanol for 15 minutes. Cells were then incubated with a 1/100 dilution of anti-phospho-ERK1/2 (Ser 217/221) rabbit monoclonal antibodies (Cell Signaling Technology) in antibody binding buffer containing 0.05% Triton X-100, for 1 hour at 37°C, followed by a 30-minute incubation with a 1/100 dilution of a fluorescein isothiocyanate (FITC)-conjugated mouse anti-rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Fixed cells stained only with FITC-conjugated mouse anti-rabbit polyclonal antibody were used as isotypic control. Samples were then analyzed using a C6 flow cytometer (BD Accuri). The presence of phospho-ERK1/2 was analyzed and compared with that of the isotopic control. Positivity for the presence of phospho-ERK1/2 was determined using the ratio of fluorescence intensity (RFI) between the mean fluorescence intensity of the stained cells and the mean fluorescence intensity of the isotypic control. RFI ≥ 2 was considered positive.
Analysis of Cell Cytotoxicity
Determination of apoptotic versus nonapoptotic cell death was carried out using an Annexin V-FITC and PI-labeled apoptosis/necrosis detection kit (Abcam, Cambridge, MA) and an FITC-conjugated active caspase inhibitor (ApoStat Apoptosis Detection Kit; R&D Systems, Abingdon, England) on flow cytometry. Briefly, 104 cells/well were plated onto 100 µl of media in a flat-bottom, 96-well plate and were incubated with either 100 µl of medium alone (control cells) or medium containing three different concentrations of LeTx (as described above under cell cycle analysis) for 24 and 48 hours at 37°C/5% CO2. Cells were then harvested and incubated with an FITC-conjugated annexin V antibody (2.5 mg/ml and PI (5 mg/ml) in antibody binding buffer for 45 minutes at 37°C or incubated with 0.5 µg/ml apostat for 30 minutes and then harvested. Cells were then read using a C6 flow cytometer. Annexin V/PI data were analyzed on FL1-H versus FL2-H scatter plot and active caspases were detected on FL1-H. Unstained cells were used as negative control. Cells had to show positive annexin V staining, negative PI staining, and positive active caspase staining to be considered apoptotic, whereas cells positive for both annexin V and PI staining and negative for active caspase staining were considered nonapoptotic.
Results
Cytotoxicity of LeTx
We tested the cytotoxicity of LeTx on a panel of 11 human AML cell lines using an XTT proliferation inhibition assay. Seven of 11 AML cell lines (64%) were sensitive to the LF-mediated inhibition of the MAPK pathway with IC50 = 13 to 94 pM and percent cell kill at highest concentration >75%. The remaining four cell lines were not sensitive to the LF-mediated inhibition of the MAPK pathway (IC50 > 10,000 pM and percent cell kill at highest concentration ≤ 40%) (Table 1 and Figure 1E). To demonstrate that resistance to LeTx was due to resistance to the LF-mediated inhibition of the MAPK pathway and not to the inability of LF to translocate into the cytosol of targeted cells, we tested the cytotoxicity of a combination of PrAg and FP59 to AML cell lines. FP59 is a fusion of the PrAg binding domain of LF and the catalytic domain of P. aeruginosa exotoxin A. Binding to PrAg and translocation of FP59 into the cytosol are identical to those of LF; however, FP59 does not target the MAPK pathway but rather ADPribosylates elongation factor 2 leading to the inhibition of protein synthesis and subsequent cell death. The combination of PrAg and FP59, therefore, induces MAPK-independent cytotoxicity to all cells that express the anthrax toxin receptors. PrAg/FP59 was cytotoxic to all the AML cell lines tested with IC50 = 0.7 to 18 pM and percent cell death >90%, indicating that differential sensitivity of AML cells to LeTx is independent of the ability of LF to translocate into the cytosol (Table 1 and Figure 1, A–D) and dependent on the addiction of cells to the MAPK pathway. In addition, intracellular staining and flow cytometry analysis of phospho-ERK1/2 in sensitive cell lines before and after a 10-hour incubation with LeTx revealed the absence of phosphoERK1/2 in cells treated with LeTx, compared to nontreated cells, thus indicating that cytotoxicity of LeTx to AML cells is mediated through the proteolytic cleavage of MEKs by LF and subsequent inhibition of the MAPK pathway as evidenced by the absence on phospho-ERK1/2 in LeTx-treated cells (data not shown). Moreover, because LeTx inhibits all branches of the MAPK pathway, it is possible that cytotoxicity of LeTx to AML cell lines is due to the simultaneous inhibition of all three branches of the MAPK pathway and not only the MEK1/2-ERK1/2 branch. Therefore, to determine the contribution of the MEK1/2-ERK1/2 branch to the cytotoxicity of LeTx, we tested the sensitivity of our panel of AML cell lines to U0126, the specific MEK1/2 inhibitor. Cytotoxicity of U0126 mimicked that of LeTx with the same pattern seen with both LeTx and U0126 across the panel of AML cell lines (Table 1 and Figure 1F). The four cell lines that were resistant to LeTx-induced cytotoxicity were also resistant to U0126 (IC50 > 1000 µM and percent cell kill at the highest concentration ≤ 35%), thus confirming that these cell lines do not rely on the MAPK pathway for survival and are resistant to both the specific inhibition of MEK1/2 and the inhibition of the entire MAPK pathway. The seven AML cell lines that were sensitive to LeTx-induced cytotoxicity were also sensitive to the specific MEK1/2 inhibitor U0126 (IC50 = 1.3–2.7 µM and percent cell kill at the highest concentration >60%), thus indicating that cytotoxicity of LeTx is mediated through the inhibition of the MEK1/2-ERK1/2 branch of the MAPK pathway.
Table 1.
Sensitivity of Human AML Cell Lines to LeTx (PrAg/LF), U0126, and the Protein Synthesis Inhibitor PrAg/FP59.
| Cell Line | LeTx (PrAg/LF) (IC50; pM) | PrAg/FP59 (IC50; pM) | U0126 (IC50; µM) |
| HL60 | 13.0 | 0.7 | 2.6 |
| TF1-VSrc | 15.0 | 3.0 | 2.0 |
| TF1-VRaf | 16.0 | 4.0 | 1.3 |
| Mono-Mac-6 | 39.0 | 13.0 | 2.1 |
| SigM5 | 40.0 | 17.0 | 1.9 |
| ML-2 | 81.0 | 7.0 | 1.5 |
| TF1-HaRas | 94.0 | 0.4 | 2.7 |
| ML-1 | >10,000 | 6.0 | >1,000 |
| U937 | >10,000 | 1.0 | >1,000 |
| KG-1 | >10,000 | 1.0 | >1,000 |
| Mono-Mac-1 | >10,000 | 2.0 | >1,000 |
Figure 1.

Nonlinear regression curves of LeTx (PrAg/LF) (square) and PrAg/FP59 (triangle) on human AML cell lines. HL60 (A) and TF1-vRaf (B) cell lines are sensitive to both LeTx and PrAg/FP59. Mono-Mac-1 (C) and U937 (D) are only sensitive to PrAg/FP59 but not to PrAg/LF, indicating resistance to the LF-mediated inhibition of the MAPK pathway. (E) Compilation of LeTx nonlinear regression curves on all AML cell lines tested. (F) Nonlinear regression curves of the specific MEK1/2 inhibitor U0126 on TF1-vRaf (square), HL60 (triangle), Mono-Mac-1 (inverted triangle), and U937 (diamond) cell lines. The LeTx-sensitive cell lines TF1-vRaf and HL60 are sensitive to U0126, whereas the LeTx-resistant cell lines Mono-Mac-1 and U937 are resistant to U0126.
Cell Cycle Effect of LeTx
To determine whether, in addition to its cytotoxicity, LeTx induces cell cycle arrest in AML cells, we determined the cell cycle status of our panel of AML cell lines following 24- and 48-hour incubation with three different concentrations of the LeTx. One of the four AML cell lines that were not sensitive to the cytotoxicity of LeTx (ML1) showed dose-dependent cell cycle arrest at 24 hours (data not shown) and 48 hours following treatment with LeTx, whereas the other three (U937, Mono-Mac-1, and KG-1) did not show any effect of LeTx treatment on cell cycle (Figure 2, A and B). The fraction of cells in the G0/G1 phase of ML1 increased from approximately 34% of the total cell population in control cells to approximately 60% of the total cell population following incubation with 10,000 pM of LeTx for 48 hours. This was associated with a corresponding decrease in the percentage of cells in the G2/M phase from approximately 19% of the cell population in control cells to approximately 8% of the cell population in treated cells (Figure 2A).
Figure 2.

Cell cycle analysis of AML cell lines following treatment with LeTx. Control cells are represented in the left panels, and cells treated with 10 nM LeTx for 48 hours are represented in the right panels. Cells are gated on width versus forward scatter (R1). Cells in G0/G1 are gated M2, those in G2/M are gated M3, and those in pre-G0/G1 (dead) are gated M4 or M5. One of the cell lines that did not show a cytotoxic response to LeTx, ML1 (A), did show cell cycle arrest, whereas the other, U937 (B), did not. Similarly, one of the LeTxsensitive cell lines ML2 (C) showed cell cycle arrest in addition to cell death, whereas the other, TF1-vRaf (D), showed complete cell death following treatment.
Furthermore, cell cycle arrest was observed in four of the seven cell lines that showed a cytotoxic response to the LF-mediated inhibition of the MAPK pathway (ML2, HL60, TF1-HaRas, and TF1-vSrc) at 24 hours (data not shown) and 48 hours following treatment with LeTx. A dose-dependent increase in the percentage of surviving cells in the G0/G1 phase was observed (42%, 58%, 41%, and 40% in control cells versus 54%, 73%, 67%, and 54% in treated cells) at 48 hours, in ML2 (Figure 2C), HL60, TF1-HaRas, and TF1-vSrc cells, respectively. This was associated with a corresponding decrease in the percentage of cells in the G2/M phase, by approximately 10%, in treated versus control cells in all four cell lines. The remaining three LeTx-sensitive cell lines [TF1-vRaf (Figure 2D), SigM5, and Mono-Mac-6] showed complete cell death with more than 90% of cells in the pre-G0/G1 peak and no difference in the percentage of cells in the G0/G1 phase between control cells and surviving cells at the highest concentration of LeTx at both 24-hour incubation (data not shown) and 48-hour incubation. This indicates that, in addition to significant cytotoxicity to the majority of AML cell lines, LeTx induces cell cycle arrest in a subset of AML cells (approximately 45%) irrespective of their cytotoxic response to the LF-mediated inhibition of the MAPK pathway.
Inhibition of PI3K
To determine whether AML cell lines resistant to the LF-mediated inhibition of the MAPK pathway are susceptible to the inhibition of the PI3K/Akt pathway, we tested the sensitivity of the LeTx-resistant cell lines (ML1, Mono-Mac-1, U937, and KG-1) to the small molecular weight PI3K inhibitor LY294002. Incubation with LY294002 at 20 and 50 µM for 48 hours induced significant cytotoxicity in all four cell lines with cell viability decreasing by approximately 40% to 50% at the 20 µM concentration and 60% to 80% at the 50 µM concentration compared to untreated cells (P < .05; Figure 3A). Co-incubation of these cells with both LeTx (10 nM) and LY294002 (20 and 50 µM) led to only a slight decrease in cell viability, approximately 10%, compared to LY294002 alone. Although the decreases in cell viability observed following co-incubation with both LeTx and LY294002 were statistically significant for the U937 cell line at the 20 µM concentration, the ML1 cell line at the 50 µM concentration, and the Mono-Mac-1 and KG-1 cell lines at both the 20 and 50 µM concentrations (P < .05), compared to LY294002 alone, they were not considered functionally significant because co-incubation only accounted for an additional decrease in cell viability of no more than 10% compared to LY294002 alone (Figure 3A). Moreover, cytotoxicity of the combination of LY294002 and LeTx was similar to the cytotoxicity of either LeTx alone or LY294002 alone as evidenced by similar nonlinear regression curves and similar IC50 values, at 48 hours, in a proliferation inhibition assay (Figure 3, B–D). Hence, cell lines that are not sensitive to LeTx are sensitive to the inhibition of the PI3K/Akt pathway with the simultaneous inhibition of both pathways not resulting in increased cell cytotoxicity. This shows the existence of two distinct populations of AML cell lines sensitive to the inhibition of either the MAPK pathway or the PI3K/Akt pathway with no additive or synergistic effect observed when both pathways are inhibited simultaneously.
Figure 3.
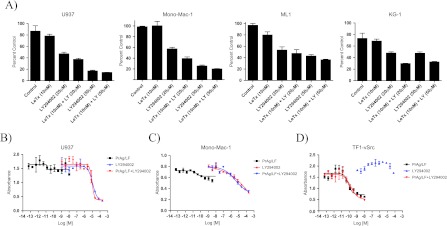
Sensitivity of LeTx-resistant AML cell lines to LY294002 alone and in combination with LeTx. (A) LeTx-resistant cell lines incubated with LY294002 at 20 and 50 µM for 48 hours were sensitive to both concentrations of LY294002, whereas co-incubation with LeTx (10 nM) did not lead to any significant increase in cell sensitivity. Nonlinear regression curves of U937 (B), Mono-Mac-1 (C), and TF1-vSrc (D) cells incubated with LeTx (square), LY294002 (triangle), or a combination of both (inverted triangle). The combination of LeTx and LY294002 did not lead to increased cytotoxicity compared to either LeTx or LY294002 alone.
Analysis of MAPK Activation
We examined the activation level of the MAPK pathway in AML cell lines by determining the phosphorylation status of phospho-ERK1/2, using single-cell intracellular staining on flow cytometry. A representative sample of four AML cell lines was tested and the presence or absence of phospho-ERK1/2 was linked to the cell response to the LF-mediated inhibition of the MAPK pathway. Both of the cell lines that showed a cytotoxic response to the LF-mediated inhibition of the MAPK pathway (ML-2 and TF1-vRaf) had an active MEK1/2-ERK1/2 pathway as evidenced by the presence of phospho-ERK1/2 (RFI = 2.14 and 2.32 for ML-2 and TF1-vRaf, respectively, compared to isotype control; Figure 4, A and B). However, the two cell lines that did not show a cytotoxic response to the LF-mediated inhibition of the MAPK pathway (U937 and ML-1) were not positive for the presence of phospho-ERK1/2, hence had an inactive MEK1/2-ERK1/2 pathway (RFI = 1.41 and 1.51 for U937 and ML-1, respectively, compared to isotype control; Figure 4, C and D). This indicates that phospho-ERK1/2 levels may serve as a useful marker for the sensitivity of AML cells to the inhibition of the MAPK pathway.
Figure 4.

Single-cell intracellular staining of phospho-ERK1/2 in four AML cell lines using flow cytometry. TF1-vRaf (A) and ML-2 (B) cells, which showed a cytotoxic response to the LF-mediated inhibition of the MAPK pathway, were positive for the presence of phospho-ERK1/2 as evidenced by an RFI > 2 between cells stained for phospho-ERK1/2 (gray) and cells incubated with an isotype control (black). Cells are gated on width versus forward scatter (R2 and R3). ML-1 (C) and U937 (D) cells, which were resistant to the cytotoxicity of LeTx, were negative for phospho-ERK1/2 with RFI < 1.5.
Analysis of Cell Death
To determine the mechanism of the cell death observed following the LF-mediated inhibition of the MAPK pathway in AML cells, we tested for caspase activation and annexin V/PI staining in our panel of AML cell lines following treatment with three different concentrations of LeTx (10,000, 300, and 4.5 pM) for 24 and 48 hours. The four cell lines that did not show a cytotoxic response following treatment with LeTx in the proliferation inhibition assay stained negatively with both PI and Annexin V, confirming the absence of cell death following LF-mediated inhibition of the MAPK pathway in these cell lines. However, in all seven LeTx-sensitive cell lines (TF1-vRaf, TF1-vSrc, TF1-HaRas, HL60, SigM5, Mono-Mac-6, and ML-2), an increase in the percentage of cells stained with both annexin V and PI was observed, at both 24 and 48 hours, in cells treated at the highest concentration of LeTx (10 nM) compared to controls, indicating either necrotic or late-stage apoptotic cell death (Figure 5). However, staining for active caspases revealed a total absence of caspase activation in all AML cell lines tested, following treatment with LeTx for 24 and 48 hours (Figure 5). The absence of caspase activation, in addition to the loss of membrane integrity as evidenced by positive PI staining, indicates that LeTx-induced cytotoxicity in AML cells is mediated through caspase-independent, nonapoptotic mechanisms.
Figure 5.

Analysis of the mechanism of LeTx-mediated cytotoxicity in TF1-vRaf cells using annexin V/PI (A) and active caspase staining (B). TF1-vRaf cells incubated with 10 nM LeTx for 24 hours (right panel) stained positively with both annexin V (FL1-H) and PI (FL2-H). Incubation of LeTx-treated TF1-vRaf cells with a cell permeable, FITC-conjugated active caspase inhibitor revealed the absence of active caspases following incubation with LeTx (gray).
Discussion
In this study, we have shown that LeTx is highly cytotoxic to a majority of human AML cell lines through the LF-mediated inhibition of the MAPK pathway. Limited information exists regarding the importance of the MAPK pathway in AML cells and the potential targeting of AML through the inhibition of the MAPK pathway has not been sufficiently investigated so far [19–21]. We have demonstrated that differential sensitivity of AML cell lines to LeTx was not due to the inability of the LF moiety to translocate into the cytosol but rather to the differential sensitivity of AML cell lines to the LF-mediated inhibition of the MAPK pathway. When we replaced the catalytic domain of LF with the catalytic domain of P. aeruginosa exotoxin A, which inhibits protein synthesis rather than the MAPK pathway, differential sensitivity was lost and all AML cell lines tested were sensitive to the combination of PrAg and FP59, demonstrating that differential sensitivity of AML cells to LeTx was due to the differential response of AML cell lines to the LF-mediated inhibition of the MAPK pathway. In addition, we have shown that the pattern of cytotoxicity of LeTx on our panel of AML cell lines was mimicked by the specific, small molecular weight MEK1/2 inhibitor U0126, indicating that cytotoxicity of LeTx is mainly mediated through the inhibition of the MEK1/2-ERK1/2 branch of the MAPK pathway. Moreover, we have shown that incubation of AML cells with LeTx leads to a marked inhibition of the MEK1/2-ERK1/2 branch of the MAPK pathway as evidenced by the absence of phosphorylated ERK1/2 in cells incubated with LeTx for 10 hours compared to nontreated cells.
The four AML cell lines that proved to be resistant to the inhibition of the MAPK pathway by LeTx were sensitive to the inhibition of the PI3K/Akt pathway by LY294002, a small molecular weight PI3K inhibitor. Interestingly, co-incubating AML cell lines with both LeTx and LY294002 did not show any significant increase in cytotoxicity compared to LY294002 alone. Therefore, the panel of AML cell lines tested consisted of two distinct populations, one sensitive to the inhibition of the MAPK pathway and the other sensitive to the inhibition of the PI3/Akt pathway with the simultaneous inhibition of both pathways not leading to any additive or synergistic effects. These findings are surprising because the inhibition of both the MEK1/2-ERK1/2 pathway and the PI3K/Akt pathway has been shown to have additive or synergistic effects in a number of tumor types, including hepatocellular carcinoma, colorectal cancer, pancreatic adenocarcinoma, and melanoma [26–29]. Moreover, it has been shown that resistance to MEK inhibitors is mediated through the activation of the PI3k/Akt pathway in a number of tumors [30]. Inhibition of both the MEK1/2-ERK1/2 pathway and the PI3K/Akt pathway in AML cells has not been investigated to date. Our results demonstrate that AML cells are sensitive to the inhibition of either the MEK1/2-ERK1/2 pathway or the PI3K/Akt pathway with no additive or synergistic effect for the simultaneous inhibition of both pathways.
In addition to its cytotoxic effect on a majority of AML cell lines, we have shown that LeTx also induces cell cycle arrest in a large subset of cell lines, independently of their sensitivity to the cytotoxic effects of the LF-mediated inhibition of the MAPK pathway. One of the four cell lines that did not show a cytotoxic response to LeTx, in addition to four of the seven cell lines that did show a cytotoxic response, had an increase in the fraction of cells in the G0/G1 phase along with a decrease in the fraction of cells in the G2/M and S phases in surviving cells at the highest concentration compared to controls.
We then determined the activity of the Ras-Raf-MEK1/2-ERK1/2 pathway in AML cell lines by assessing phospho-ERK1/2 levels using single-cell intracellular staining and comparing it to the cytotoxic response of these cells to the LF-mediated inhibition of the MAPK pathway. Cell lines in which the LF-mediated inhibition of the MAPK pathway induced cytotoxicity were positive for phosphoERK1/2 indicating an active Ras-Raf-MEK1/2-ERK1/2 pathway, whereas cell lines in which the inhibition of this pathway did not induce cytotoxicity were negative for phospho-ERK1/2. This indicates that sensitivity of AML cells to the LF-mediated inhibition of the MAPK pathway is determined by the activity of the MAPK pathway and that phospho-ERK1/2 levels may be used as a marker to predict sensitivity of AML cells to the inhibition of the MAPK pathway. Predicting a cell response to the inhibition of a signaling pathway, such as the MAPK pathway, may be more complicated than a simple determination of the activity of the pathway; however, in the absence of alternatives, this remains the only available means of assessing the importance of a particular pathway to the survival of cells.
Analysis of the mechanism of cell death in AML cell lines following LF-mediated inhibition of the MAPK pathway revealed that LeTx induces caspase-independent, nonapoptotic cell death in AML cells. Cells treated with LeTx for 24 hours were positive for both Annexin V and PI while being negative for the presence of active caspases. LeTx has been shown to induce caspase-dependent, apoptotic cell death in a number of tumor types, including melanoma [4]. However, LeTx-mediated inhibition of the MAPK pathway has not been investigated in AML cells and the type of cell death induced by the LF-mediated inhibition of this pathway may be dependent on the tumor type. Furthermore, though some studies have shown that Ras inhibitors and MEK inhibitors do induce apoptosis in AML cell lines, the nonapoptotic cell death induced by LeTx in AML cells is not surprising knowing the different mechanism of action of LeTx that catalytically cleaves all MEKs leading to the complete inhibition of all branches of the MAPK pathway [31–33].
In this study, we have shown that a majority of AML cell lines are sensitive to the LF-mediated inhibition of the MAPK pathway, hence confirming the potential for selectively targeting this pathway in AML. Furthermore, we have demonstrated that LeTx-induced cytotoxicity in AML cells, following inhibition of the MAPK pathway, is nonapoptotic and is dependent on phospho-ERK1/2 levels in targeted cells.
Acknowledgments
The authors thank Rasem Fattah for protein purification.
Footnotes
This work was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (Bethesda, MD). The authors have no conflict of interest to declare.
References
- 1.American Cancer Society, author. Cancer Facts and Figures 2012. Atlanta, GA: American Cancer Society; 2012. [Google Scholar]
- 2.Bennett JM, Kouides PA, Forman SJ. The myelodysplastic syndromes: morphology, risk assessment, and clinical management. Int J Hematol. 2002;2:228–238. doi: 10.1007/BF03165122. [DOI] [PubMed] [Google Scholar]
- 3.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Curr Opin Chem Biol. 1999;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koo HM, VanBrocklin M, McWilliams MJ, Leppla SH, Duesbery NS, Vande Woude GF. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc Natl Acad Sci USA. 2002;99:3052–3057. doi: 10.1073/pnas.052707699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abi-Habib RJ, Urieto JO, Liu SH, Leppla SH, Duesbery NS, Frankel AE. Braf status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol Cancer Ther. 2005;4:1303–1310. doi: 10.1158/1535-7163.MCT-05-0145. [DOI] [PubMed] [Google Scholar]
- 6.Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-Raf is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 7.Lee JT, Jr, McCubrey JA. The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia. 2002;16:486–507. doi: 10.1038/sj.leu.2402460. [DOI] [PubMed] [Google Scholar]
- 8.Hilger RA, Scheulen ME, Strumberg D. The Ras-Raf-MEK-ERK pathway in the treatment of cancer. Onkologie. 2002;25:511–518. doi: 10.1159/000068621. [DOI] [PubMed] [Google Scholar]
- 9.Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res. 2004;64:6461–6468. doi: 10.1158/0008-5472.CAN-04-0866. [DOI] [PubMed] [Google Scholar]
- 10.Milella M, Kornblau S, Estrov Z, Carter B, Lapillonne H, Konopleva M, Zhao S, Estey E, Andreef M. Therapeutic targeting of the Mek/Mapk signal transduction module in acute myeloid leukemia. J Clin Invest. 2001;108:851–859. doi: 10.1172/JCI12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 12.Scobie HM, Rainey JA, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrami L, Liu S, Cosson P, Leppla SH, Van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abrami L, Leppla SH, Van der Goot FG. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol. 2006;172:309–320. doi: 10.1083/jcb.200507067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melnyk PA, Collier RJ. A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc Natl Acad Sci USA. 2006;103:9802–9807. doi: 10.1073/pnas.0604000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paul KD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 17.Chopra AP, Boone SA, Liang X, Duesbery NS. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J Biol Chem. 2003;278:9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- 18.Abi-Habib RJ, Singh R, Leppla SH, Greene JJ, Ding Y, Berghuis B, Duesbery NS, Frankel AE. Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin Cancer Res. 2006;12:7437–7443. doi: 10.1158/1078-0432.CCR-06-2019. [DOI] [PubMed] [Google Scholar]
- 19.Zaidi SK, Dowdy CR, van Wijnen AJ, Lian JB, Raza A, Stein JL, Croce CM, Stein GS. Altered Runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a miR-24/MKP-7/MAPK network. Cancer Res. 2009;69:8249–8255. doi: 10.1158/0008-5472.CAN-09-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricciardi M, Scerpa M, Bergamo P, Ciuffreda L, Petrucci M, Chiaretti S, Tavolaro S, Mascolo MG, Abrams SL, Steelman LS, et al. Therapeutic potential of MEK inhibition in acute myelogenous leukemia: rationale for “vertical” and “lateral” combination strategies. J Mol Med (Berl) 2012;90(10):1133–1144. doi: 10.1007/s00109-012-0886-z. [DOI] [PubMed] [Google Scholar]
- 21.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia. 2005;19:1350–1354. doi: 10.1038/sj.leu.2403828. [DOI] [PubMed] [Google Scholar]
- 22.Ramirez DM, Leppla SH, Schneerson R, Shiloach J. Production, recovery and immunogenicity of the protective antigen from a recombinant strain of Bacillus anthracis. J Ind Microbiol Biotechnol. 2002;28:232–238. doi: 10.1038/sj/jim/7000239. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Bugge TH, Leppla SH. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J Biol Chem. 2001;276:17976–17984. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- 24.Ramage J, Vallera DA, Black J, Aplan P, Kees U, Frankel AE. The diphtheria toxin/urokinase fusion protein (DTAT) is selectively toxic to CD87 expressing leukemic cells. Leuk Res. 2003;27:79–84. doi: 10.1016/s0145-2126(02)00077-2. [DOI] [PubMed] [Google Scholar]
- 25.Bardet V, Tamburini J, Ifrah N, Dreyfus F, Mayeux P, Bouscary D, Lacombe C. Single cell analysis of phosphoinositide 3-kinase/Akt and ERK activation in acute myeloid leukemia by flow cytometry. Haematologica. 2006;91:757–764. [PubMed] [Google Scholar]
- 26.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 27.Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Evers BM. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J Surg Res. 2012;176:542–548. doi: 10.1016/j.jss.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 28.Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR. The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer. 2012;106:1386–1394. doi: 10.1038/bjc.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A, et al. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Mol Cancer Ther. 2012;11:1193–1202. doi: 10.1158/1535-7163.MCT-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 31.James JA, Smith MA, Court EL, Yip C, Ching Y, Willson C, Smith JG. An investigation of the effects of the MEK inhibitor U0126 on apoptosis in acute leukemia. Hematol J. 2003;4:427–432. doi: 10.1038/sj.thj.6200327. [DOI] [PubMed] [Google Scholar]
- 32.Milella M, Estrov Z, Kornblau SM, Carter BZ, Konopleva M, Tari A, Schober WD, Harris D, Leysath CE, Lopez-Berestein G, et al. Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood. 2002;99:3461–3464. doi: 10.1182/blood.v99.9.3461. [DOI] [PubMed] [Google Scholar]
- 33.Morgan MA, Dolp O, Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood. 2001;97:1823–1834. doi: 10.1182/blood.v97.6.1823. [DOI] [PubMed] [Google Scholar]