

ABSTRACT
A cluster of human plague cases occurred in the seaport city of Mahajanga, Madagascar, from 1991 to 1999 following 62 years with no evidence of plague, which offered insights into plague pathogen dynamics in an urban environment. We analyzed a set of 44 Mahajanga isolates from this 9-year outbreak, as well as an additional 218 Malagasy isolates from the highland foci. We sequenced the genomes of four Mahajanga strains, performed whole-genome sequence single-nucleotide polymorphism (SNP) discovery on those strains, screened the discovered SNPs, and performed a high-resolution 43-locus multilocus variable-number tandem-repeat analysis of the isolate panel. Twenty-two new SNPs were identified and defined a new phylogenetic lineage among the Malagasy isolates. Phylogeographic analysis suggests that the Mahajanga lineage likely originated in the Ambositra district in the highlands, spread throughout the northern central highlands, and was then introduced into and became transiently established in Mahajanga. Although multiple transfers between the central highlands and Mahajanga occurred, there was a locally differentiating and dominant subpopulation that was primarily responsible for the 1991-to-1999 Mahajanga outbreaks. Phylotemporal analysis of this Mahajanga subpopulation revealed a cycling pattern of diversity generation and loss that occurred during and after each outbreak. This pattern is consistent with severe interseasonal genetic bottlenecks along with large seasonal population expansions. The ultimate extinction of plague pathogens in Mahajanga suggests that, in this environment, the plague pathogen niche is tenuous at best. However, the temporary large pathogen population expansion provides the means for plague pathogens to disperse and become ecologically established in more suitable nonurban environments.
IMPORTANCE
Maritime spread of plague led to the global dissemination of this disease and affected the course of human history. Multiple historical plague waves resulted in massive human mortalities in three classical plague pandemics: Justinian (6th and 7th centuries), Middle Ages (14th to 17th centuries), and third (mid-1800s to the present). Key to these events was the pathogen’s entry into new lands by “plague ships” via seaport cities. Although initial disease outbreaks in ports were common, they were almost never sustained for long and plague pathogens survived only if they could become established in ecologically suitable habitats. Although plague pathogens’ ability to invade port cities has been essential for intercontinental spread, these regions have not proven to be a suitable long-term niche. The disease dynamics in port cities such as Mahajanga are thus critical to plague pathogen amplification and dispersal into new suitable ecological niches for the observed global long-term maintenance of plague pathogens.
Introduction
Yersinia pestis, the etiologic agent of plague, has demonstrated a remarkable ability to spread over long distances and cause intense outbreaks interrupted by long periods of silence or reduced activity. Molecular genetic investigations have indicated that Y. pestis spread multiple times from foci in central Asia in greatly widening swaths as human-mediated transport became more efficient (1–3). Plague attained its current global distribution during the third pandemic, which began in 1855 in the Chinese province of Yünnan, when it was introduced into many previously unaffected countries, including Madagascar, via infected rats on steam ships. Thus, the maritime spread of plague that led to much of its current global distribution was highly dependent upon an initial outbreak in an urban seaport. Key to this were the “plague ships” that introduced rats and flea vectors into seaport cities. Although initial disease outbreaks in ports were common, they were never sustained (4). Rather, in most areas, plague went extinct in the ports and survived only if it could become ecologically established in more suitable rural habitats, where it could establish a sustainable rodent-to-flea-to-rodent transmission cycle. Although the ecology of plague in port cities is essential for its spread, these cities have not proven to provide a suitable long-term niche. The disease dynamics of plague in port cities is largely unknown but represents a critical, if tenuous, step between arrival via plague ships or other human-mediated means and eventual long-term ecological establishment in more suitable areas outside port cities.
Plague was introduced into Madagascar during the third pandemic and remains an important human health threat in that country. Plague was first introduction into Madagascar in the coastal city Toamasina in 1898 (5), likely by a ship from India (2). This was followed by outbreaks in other coastal cities, including Mahajanga. Plague then spread to the central highlands, reaching the capital city of Antananarivo in 1921, probably via the railroad linking Toamasina and Antananarivo (5). Plague then disappeared from the coast but became established in the highlands, where it remains to this day (5, 6). It is a significant human health threat, and hundreds of cases occur each year, making Madagascar one of the top three countries in the world for human plague cases between 1995 and 2009 (7).
There are three recognized plague foci in Madagascar, two traditional and one that has recently re-emerged. The two traditional foci consist of two large areas in the central and northern highlands above 800 m in elevation (6). Plague was introduced into these regions by 1921 and has continued to cycle there ever since (5, 6). The third focus has recently re-emerged and consists of the port city of Mahajanga, located ~400 km by air from Antananarivo (6). Plague was first introduced into Mahajanga during a 1902 outbreak, followed by additional outbreaks in 1907 and between 1924 and 1928 (5). After these outbreaks, plague disappeared from Mahajanga for 62 years before reappearing during a large 1991 outbreak (8); additional outbreaks followed between 1995 and 1999 (9–11). These outbreaks were very large, accounting for ~30% of the reported human plague cases in Madagascar during this time period (9).
There are marked differences in the ecology of plague between the highland foci and the Mahajanga focus. Two flea vectors, Xenopsylla cheopis and Synopsyllus fonquerniei, found primarily inside and outside houses, respectively, are important in the highlands (12, 13). X. cheopis is less abundant and S. fonquerniei is absent below 800 m (12, 13), leaving X. cheopis as the only vector in Mahajanga (9, 14). The black rat (Rattus rattus), the principal Malagasy plague host, is found in abundance all over the island and is responsible for maintaining plague in the highlands (5, 6, 12, 13, 15). In addition to R. rattus, the brown rat (R. norvegicus) also plays a role in the highlands as the dominant host in Antananarivo (6, 13, 14). Though both R. rattus and R. norvegicus may be found in Mahajanga (6, 10, 13), the important host during the most recent Mahajanga outbreaks is thought to have been the Asian shrew (Suncus murinus) (12, 14). In the highlands, the plague season stretches from October to April, during the warm, rainy season (16), with an onset when flea populations are at their maximum and rat populations are at their minimum (12, 13). In contrast, the Mahajanga outbreaks all occurred between July and November, during the cool, dry season (16), with an onset when flea abundance on S. murinus was at its maximum (12, 14).
Unlike other known plague foci, the Mahajanga plague focus likely represents a true reintroduction of plague into a plague-free area rather than a case of reemergence of human cases in a silent but still active plague focus. The restriction of plague to the Malagasy highlands (6) is thought to be linked to the absence of S. fonquerniei below 800 m (12, 13), and there is no evidence to counter this theory. Despite the presence of a highly susceptible host (R. rattus) and an effective vector (X. cheopis) in other parts of the island (12, 13) and despite active plague surveillance (6), there have been no reports of any rat epizootics (die-offs) in the coastal regions after plague “disappeared” from these areas, although rat deaths were observed before each Mahajanga outbreak in the 1990s (6, 10). In addition, rats from the coastal regions show no development of plague resistance, in contrast to rats from the highland plague foci (17). Together, these findings argue that the coast, including Mahajanga, was truly plague free from 1929 to 1990 and that plague was reintroduced into the previously extinguished Mahajanga focus in 1991 (14).
Plague was likely reintroduced into Mahajanga from the central highlands. In fact, there may have been multiple plague transfers both from the central highlands to Mahajanga and from Mahajanga back to the central highlands, although one introduction appears to have become established and undergone local cycling in Mahajanga (15). Isolates from the Mahajanga outbreaks are thus of great interest for exploration of the evolution of Y. pestis over a short (~1-decade) time scale but in the same geographic location. We further investigated the molecular evolution of Y. pestis from Mahajanga by whole-genome sequencing of four Mahajanga Y. pestis isolates, discovering single-nucleotide polymorphisms (SNPs) by using those genomes, screening those SNPs, and then performing a high-resolution 43-locus multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) of 44 Mahajanga isolates and an additional 218 Malagasy isolates from the highland foci.
RESULTS
Our SNP discovery and analysis efforts expanded on previously published work on Y. pestis in Madagascar. A previously published SNP phylogeny of Y. pestis in Madagascar based upon 56 SNPs contained two major groups and 12 subgroups related to those two groups (Fig. 1) (15). We sequenced the whole genomes of four isolates from Mahajanga and discovered 22 new SNPs from these genomes. As these isolates were members of the k node in the previous SNP analysis, our efforts led to the discovery of a new lineage (s lineage, Fig. 1), rooted in the k node and terminating in a node (s9, Fig. 1) containing the two most recently isolated, sequenced Mahajanga strains, 154/98 B and 17/99 B. By screening these 22 SNPs across a large panel of DNAs, we identified nine new nodes within this lineage (Fig. 1). This type of lineage and node discovery (i.e., nodes along a linear phylogeny rooted in the node from which the sequenced strain was chosen and terminating in the new sequenced strain) is expected whenever whole-genome sequences (WGSs) are used for SNP discovery because of the phylogenetic discovery bias inherent in this method (18, 19). Since there is little to no evidence of recombination in the diversification of Y. pestis (2), these SNPs can be used in parsimony analyses to determine the evolutionary history of Y. pestis.
FIG 1 .

SNP phylogeny of 262 Malagasy Y. pestis isolates. Nodes (lowercase letters) were named as in reference 15 and include all of the nodes described there and a new lineage containing nine nodes (s1 to s9) described here. Black and gray outlines indicate previously identified nodes (2, 15) that were and were not, respectively, represented by isolates in this study. The nine new nodes are colored to indicate which nodes were found predominantly in the central highlands (yellow), the node that was likely introduced into Mahajanga from the central highlands (orange), and which nodes were likely derived in Mahajanga (red). The numbers of isolates in nodes with more than one isolate are indicated as are the numbers of SNPs on branches (red numbers) with more than one SNP. The nodes containing the sequenced Mahajanga strains (53/91, 64/91, 154/98 B, and 17/99 B) and the two previously sequenced Malagasy strains (MG05-1020 and IP275) are labeled with the strain names.
Our SNP analysis also provided support for a previous MLVA-based analysis. The s lineage in our SNP analysis corresponded to subclade I.A. in a previous MLVA-based analysis (15), with all subclade I.A. isolates possessing the derived state for SNP Mad-57 (see Table S1 in the supplemental material). This confirmed the previous designation of subclade I.A. as a robust genetic group despite only weak bootstrap support in the previous study (15) and supports the use of MLVA to identify robust genetic groups even when statistical support may be weak, similarly to other studies (20, 21). Likewise, our SNP analysis also supported and expanded on the proposed origin and spreading pattern suggested by the previous MLVA-based analysis. First, the initial node in our SNP analysis, node s1, contained a single isolate from the Ambositra district (Fig. 2; see Table S2 in the supplemental material), the proposed origin point of subclade I.A. in the previous MLVA-based analysis (15). Second, the MLVA-based analysis suggested that following its origin, subclade I.A. then continued to exist in the Ambositra district, spread to and became established in a large area including and surrounding Antananarivo, spread to and became established in Mahajanga, and was at least introduced into the Fianarantsoa district, though it may not have become established there (15). Supporting this, our geographic analysis of the new s lineage nodes in our SNP analysis suggested that, following its origin in the Ambositra district, the s lineage continued to exist in the Ambositra district, as indicated by the node s2 isolates found there (Fig. 2; see Table S2). The lineage then appears to have been transferred to the northwestern portion of the central highlands, where it spread in a predominantly west-to-east direction, becoming established in the Soavinandriana, Miarinarivo, Arivonimamo, and ultimately Antananarivo and Manjakandriana districts, as indicated by the geographic distribution of nodes s2 to s5 (Fig. 2; see Table S2). From Antananarivo, one of the few urban areas in Madagascar, it likely spread to Mahajanga (see below) and was at least introduced into the Antanifotsy and Fianarantsoa districts, though it may not have become established there (Fig. 2). This pattern supports the results of the previous MLVA-based analysis (15) but also provides additional insight into the direction of spreading because of the directionality provided by the SNP analysis.
FIG 2 .

Geographic distribution of nodes s1 to s9. The s lineage portion of the SNP phylogeny from Fig. 1 is shown, as well as a map of Madagascar indicating the geographic distribution of isolates from this lineage. Light-gray-shaded polygons indicate Madagascar districts where Y. pestis isolates used in this study were obtained. Districts where isolates from the s lineage were found are labeled by letters as follows: A, Soavinandriana; B, Miarinarivo; C, Arivonimamo; D, Antananarivo; E, Manjakandriana; F, Antanifotsy; G, Ambositra; H, Fianarantsoa; I, Mahajanga. Colors within the mapped circles and squares correspond to the node color designations in the SNP phylogeny. Divisions within circles indicate that multiple nodes were found at that location. Circles represent isolates where the city or commune of origin is known. Squares represent isolates where only the district of origin is known and are placed within their corresponding districts near cities or communes containing the same node(s) where possible. Circles, squares, and pie chart slices in the map are numbered on the basis of the node number in the SNP phylogeny for the isolates represented by those shapes. A large arrow indicates the likely geographic source and direction of travel of the Y. pestis strain that was introduced into and became established in Mahajanga.
Most of the Mahajanga isolates were very closely related, suggesting that a single introduction became established in Mahajanga and then underwent local cycling and differentiation. Consistent with a previous analysis (15), 42 of the 44 Mahajanga isolates belonged to the s lineage. Forty of these belonged to nodes s5 through s9 in the SNP analysis (see Table S2 in the supplemental material). Thirty-nine of these were very closely related on the basis of a combination of SNPs and MLVA, with most MLVA differences involving only a single repeat change at a single VNTR locus (Fig. 3). Furthermore, these 39 Mahajanga isolates were genetically distinct from 16 central-highland isolates that were also found in node s5, with the genetically closest central-highland isolates differing at a minimum of three or four VNTR loci from the Mahajanga node s5 isolates (data not shown). The close genetic relationships among these 39 Mahajanga isolates and their distinction from the genetically nearest central-highland isolates strongly suggest that these isolates are the product of a single successful introduction from the central highlands to Mahajanga that underwent local cycling and differentiation during the outbreaks of 1991 to 1999. These isolates are here referred to as the Mahajanga subpopulation.
FIG 3 .
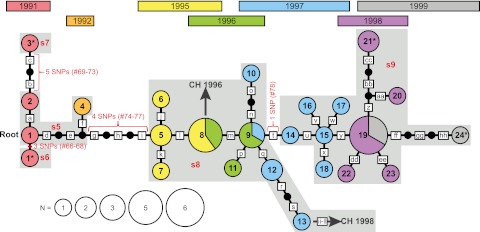
Maximum-likelihood phylogeny of Mahajanga Y. pestis isolates. A maximum-likelihood phylogeny based upon MLVA data is presented for 39 Y. pestis isolates believed to have originated from local cycling in Mahajanga. Numbered circles indicate MLVA genotypes. Genotype circles are color coded by the year of isolation and sized according to the number of isolates with each genotype. An asterisk marks the genotype circles containing each of the four Mahajanga strains sequenced. Small black circles indicate theoretical intermediate MLVA genotypes that were not observed in our isolate set. Red brackets or bars indicate the locations of SNP mutations and are labeled with the number of SNPs and the SNP ID numbers presented in Table S1 in the supplemental material; the brackets span the entire branch linking two SNP-defined nodes to reflect the fact that the exact order of these SNP mutations among the theoretical intermediate genotypes depicted is unknown. The corresponding SNP nodes from Fig. 1 are indicated by light-gray-shaded areas labeled with the name of that node in red. Dark gray arrows pointing to a “CH” and a year indicate those points along the Mahajanga phylogeny where isolates appear to have been transferred to the central highlands; the years of isolation of the central-highland isolate are shown. Boxed letters indicate individual VNTR mutations. Assuming a root at genotype 1 and moving from left to right, these mutations were as follows, where a plus or minus sign indicates an insertion or deletion, respectively, followed by the number of repeats involved in the mutation: a, q, r, u, v, and gg, M19 + 1; b, M22 + 2; c, M19 + 2; d, f, s, and aa, M27 − 1; e and j, M19 − 2; g, M19 − 6; h and bb, M22 − 1; i, m, and dd, M23 + 1; k and n, M12 − 1; l and p, M25 − 1; o, M19 − 1; t, M58 + 1; w, M25 + 2; x, M28 + 1; y, M79 + 1; z, M25 + 1; cc, M31 − 1; ee, M27 + 1; ff, M12 − 2; hh, M28 − 1. Note that the ordering of VNTR mutations and theoretical genotypes along those branches with multiple VNTR mutations is arbitrary, as the exact order of the VNTR mutations is unknown.
The remaining five Mahajanga isolates appear to represent separate transfers from the central highlands to Mahajanga. Two Mahajanga isolates (364/97 S and 2/92) belonged to different genetic subpopulations (k and r), suggesting at least two additional transfers to Mahajanga. Two more Mahajanga isolates (52/91 and 103/97 S) belonged to the s lineage but belonged in earlier nodes (s2 and s3) in that lineage than the Mahajanga subpopulation isolates (see Table S2 in the supplemental material). The other isolates in these nodes were most commonly found in the Ambositra district and in the northwestern portion of the central highlands (predominantly the Soavinandriana and Miarinarivo districts) (Fig. 2; see Table S2), suggesting that the node s2 and s3 Mahajanga isolates were due to two more transfers to Mahajanga. The available temporal data support the above hypotheses, as none of the above four Mahajanga isolates was the earliest isolate in its respective node (see Table S2). This, together with the fact that Mahajanga is a recently re-emerged plague focus, supports the idea that these isolates all represent transfers to Mahajanga rather than the reverse. Finally, a fifth Mahajanga isolate, 93/98 S, although it belonged to the same node (s5) as three of the Mahajanga subpopulation isolates (see Table S2), was more closely related to central-highland isolates in that node than to the other Mahajanga isolates in that node. In addition, this isolate was isolated in 1998, 6 to 7 years later than the other Mahajanga node s5 isolates (see Table S2), suggesting that it was not related to the initial introduction of Y. pestis to Mahajanga in 1991 and was more likely due to another transfer from the central highlands to Mahajanga.
The maximum-likelihood analysis of the 39 Mahajanga isolates determined to represent the Mahajanga subpopulation revealed a striking temporal pattern that reinforced the identification of these isolates as a locally differentiating subpopulation (Fig. 3). The overall maximum-likelihood phylogeny appears linear when examined with regard to the isolation years of the different genotypes. However, the phylogeny is not completely linear, as several genotypes branch off from the “backbone” of the linear portion of the maximum-likelihood phylogeny, giving the overall phylogeny the appearance of a series of “star” phylogenies strung together (Fig. 3). This pattern suggests that significant bottlenecks likely occurred during the successive years of the Mahajanga outbreaks of 1991 to 1999. Specifically, the large number of Y. pestis generations characteristic of a plague outbreak likely led to an increase in diversity during each outbreak. However, much of this diversity would have been eliminated when the seasonal outbreak came to an end, with representatives of only a few genotypes surviving to begin the next outbreak in the following year. This would account for the patterns observed in the maximum-likelihood phylogeny in Fig. 3; the linear backbone of the phylogeny represents the string of genotypes that emerged and survived to start each subsequent outbreak, whereas the branches off of this linear backbone represent genotypes that emerged but did not survive.
There appears to have been at least one transfer from Mahajanga back to the central highlands. Two node s8 isolates were found in Antananarivo in 1996 (30/96 B) and 1998 (181/98 S) (see Table S2 in the supplemental material). These isolates are almost certainly due to a transfer from Mahajanga to Antananarivo, given that, aside from these two isolates, all of the isolates belonging to nodes s6 through s9 were from Mahajanga (see Table S2), strongly suggesting that node s8 arose in Mahajanga and not elsewhere. Further supporting this idea, the earliest node s8 isolates came from Mahajanga in 1995 and the two Antananarivo node s8 isolates were from 1996 and 1998 (see Table S2), at least 1 year later than the s8 genotype must have appeared. Whether these two node s8 isolates in Antananarivo were the result of one or two transfers from Mahajanga to Antananarivo is not clear, though the maximum-likelihood analysis suggests that there were two transfer events. Specifically, rather than being more closely related to each other, isolate 30/96 B possesses the same MLVA genotype as genotype 8 in the maximum-likelihood analysis, whereas isolate 181/98 S is most closely related to genotype 13 in the maximum-likelihood analysis, although it does differ from genotype 13 at four VNTR loci (Fig. 3).
In addition to the above, there may have been another transfer from Mahajanga to the central highlands, depending upon the geographic origin of node s5. The earliest node s5 isolate was from Mahajanga (see Table S2 in the supplemental material), suggesting that Mahajanga may have been the geographic origin point of node s5. If this was the case, then there would have to have been a transfer from Mahajanga to the central highlands to account for the node s5 isolates in the central highlands. However, it is more parsimonious to assume that node s5 originated in the central highlands and was then transferred to Mahajanga, as this would have required fewer overall transfer events. Indeed, one of the central-highland node s5 isolates was isolated in 1992 (see Table S2 in the supplemental material), just a year later than the earliest Mahajanga node s5 isolates, suggesting that node s5 existed at both locations at nearly the same time and could very likely have originated in the central highlands.
The number of SNP mutations expected during the Mahajanga outbreaks was very close to the number of SNP mutations observed. There were 34 VNTR mutations among the Mahajanga subpopulation isolates (Fig. 3). Using the cumulative 43-locus VNTR mutation rate of 1.1 × 10−3 mutations per generation for the 43 VNTR loci examined (22), we estimated that the probability of 34 VNTR mutations was maximized at 30,357 (95% confidence interval, 23,566 to 38,343) generations. Using this estimate of 30,357 generations, the estimated SNP mutation rate of 1.7 × 10−10 mutations per nucleotide per generation (2) and the number of nucleotides in the Y. pestis genome minus 388 kb of repetitive sequence (4.27 Mbp) (2, 23), we expected 22 SNPs to occur during the Mahajanga outbreaks. This expected number of SNPs was exceptionally close to the 13 SNPs observed, especially when considering the fact that additional SNPs would likely be discovered if additional isolates were sequenced. There appears to be a concordance between the estimated mutation rates in VNTRs and SNPs in this population.
DISCUSSION
Y. pestis is one of the most successful pathogens in history. It has been linked to three historical pandemics during which it spread to every inhabited continent, killed hundreds of millions of people, and established stable ecological foci on every inhabited continent except Australia (4). This success was dependent on at least three key factors: (i) the ability of Y. pestis to travel long distances, usually facilitated by humans; (ii) the ability of Y. pestis to cause large outbreaks at the locations where it was introduced; and (iii) the ability of Y. pestis to eventually become ecologically established in long-term foci. Our study of the Mahajanga plague outbreaks of 1991 to 1999 provides insight into these factors and serves as a model of how, during the previous pandemics, Y. pestis spread from unstable port city populations to become ecologically established in long-term foci.
Y. pestis has demonstrated tremendous dispersal ability, affecting the entire “known world” during each of the three historical pandemics (4). Y. pestis is the undisputed etiologic agent of the third pandemic (4), and ancient DNA and protein analyses have provided compelling molecular evidence of the involvement of Y. pestis in the first two pandemics (24–36), confirming its dispersal ability even before the advent of steam-powered ships. Additional molecular studies of both extant strains and ancient DNA have suggested that the “three pandemics” were actually made up of multiple epidemic waves that spread from the geographical origin of Y. pestis in central Asia (2, 29), suggesting that long-distance dispersal of Y. pestis was not an uncommon event. Y. pestis dispersal events can be correlated with historically documented human travel (2), emphasizing the importance of the human-mediated transport of this pathogen. Indeed, the advent of steam-powered shipping was directly responsible for the dissemination of Y. pestis to every inhabited continent in just a few years during the third pandemic (4).
Frequent human-mediated dispersal of Y. pestis can clearly be seen in our Mahajanga study. Specifically, several successful transfer events appear to have occurred during the evolution of the s lineage described here, including at least one long-distance transfer from the presumed geographic origin point in the Ambositra district to the northwestern central highlands, additional transfers over unknown distances as the s lineage spread east, and several additional long-distance transfers from Antananarivo to other locations, including Mahajanga. Overall, there is genetic evidence of at least six probable transfers from the central highlands to Mahajanga and two likely transfers from Mahajanga back to the central highlands. Indeed, 5 (11%) of the 44 Mahajanga isolates appeared to be due to transfer events unrelated to the transfer event that resulted in the establishment of the Mahajanga subpopulation described here. Most of these transfers, particularly those over long distances, were likely due to inadvertent human-mediated transport of infected rats and their fleas together with legitimate shipments. Although these human-mediated long-distance transfers could conceivably occur between any two locations, the establishment of the s lineage in Antananarivo, one of the few urban areas in Madagascar, appears to have been particularly important in facilitating long-distance transfers of this lineage throughout Madagascar (to the Antanifotsy, Fianarantsoa and Mahajanga districts; Fig. 2). The increase in observed long-distance transfers once the s lineage reached Antananarivo is likely due to the increased commerce and ease of travel associated with an urban area. Indeed, the Mahajanga-Antananarivo transfers likely occurred via RN4, a relatively well-maintained highway that links Antananarivo and Mahajanga and can be traveled in ~10 h (according to the BBC Worldwide Lonely Planet website).
Y. pestis has routinely caused very large outbreaks when introduced into a susceptible population, whether human or rodent. Historically, Y. pestis has been linked to multiple epidemics during the first two pandemics, the most devastating of which killed ~30 to 40% of the European population and became known as the black death (4). During the third pandemic, Y. pestis caused outbreaks in multiple major port cities during its initial spread (4, 37–40) and currently causes large outbreaks among susceptible epizootic rodent populations, such as prairie dogs in North America (4, 41). Similarly, when Y. pestis was reintroduced into Mahajanga, it caused large human outbreaks in that port city, which had not had any human plague cases for 62 years (8–11).
Y. pestis has also been remarkably adept at becoming ecologically established in new locations, but with curious limitations. The clearest example of this ability can be seen during the third pandemic, during which Y. pestis established much of its current worldwide distribution and became successfully ecologically established on every inhabited continent except Australia (4). However, despite its great success, Y. pestis did not become ecologically established at every location into which it was introduced during the third pandemic. For example, although it caused outbreaks in port cities in the southern United States, Hawaii, and Australia (38–40), Y. pestis was unable to become ecologically established in these areas, which are now plague free (4, 40). Nor is this phenomenon limited to the third pandemic. Despite cycles of epidemics from the mid-14th century to the late 17th century, there are no contemporary ecological plague foci in western Europe (4). Ancient DNA analyses have even suggested that the repeated epidemic waves in medieval Europe may have been caused by different genotypes that emerged separately from central Asia (29), suggesting that there may not have been stable ecologically established foci in Europe during medieval times either.
The Mahajanga plague outbreaks of 1991 to 1999 provide a modern example of this phenomenon of outbreaks without the establishment of permanent local plague foci. Mahajanga was plague free for 62 years before the outbreaks of 1991 to 1999 (10, 14). Following these outbreaks, the plague pathogen appears to have again gone extinct from Mahajanga on the basis of the absence of human plague cases (14). Our molecular analyses of the outbreak isolates suggest that these outbreaks were due predominantly to the introduction and establishment of a single genotype in Mahajanga, although other genotypes were introduced and present in Mahajanga at least transitorily. In addition, the maximum-likelihood analysis of the Mahajanga subpopulation isolates revealed a telling phylotemporal pattern: population diversity was generated during each plague outbreak but was lost when each outbreak subsided, with representatives of only one to a few genotypes surviving to cause the next outbreak (Fig. 3). A schematic of this process and the corresponding number of observed confirmed and presumptive human plague cases in Mahajanga during the 1991-to-1999 outbreaks is presented in Fig. 4. The generation of diversity with larger population sizes is expected (42). Of greater interest is the observed loss of genetic diversity following each outbreak observed here. This suggests that the ecological establishment of Y. pestis in Mahajanga was tenuous at best and likely highly dependent upon local conditions. The tenuous nature of the Mahajanga niche is reinforced by the fact that only one of the several introductions of Y. pestis into Mahajanga led to a lineage that persisted for any length of time. The 1991-to-1999 Mahajanga outbreaks were centered in and around the main marketplace of the Marolaka district, in an area densely populated with very poor people (9–11). The large amount of rubbish generated by the market and poor conditions provided an excellent habitat for the rodent reservoirs of Y. pestis. The population genetic analysis suggests that the ecological niche was small, at best, and once the hygiene of these areas was improved, Y. pestis could be eliminated.
FIG 4 .
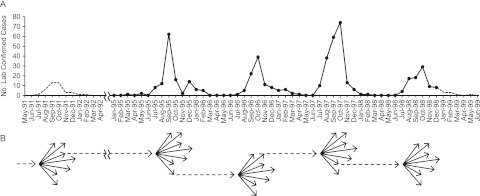
Schematic of the Mahajanga outbreaks. (A) Graph of the number of laboratory-confirmed human plague cases in Mahajanga from May 1991 through June 1999. Solid lines and points indicate actual numbers of confirmed and presumptive human plague cases derived from reference 9. Dotted lines represent the estimated numbers of confirmed and presumptive human plague cases on the basis of either the average percentage of cases per month observed from 1995 to 1998 multiplied by the total number of laboratory-confirmed cases reported for the 1991-1992 outbreak (11) for May 1991 through April 1992 or the average number of cases per month observed from 1995 to 1998 for January 1999 through June 1999. (B) Schematic of the expansions and contractions that occurred in the Y. pestis subpopulation in Mahajanga during the 1991-to-1999 outbreaks. Each dotted arrow represents the founding genotype of an outbreak, and each arrow cluster indicates the population expansion and the corresponding increase in genetic diversity that occurred during each outbreak. As Y. pestis was apparently eliminated from Mahajanga following the 1998-1999 outbreak, no dotted arrow leads from that arrow cluster.
The environmental conditions that allow the tenuous establishment of Y. pestis in a particular location can be described as an ephemeral niche. If a given ephemeral niche can be disrupted, Y. pestis can be driven to extinction, which is what occurred in Mahajanga, Madagascar. However, long-term ecological establishment can occur when Y. pestis is able to transition from an ephemeral niche to a permanent niche. That said, this can occur only when appropriate ecological conditions exist, such as in western North America during the third pandemic, when Y. pestis became ecologically established in susceptible native rodent populations adjacent to the affected port city of San Francisco (39). In contrast, although Y. pestis was introduced into the southern United States, Hawaii, and Australia during the third pandemic (38–40), it did not become permanently ecologically established at those locations (4, 40), most likely because of the lack of suitable ecological conditions (4). For example, the presence of very few native rodents in Australia likely prevented the establishment of long-term foci there (43). The very large populations and genetic diversity generated during the initial outbreaks caused when Y. pestis is introduced into a new area increase the chances for the transition of Y. pestis from an ephemeral to a permanent niche, provided that the appropriate ecological conditions exist. This process is similar to what has been described for classic plague epidemiology among rodent populations where epizootics (large-scale die-offs) that occur among susceptible rodents exposed to plague serve to amplify and spread Y. pestis in the environment (4, 42, 44), whereas the generally more cryptic enzootic (reservoir) hosts serve to maintain Y. pestis in the environment (4, 42, 44, 45). In this case, the epizootic hosts represent the ephemeral niche, whereas the enzootic hosts represent the permanent niche.
Certain Y. pestis genotypes appear to be particularly successful at spreading and becoming established in new areas. For example, most of the geographic range of Y. pestis is made up of members of a single group in branch 1 of the worldwide Y. pestis phylogeny, group 1.ORI, which corresponds to the classical Orientalis biovar. The other major groups in the Y. pestis worldwide phylogeny have much smaller geographic distributions (2). In Madagascar, the s lineage appears to be a similarly successful clone that was able to spread from its geographical origin in the Ambositra district throughout much of the central highlands and to Mahajanga. Whether the success of these clones was due to chance or a specific adaptive advantage has yet to be determined but merits further investigation, particularly since this phenomenon has been observed in other geographically widespread pathogens (43). Regardless, the genetic diversity generated during large outbreaks/epizootic events provides an opportunity for the emergence of a new, highly fit clone.
Our Mahajanga analysis has provided a model for the process of ecological Y. pestis establishment by allowing us to observe the introduction, establishment, and presumed extinction of Y. pestis in a modern port city. Under this model, one to a few fit or lucky genotypes can be transferred to an ephemeral niche, where they can cause a large outbreak, generating a large Y. pestis population and considerable genetic diversity. Much of this genetic diversity is then eliminated because of the bottlenecks created as the outbreak wanes. By chance and/or due to an adaptive advantage, a few of these isolates, some of which may carry genetic mutations, can survive and disperse to transition to a permanent niche if the appropriate ecological conditions exist. In contrast, if suitable permanent niche conditions do not exist, Y. pestis can be driven to extinction by disruption of the ephemeral niche. This process has implications for the phylogeography of Y. pestis across multiple geographic scales since some mutations, whether due to genetic drift or to adaptation, can become fixed in a particular subpopulation because of the bottlenecks that occur during any transition from an ephemeral niche to a permanent niche. In many cases, these subpopulations are likely to be geographically segregated. Indeed, relatively recent phylogenies of Y. pestis on worldwide (2, 3), regional (15), and local (42) scales are all consistent with this process.
MATERIALS AND METHODS
Y. pestis DNAs.
DNA was obtained from 44 Mahajanga Y. pestis isolates and an additional 218 geographically diverse isolates from the highland plague foci (see Table S2 in the supplemental material). These 262 DNAs were previously reported on by Vogler et al. (15) in an island-wide study of Y. pestis in Madagascar. DNAs consisted of simple heat lysis preparations or whole-genome amplification (WGA; Qiagen, Valencia, CA) products generated from the heat lysis preparations. Most of the isolates were collected by the Malagasy Central Laboratory for Plague, supervised by the Institut Pasteur de Madagascar, and were isolated primarily from human cases, with a few isolated from other mammals or fleas. A few other isolates were from other institutions (still originally collected by the Malagasy Central Laboratory for Plague) or represent publically available WGSs (see Table S2).
Whole-genome sequencing.
We sequenced four Mahajanga isolates, two (53/91 and 64/91) from 1991, one (154/98 B) from 1998, and one (17/99 B) from 1999 (see Table S2 in the supplemental material). These strains were chosen because of their positioning at the tips of a maximum-likelihood phylogeny generated from MLVA data (see below) and also to span the entire time frame of the Mahajanga plague outbreaks. Genomic DNA from each isolate for sequencing was amplified with a WGA kit (Qiagen, Valencia, CA) by using template DNA from the original heat lysis DNA preparations. Five micrograms of WGA DNA was sheared to an average fragment size of 500 bp with a SonicMan high-throughput sonication system (Matrical BioScience, Spokane, WA). Bar-coded libraries for paired-end Illumina whole-genome sequencing were prepared with a NEBNext DNA Sample Prep Master Mix Set 1 kit (New England Biolabs, Ipswich, MA) and a Multiplexing Sample Preparation Oligonucleotide kit (Illumina, San Diego, CA). Libraries were validated by quantitative PCR with a KAPA Library Quantification kit (KAPA Biosystems, Boston, MA) on a 7900 Real-Time PCR system (Life Technologies, Carlsbad, CA). Libraries were sequenced with an Illumina Genomic Analyzer IIx with Paired End Module and Cluster Station using the manufacturer’s protocol to produce 100-bp paired-end reads. Image analysis for base calling and alignments was performed as previously described (46).
SNP discovery.
To identify putative SNPs, the four Mahajanga isolate WGSs were aligned with CO92 (GenBank accession no. AL590842) (23) and compared to each other and to two previously available Malagasy WGSs (MG05-1020 [GenBank accession no. AAYS00000000] and IP275 [GenBank accession no. AAOS00000000]) (2). Alignment of the Mahajanga WGS sequence reads was performed with BFAST v. 0.6.4e with a key width of 14 and a previously described index set (47). SolSNP (retrieved on 6 April 2011 from http://solsnp.sourceforge.net) was used to detect putative SNPs in the alignments relative to the reference sequence with the following specifications: minimum coverage, 5; minimum base quality, 10; minimum mapping quality, 10; filter, 0.85. The MUMmer nucmer module (48) was used to align the Malagasy WGSs with the reference sequence, and the show-snps module was used to perform pairwise comparisons for the identification of SNPs. Custom PERL and Java Scripts were used to tabulate SNPs common to both analyses. SNPs with an adjacent SNP less than 30 bp away were removed from the analysis. Finally, SNPs were required to be from a region common to all seven of the genomes analyzed. Putative SNPs identified by the above pipeline were then confirmed visually by examining the reads containing those putative SNPs for the four Mahajanga isolate WGSs using Integrative Genomics Viewer (49).
SNP screening.
Melt-MAMA assays were designed as previously described (50) around 22 SNPs identified from the above and screened across all 262 Malagasy DNAs. SNP locations, primer sequences, primer concentrations, and other information for these assays are presented in Table S1 in the supplemental material. Primers were designed using NetPrimer software (Premier Biosoft, Palo Alto, CA). Each 5 µl of Melt-MAMA reaction mixture contained 1× SYBR Green PCR Master Mix (Applied Biosystems), derived and ancestral allele-specific MAMA primers, a common reverse primer (for primer concentrations, see Table S1), water, and 1 µl of diluted template DNA. Template DNAs were diluted 1/10 for heat lysis preparations or 1/50 for WGAs. All assays were performed with an Applied Biosystems 7900HT Fast Real-Time PCR System with SDS software v2.4. Thermal cycling conditions for the Melt-MAMA assays were as follows: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Melt-MAMA results were interpreted as previously described (50).
MLVA.
All 262 Malagasy isolates were also genotyped with a 43-marker MLVA system as previously described (42).
Phylogenetic analyses.
An SNP phylogeny was generated for all 262 isolates by using data from the 22 SNPs analyzed here and previously reported data from 56 additional SNPs (15) (Fig. 1). The geographic distributions of new nodes identified in this phylogeny (s lineage nodes, Fig. 1) were then geographically mapped to determine phylogeographic patterns (Fig. 2). This information was then used to assist in the selection of isolates to include in the analysis described below.
A second, MLVA-based phylogeny was created for 39 Mahajanga isolates by using the maximum-likelihood method outlined by Vogler et al. (22) (Fig. 3). This maximum-likelihood phylogenetic analysis uses VNTR mutation rates and a general VNTR mutation model to determine the probabilities of different mutations that are, in turn, used to determine the most likely phylogeny (22). The 39 Mahajanga isolates used in this analysis all belonged to nodes s5 through s9 in the SNP analysis (see above) and were assumed to represent the locally differentiating Y. pestis population in Mahajanga during the 1991-to-1999 plague outbreaks (see Table S2 in the supplemental material). Five other Mahajanga isolates were not included in this analysis (see Table S2) since their genotypes suggested that they were due to separate introductions to Mahajanga (see above).
Calculation of generations.
We compared the number of SNP mutations expected to the number of SNP mutations observed among the 39 Mahajanga isolates used to generate the maximum-likelihood phylogeny. We first estimated the number of generations during the Mahajanga outbreaks by using the number of observed VNTR mutations in this data set (Fig. 3) and previously described methods (22, 42, 51). We then used this estimated number of generations and the estimated SNP mutation rate of 1.7 × 10−10 mutations per nucleotide per generation from Morelli et al. (2) to estimate the number of expected SNP mutations for the Mahajanga outbreaks. Finally, we compared this number to the number of SNP mutations actually observed in our data set.
Nucleotide sequence accession number.
The sequence read archives for all four newly sequenced Mahajanga strains have been deposited in the GenBank database and assigned accession number SRP017903.
SUPPLEMENTAL MATERIAL
SNP assay primers and probes used in this study.
Isolates used in this study.
ACKNOWLEDGMENTS
This work was funded by the United States Department of Homeland Security (DHS) Science and Technology Directorate (award HSHQDC-10-C-00139), the Cowden Endowment in Microbiology and the Technology and Research Initiative Fund at Northern Arizona University, the Malagasy Ministry of Health (contract Nu01/95 IDA 2252-MAG) (F.C., S.C.), and the French Ministry of Cooperation (FAC Nu 94008 300) (F.C., S.C.).
The use of products and/or names does not constitute endorsement by the United States DHS.
Footnotes
Citation Vogler AJ, Chan F, Nottingham R, Andersen G, Drees K, Beckstrom-Sternberg SM, Wagner DM, Chanteau S, Keim P. 2013. A decade of plague in Mahajanga, Madagascar: insights into the global maritime spread of pandemic plague. mBio 4(1):e00623-12. doi:10.1128/mBio.00623-12.
REFERENCES
- 1. Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, Vogler AJ, Wagner DM, Allender CJ, Easterday WR, Chenal-Francisque V, Worsham P, Thomson NR, Parkhill J, Lindler LE, Carniel E, Keim P. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 101:17837–17842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morelli G, Song Y, Mazzoni CJ, Eppinger M, Roumagnac P, Wagner DM, Feldkamp M, Kusecek B, Vogler AJ, Li Y, Cui Y, Thomson NR, Jombart T, Leblois R, Lichtner P, Rahalison L, Petersen JM, Balloux F, Keim P, Wirth T, Ravel J, Yang R, Carniel E, Achtman M. 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 42:1140–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, Weinert LA, Wang Z, Guo Z, Xu L, Zhang Y, Zheng H, Qin N, Xiao X, Wu M, Wang X, Zhou D, Qi Z, Du Z, Wu H, Yang X, Cao H, Wang H, Wang J, Yao S, Rakin A, Li Y, Falush D, Balloux F, Achtman M, Song Y, Wang J, Yang R. 2013. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 110:577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brygoo ER. 1966. Epidémiologie de la peste à Madagascar. Arch. Inst. Pasteur Madagascar 35:9–147 [Google Scholar]
- 6. Chanteau S, Ratsifasoamanana L, Rasoamanana B, Rahalison L, Randriambelosoa J, Roux J, Rabeson D. 1998. Plague, a reemerging disease in Madagascar. Emerg. Infect. Dis. 4:101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Health Organization. WHO 2009. Human plague: review of regional morbidity and mortality, 2004–2009. Wkly. Epidemiol. Rec. 85:40–45 [PubMed] [Google Scholar]
- 8. Laventure S, Andrianaja V, Rasoamanana B. 1991. Epidémie de peste à Majunga en 1991. Rapport de mission de l’Institut Pasteur de Madagascar, 1–26 [Google Scholar]
- 9. Boisier P, Rahalison L, Rasolomaharo M, Ratsitorahina M, Mahafaly M, Razafimahefa M, Duplantier JM, Ratsifasoamanana L, Chanteau S. 2002. Epidemiologic features of four successive annual outbreaks of bubonic plague in Mahajanga, Madagascar. Emerg. Infect. Dis. 8:311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boisier P, Rasolomaharo M, Ranaivoson G, Rasoamanana B, Rakoto L, Andrianirina Z, Andriamahefazafy B, Chanteau S. 1997. Urban epidemic of bubonic plague in Majunga, Madagascar: epidemiological aspects. Trop. Med. Int. Health 2:422–427 [PubMed] [Google Scholar]
- 11. Rasolomaharo M, Rasoamanana B, Andrianirina Z, Buchy P, Rakotoarimanana N, Chanteau S. 1995. Plague in Majunga, Madagascar. Lancet 346:1234. [DOI] [PubMed] [Google Scholar]
- 12. Duplantier JM. 2001. The black rat’s role in spreading human plague in Madagascar. Inst. Rech. Dev. Sci. Bull. 131:1–3 [Google Scholar]
- 13. Duplantier JM, Rakotondravony D. 1999. The rodent problem in Madagascar: agricultural pest and threat to human health, p 441–459 In Singleton G, Hinds L, Leirs H, Zhang Z. (ed.), Ecologically based management of rodent pests. Australian Centre for International Agricultural Research, Canberra, Australia [Google Scholar]
- 14. Duplantier JM, Duchemin JB, Chanteau S, Carniel E. 2005. From the recent lessons of the Malagasy foci towards a global understanding of the factors involved in plague reemergence. Vet. Res. 36:437–453 [DOI] [PubMed] [Google Scholar]
- 15. Vogler AJ, Chan F, Wagner DM, Roumagnac P, Lee J, Nera R, Eppinger M, Ravel J, Rahalison L, Rasoamanana BW, Beckstrom-Sternberg SM, Achtman M, Chanteau S, Keim P. 2011. Phylogeography and molecular epidemiology of Yersinia pestis in Madagascar. PLoS Negl. Trop. Dis. 5:e1319 http://dx.doi.org/10.1371/journal.pntd.0001319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chanteau S, Ratsitorahina M, Rahalison L, Rasoamanana B, Chan F, Boisier P, Rabeson D, Roux J. 2000. Current epidemiology of human plague in Madagascar. Microbes Infect. 2:25–31 [DOI] [PubMed] [Google Scholar]
- 17. Tollenaere C, Rahalison L, Ranjalahy M, Duplantier JM, Rahelinirina S, Telfer S, Brouat C. 2010. Susceptibility to Yersinia pestis experimental infection in wild Rattus rattus, reservoir of plague in Madagascar. Ecohealth 7:242–247 [DOI] [PubMed] [Google Scholar]
- 18. Pearson T, Busch JD, Ravel J, Read TD, Rhoton SD, U’Ren JM, Simonson TS, Kachur SM, Leadem RR, Cardon ML, Van Ert MN, Huynh LY, Fraser CM, Keim P. 2004. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc. Natl. Acad. Sci. U. S. A. 101:13536–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pearson T, Okinaka RT, Foster JT, Keim P. 2009. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 9:1010–1019 [DOI] [PubMed] [Google Scholar]
- 20. Li Y, Cui Y, Hauck Y, Platonov ME, Dai E, Song Y, Guo Z, Pourcel C, Dentovskaya SV, Anisimov AP, Yang R, Vergnaud G. 2009. Genotyping and phylogenetic analysis of Yersinia pestis by MLVA: insights into the worldwide expansion of Central Asia plague foci. PLoS One 4:e6000 http://dx.doi.org/10.1371/journal.pone.0006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riehm JM, Vergnaud G, Kiefer D, Damdindorj T, Dashdavaa O, Khurelsukh T, Zöller L, Wölfel R, Le Flèche P, Scholz HC. 2012. Yersinia pestis lineages in Mongolia. PLoS One 7:e30624 http://dx.doi.org/10.1371/journal.pone.0030624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vogler AJ, Keys CE, Allender C, Bailey I, Girard J, Pearson T, Smith KL, Wagner DM, Keim P. 2007. Mutations, mutation rates, and evolution at the hypervariable VNTR loci of Yersinia pestis. Mutat. Res. 616:145–158 [DOI] [PubMed] [Google Scholar]
- 23. Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeño-Tárraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PC, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413:523–527 [DOI] [PubMed] [Google Scholar]
- 24. Bianucci R, Rahalison L, Massa ER, Peluso A, Ferroglio E, Signoli M. 2008. Technical note: a rapid diagnostic test detects plague in ancient human remains: an example of the interaction between archeological and biological approaches (southeastern France, 16th–18th centuries). Am. J. Phys. Anthropol. 136:361–367 [DOI] [PubMed] [Google Scholar]
- 25. Bos KI, Schuenemann VJ, Golding GB, Burbano HA, Waglechner N, Coombes BK, McPhee JB, DeWitte SN, Meyer M, Schmedes S, Wood J, Earn DJ, Herring DA, Bauer P, Poinar HN, Krause J. 2011. A draft genome of Yersinia pestis from victims of the black death. Nature 478:506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drancourt M, Aboudharam G, Signoli M, Dutour O, Raoult D. 1998. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: an approach to the diagnosis of ancient septicemia. Proc. Natl. Acad. Sci. U. S. A. 95:12637–12640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drancourt M, Roux V, Dang LV, Tran-Hung L, Castex D, Chenal-Francisque V, Ogata H, Fournier PE, Crubézy E, Raoult D. 2004. Genotyping, Orientalis-like Yersinia pestis, and plague pandemics. Emerg. Infect. Dis. 10:1585–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Drancourt M, Signoli M, Dang LV, Bizot B, Roux V, Tzortzis S, Raoult D. 2007. Yersinia pestis Orientalis in remains of ancient plague patients. Emerg. Infect. Dis. 13:332–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haensch S, Bianucci R, Signoli M, Rajerison M, Schultz M, Kacki S, Vermunt M, Weston DA, Hurst D, Achtman M, Carniel E, Bramanti B. 2010. Distinct clones of Yersinia pestis caused the black death. PLoS Pathog. 6:e1001134 http://dx.doi.org/10.1371/journal.ppat.1001134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pusch CM, Rahalison L, Blin N, Nicholson GJ, Czarnetzki A. 2004. Yersinial F1 antigen and the cause of black death. Lancet Infect. Dis. 4:484–485 [DOI] [PubMed] [Google Scholar]
- 31. Raoult D, Aboudharam G, Crubézy E, Larrouy G, Ludes B, Drancourt M. 2000. Molecular identification by “suicide PCR” of Yersinia pestis as the agent of medieval black death. Proc. Natl. Acad. Sci. U. S. A. 97:12800–12803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuenemann VJ, Bos K, DeWitte S, Schmedes S, Jamieson J, Mittnik A, Forrest S, Coombes BK, Wood JW, Earn DJ, White W, Krause J, Poinar HN. 2011. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the black death. Proc. Natl. Acad. Sci. U. S. A. 108:E746–E752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tran TN, Forestier CL, Drancourt M, Raoult D, Aboudharam G. 2011. Brief communication: co-detection of Bartonella quintana and Yersinia pestis in an 11th–15th burial site in Bondy, France. Am. J. Phys. Anthropol. 145:489–494 [DOI] [PubMed] [Google Scholar]
- 34. Tran TN, Signoli M, Fozzati L, Aboudharam G, Raoult D, Drancourt M. 2011. High throughput, multiplexed pathogen detection authenticates plague waves in medieval Venice, Italy. PLoS One 6:e16735 http://dx.doi.org/10.1371/journal.pone.0016735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiechmann I, Grupe G. 2005. Detection of Yersinia pestis DNA in two early medieval skeletal finds from Aschheim (upper Bavaria, 6th century A.D.). Am. J. Phys. Anthropol. 126:48–55 [DOI] [PubMed] [Google Scholar]
- 36. Wiechmann I, Harbeck M, Grupe G. 2010. Yersinia pestis DNA sequences in late medieval skeletal finds, Bavaria. Emerg. Infect. Dis. 16:1806–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barnes AM, Quan TJ. 1992. Plague, p 1285–1291 In Gorbach SL, Bartlett JG, Blacklow NR, Infectious diseases. W. B. Saunders Co., Philadelphia, PA. [Google Scholar]
- 38. Duplaix N. 1988. Fleas–the lethal leapers. Natl. Geogr. 173:672–694 [Google Scholar]
- 39. Link VB. 1955. A history of plague in United States of America. Public Health Monogr. 26:1–120 [PubMed] [Google Scholar]
- 40. Poland JD, Quan TJ, Barnes AM. 1994. Plague, p 93–112 In Beran GW, Handbook of zoonoses. Section A. Bacterial, rickettsial, chlamydial, and mycotic, 2nd ed. CRC Press, Inc., Ann Arbor, MI. [Google Scholar]
- 41. Gage KL, Lance SE, Dennis DT, Montenieri JA. 1992. Human plague in the United States: a review of cases from 1988–1992 with comments on the likelihood of increased plague activity. Border Epidemiol. Bull. 19:1–10 [Google Scholar]
- 42. Girard JM, Wagner DM, Vogler AJ, Keys C, Allender CJ, Drickamer LC, Keim P. 2004. Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc. Natl. Acad. Sci. U. S. A. 101:8408–8413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keim PS, Wagner DM. 2009. Humans and evolutionary and ecological forces shaped the phylogeography of recently emerged diseases. Nat. Rev. Microbiol. 7:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Biggins DE, Kosoy MY. 2001. Influences of introduced plague on North American mammals: implications from ecology of plague in Asia. J. Mammal. 82:906–916 [Google Scholar]
- 45. Cully JF, Jr, Williams ES. 2001. Interspecific comparisons of sylvatic plague in prairie dogs. J. Mammal. 82:894–905 [Google Scholar]
- 46. Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA, Homer N, Huentelman MJ. 2008. Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 5:887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Homer N, Merriman B, Nelson SF. 2009. BFAST: an alignment tool for large scale genome resequencing. PLoS One 4:e7767 http://dx.doi.org/10.1371/journal.pone.0007767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12 http://dx.doi.org/10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vogler AJ, Birdsell D, Price LB, Bowers JR, Beckstrom-Sternberg SM, Auerbach RK, Beckstrom-Sternberg JS, Johansson A, Clare A, Buchhagen JL, Petersen JM, Pearson T, Vaissaire J, Dempsey MP, Foxall P, Engelthaler DM, Wagner DM, Keim P. 2009. Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J. Bacteriol. 191:2474–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lowell JL, Wagner DM, Atshabar B, Antolin MF, Vogler AJ, Keim P, Chu MC, Gage KL. 2005. Identifying sources of human exposure to plague. J. Clin. Microbiol. 43:650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SNP assay primers and probes used in this study.
Isolates used in this study.