

Abstract
Six novel ruthenium(II)– and osmium(II)–arene complexes with three modified indolo[3,2-c]quinolines have been synthesized in situ starting from 2-aminoindoloquinolines and 2-pyridinecarboxaldehyde in the presence of [M(p-cymene)Cl2]2 (M = Ru, Os) in ethanol. All complexes have been characterized by elemental analysis, spectroscopic techniques (1H, 13C NMR, IR, UV–vis), and ESI mass spectrometry, while four complexes were investigated by X-ray diffraction. The complexes have been tested for antiproliferative activity in vitro in A549 (non-small cell lung), SW480 (colon), and CH1 (ovarian) human cancer cell lines and showed IC50 values between 1.3 and >80 μM. The effects of Ru vs Os and modifications of the lactam unit on intermolecular interactions, antiproliferative activity, and cell cycle are reported. One ruthenium complex and its osmium analogue have been studied for anticancer activity in vivo applied both intraperitoneally and orally against the murine colon carcinoma model CT-26. Interestingly, the osmium(II) complex displayed significant growth-inhibitory activity in contrast to its ruthenium counterpart, providing stimuli for further investigation of this class of compounds as potential antitumor drugs.
Introduction
Metal complexes with biologically active ligands have attracted some interest from researchers over the last few years. By complexation of these ligands to metal ions, physical and biological properties such as solubility, bioavailability, modes of action, and biological activity in vitro could be altered significantly. Biologically active complexes with at least one metal–carbon bond are the main subject of bioorganometallic chemistry and exhibit a variety of biological functions and effects, making them suitable for application as antimicrobial agents and anticancer drugs.1−4 Well-known examples of such anticancer drug candidates are ferrocene-based organometallics,5 i.e. the ferrocifens,6,7 which were inspired by the clinically used drug tamoxifen and its active metabolite hydroxytamoxifen.8,9 In the field of antimicrobial agents, metallorganic complexes of chloroquine and related complexes have been reported,10−12 with the ferrocene-based antimalarial drug candidate ferroquine reaching the clinical phase of development.13,14 Other anticancer drug candidates are ruthenium complexes with the naturally occurring kinase inhibitor staurosporine.15,16 Paullones, systematically named indolo[3,2-d]benzazepines (Chart 1), also are biologically active compounds able to coordinate to metal ions.
Chart 1. Paullone (Left) and the Indoloquinoline Backbone (Right) with Atom-Numbering Schemes.

The paullone backbone was initially identified as a putative cdk inhibitor in a COMPARE search (on the basis of similarities in cytotoxic profiles in 60 cancer cell lines at the U.S. National Cancer Institute),17,18 and a broad range of paullones were tested for their biological and cdk inhibitory activity. Experimental evidence indicates that these compounds are indeed moderate cdk inhibitors, although other intracellular targets, e.g. the mitochondrial malate dehydrogenase (mMDH) and glycogen synthase kinase 3β (GSK3β), have been identified as well.19−25 Alsterpaullone, the 9-nitro derivative, was found to be the most potent cdk inhibitor.26 One of the limitations encountered in the development of this class of compounds as antitumor drugs is their low aqueous solubility and bioavailability. One way to overcome these restrictions is complexation to metal ions. As the paullone backbone itself does not offer a binding site able to accommodate a metal ion, chelating moieties had to be attached at the ligand backbone. A small library of paullones able to coordinate particular metal ions has been designed and synthesized, and complexes with Ga(III),27,28 Cu(II), Ru(II), and Os(II) were reported previously.29−32 It turned out that the biological activity does not necessarily parallel the cdk inhibition profile, suggesting that other intracellular targets might be involved.33 Whereas the lactam moiety is a prerequisite for cdk inhibition of the metal-free paullones,19 antiproliferative activity of the complexes with paullones modified at the lactam moiety was found to be higher than that of the metal-based paullones with an intact lactam moiety.30 Recently, also paullones with a TEMPO free-radical unit were coordinated to ruthenium– and osmium–arene scaffolds.34
The search for novel structure–activity relationships and attempts to replace the seven-membered azepine ring of the paullones by a six-membered ring resulted in a new class of biologically active ligands, namely indolo[3,2-c]quinolines (Chart 1). The use of indoloquinoline-containing extracts has a long tradition in the cure of various diseases in Africa. Analyses of extracts of the West African climbing shrub Cryptolepsis sanguinolenta revealed that it contains indolo[3,2-b]quinoline-based alkaloids and, to a lesser extent, also indolo[3,2-c]quinolines.35 The indoloquinoline backbone is a conjugated heteroaromatic system, whereas the azepine ring of the paullones is folded. The planarity of the ligands has a strong influence on the properties of the compounds. Indoloquinolines are, for example, better DNA intercalators and show a different activity profile toward biological targets. Metal-free indoloquinolines were shown to inhibit the cell cycle in vitro either in the S phase or in the G2/M phases, depending on the cell type.36−39 Moreover, ruthenium–arene- and osmium–arene-based indoloquinolines with a chelating ethylenediamine moiety are about one order of magnitude more cytotoxic in vitro than their paullone counterparts and cause severe concentration-dependent cell cycle perturbations.40 Some complexes are prone to dissociation with liberation of the indoloquinoline ligand. This is in sharp contrast to the corresponding metal-based paullones, which are resistant to hydrolysis.40 Surprisingly, substitution of ruthenium by osmium did not lead to significantly higher stability as often reported in the literature.41,42 By introducing sp2-hybridized nitrogen donor atoms of an iminopyridine instead of the sp3-hybridized diamines, the stability of the complexes has been improved markedly, with no signs of release of the indoloquinoline ligand. In vitro, cytotoxicity was in some cases higher than that of ethane-1,2-diamine-based complexes.43,44
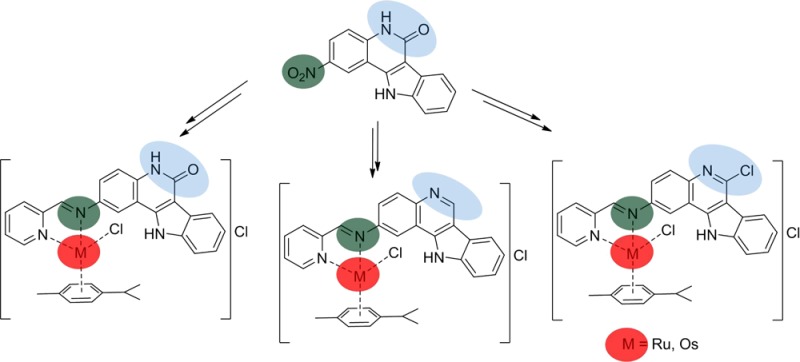
Herein, we report on the synthesis of six new complexes 1a,b–3a,b with indoloquinoline-based ligands (Scheme 1, Chart 2).
Scheme 1. Synthesis of the Indoloquinoline Backbone.

Reagents and conditions: (i) HOAc glacial, Ar, 140 °C, 4 h; (ii) N2H4·H2O, 118 °C, 135 h; (iii) POCl3, Ar, 140 °C, 26 h; (iv) N2H4·H2O, 115 °C, 27 h; (v) Fe(s), HOAc, EtOH, H2O, 35 °C, 3 h, ultrasound bath.
Chart 2. Complexes with Indoloquinoline-Based Ligandsa.

a Underlined complexes were characterized by single-crystal X-ray diffraction.
The complexes have a coordination environment similar to that of other complexes reported recently,43,44 but the binding moiety was attached at position 2 of the indoloquinoline backbone. This modification enabled for the first time an assay of antiproliferative activity of metal-based indoloquinolines with a free lactam moiety (1a,b). All complexes were characterized by 1D and 2D NMR spectroscopy, ESI mass spectrometry, and IR and UV–vis spectroscopy, while complexes 1a,b, 2a, and 3a were also studied by single-crystal X-ray diffraction. The in vitro cytotoxic activity in three human cancer cell lines, namely CH1 (ovarian carcinoma), SW480 (colon adenocarcinoma), and A549 (non-small cell lung carcinoma), is also reported. Further biological evaluation included cell cycle analyses as well as determination of cellular accumulation of the ruthenium congeners. Complexes 2a,b were also evaluated in an in vivo CT-26 murine colon carcinoma model.
Experimental Section
Characterization of the Compounds
One-dimensional 1H and 13C NMR and two-dimensional 1H–1H COSY, 1H–1H TOCSY, 1H–1H ROESY or 1H–1H NOESY, 1H–13C HSQC, and 1H–13C HMBC NMR spectra were recorded on two Bruker Avance III spectrometers at 500.32 or 500.10 MHz (1H) and 125.82 or 125.76 (13C) MHz, respectively, by using as a solvent DMSO-d6 at room temperature and standard pulse programs. 1H and 13C shifts are quoted relative to the solvent residual signals. The atom-numbering scheme used for NMR assignments is depicted in Chart S1 (Supporting Information). IR spectra were measured on a Bruker Vertex 70 FT-IR spectrometer by means of the attenuated total reflection (ATR) technique, and UV–vis spectra were recorded with a Perkin-Elmer Lambda 650 spectrophotometer equipped with a six-cell changer and a Peltier element for temperature control or with an Agilent 8453 spectrophotometer. All UV–vis experiments were performed at 25 °C. Electrospray ionization mass spectrometry (ESI-MS) was carried out with a Bruker Esquire 3000 instrument; the samples were dissolved in methanol. Elemental analyses were performed at the Microanalytical Laboratory of the University of Vienna with a Perkin-Elmer 2400 CHN elemental analyzer.
Synthesis of the Organic Compounds
Ethanol was dried using standard procedures. 2-Aminobenzylamine, phosphorus oxychloride, hydrazine hydrate, and iron powder were purchased from Sigma-Aldrich, while 5-nitroisatin and 2-pyridinecarboxaldehyde were obtained from Acros. All these chemicals were used without further purification. 2-Nitro-5,11-dihydroindolo[3,2-c]quinolin-6-one and 6-chloro-2-nitro-11H-indolo[3,2-c]quinoline were synthesized as published elsewhere.43 2-Amino-6-chloro-11H-indolo[3,2-c]quinoline was synthesized by following the procedure reported by Gamble et al.45 An ELMA Transsonic T 460/H ultrasound bath was used for acceleration of the reaction.
2-Amino-6-chloro-11H-indolo[3,2-c]quinoline
6-Chloro-2-nitro-11H-indolo[3,2-c]quinoline (1.02 g, 3.43 mmol) and iron powder (0.95 mg, 16.93 mmol) in a mixture of acetic acid (13 mL), ethanol (13 mL), and water (6.5 mL) were sonicated at 35 °C for 3 h and then filtered to separate the unreacted iron, and the solvent was removed under reduced pressure. The solid was extracted with ethyl acetate (600 mL) and was washed with a 2 M solution of NaOH. The combined organic phases were washed with brine, dried over Na2SO4, and filtered, and the solvent was removed under reduced pressure. The brown product was dried in vacuo. Yield: 0.85 g, 95%. 1H NMR (500 MHz, DMSO-d6): δ 12.75 (s, 1H, H11), 8.37 (d, 1H, 3J = 8.1 Hz, H7), 7.74 (d, 1H, 3J = 8.8 Hz, H4), 7.70 (d, 1H, 3J = 8.1 Hz, H10), 7.53–7.48 (m, 1H, H9), 7.38–7.33 (m, 2H, H1 + H8), 7.15 (dd, 1H, 3J = 8.8 Hz, 4J = 2.4 Hz, H3), 5.74 (s, 2H, H2a) ppm. 13C NMR (125 MHz, DMSO-d6): δ 147.8 (Cq, C2), 141.2 (Cq, C11a), 139.6 (Cq, C6), 139.2 (Cq, C10a), 138.0 (Cq, C4a), 129.7 (CH, C4), 126.0 (CH, C9), 121.6 (CH, C7), 121.5 (Cq, C6b), 121.2 (CH, C8), 120.5 (CH, C3), 118.6 (Cq, C11b), 112.5 (CH, C10), 111.5 (Cq, C6a), 101.0 (Cq, C1) ppm.
2-Amino-11H-indolo[3,2-c]quinoline
6-Chloro-2-nitro-11H-indolo[3,2-c]quinoline (1.00 g, 3.43 mmol) was suspended in hydrazine hydrate (10 mL) and stirred under an argon atmosphere at 115 °C for 27 h. After the mixture was cooled, the light brown precipitate was collected under suction, washed with water (2 × 10 mL), and dried in vacuo at 50 °C overnight. Yield: 0.32 g, 40%. 1H NMR (500 MHz, DMSO-d6): δ 12.32 (s, 1H, H11), 9.20 (s, 1H, H6), 8.21 (d, 1H, 3J = 7.9 Hz, H7), 7.82 (d, 1H, 3J = 8.9 Hz, H4), 7.64 (d, 1H, 3J = 8.1 Hz, H10), 7.45–7.40 (m, 1H, H9), 7.34 (d, 1H, 4J = 2.3 Hz, H1), 7.29–7.25 (m, 1H, H8), 7.11 (dd, 1H, 3J = 8.9 Hz, 4J = 2.3 Hz, H3), 5.60 (s, 2H, H2a) ppm. 13C NMR (125 MHz, DMSO-d6): δ 147.1 (Cq, C2), 140.1 (CH, C6), 139.5 (Cq, C4a), 139.2 (Cq, C10a), 139.0 (Cq, C11a), 130.8 (CH, C4), 125.4 (CH, C9), 122.6 (Cq, C6b), 120.5 (CH, C8), 120.3 (CH, C7), 119.7 (CH, C3), 119.1 (Cq, C11b), 114.5 (Cq, C6a), 112.2 (CH, C10), 101.0 (Cq, C1) ppm.
2-Amino-5,11-dihydroindolo[3,2-c]quinolin-6-one
A suspension of 2-nitro-5,11-dihydroindolo[3,2-c]quinolin-6-one (1.00 g, 7.16 mmol) in hydrazine hydrate (12 mL) was stirred at 118 °C under argon for 135 h. After the solution was cooled, it was allowed to stand at −20 °C overnight. The solid that formed was collected, washed with water (3 × 10 mL), and dried in vacuo. Yield: 0.45 g, 50%. 1H NMR (500 MHz, DMSO-d6): δ 12.35 (s, 1H, H11), 11.03 (s, 1H, H5), 8.18 (d, 1H, 3J = 7.8 Hz, H7), 7.57 (d, 1H, 3J = 8.1 Hz, H10), 7.34–7.30 (m, 1H, H9), 7.25 (d, 1H, 4J = 2.4 Hz, H1), 7.24–7.18 (m, 1H, H4 + H8), 6.86 (dd, 1H, 3J = 8.7 Hz, 4J = 2.4 Hz, H3), 5.08 (s, 2H, H2a) ppm. 13C NMR (125 MHz, DMSO-d6): δ 159.7 (Cq, C6), 143.8 (Cq, C2), 141.1 (Cq, C11a), 138.2 (Cq, C10a), 130.2 (Cq, C4a), 125.1 (Cq, C6b), 124.1 (CH, C9), 121.2 (2CH, C7 + C8), 118.6 (CH, C3), 117.3 (CH, C4), 113.1 (Cq, C11b), 112.0 (CH, C10), 107.0 (Cq, C6a), 104.7 (Cq, C1) ppm.
Synthesis of the Organometallic Compounds
General Procedure
A mixture of the corresponding 2-amino-11H-indolo[3,2-c]quinoline (1 equiv), 2-pyridinecarboxaldehyde (1.1 equiv), and the metal–arene dimer [M(p-cymene)Cl2]2, where M = RuII, OsII (0.5 equiv), in dry EtOH was stirred at room temperature under an argon atmosphere for 24 h. The precipitate that formed was filtered off, washed with ethanol and diethyl ether, and dried in vacuo at 45–50 °C.
(η6-p-Cymene)[(5,11-dihydroindolo[3,2-c]quinolin-6-on-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoruthenium(II) Chloride (1a)
2-Amino-5,11-dihydroindolo[3,2-c]quinolin-6-one (0.15 g, 0.60 mmol), 2-pyridinecarboxaldehyde (63 μL, 0.66 mmol), [Ru(p-cymene)Cl2]2 (0.18 g, 0.30 mmol), and EtOH (5 mL). Yield: 0.26 g, 67% (orange powder). Anal. Calcd for C31H28N4Cl2ORu·1.7H2O (Mr 675.18): C, 55.15; H, 4.69; N, 8.30. Found: C, 55.16; H, 4.82; N, 8.22. ESI-MS (methanol), positive: m/z 609 [M – Cl]+. FT-IR (ATR, selected bands, λmax): 3375, 3100, 3039, 2973, 2878, 1643, 1588, 1477, 1454, 1392, 1337, 787, 749, 660, 606 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.14 (s, 1H, H11), 11.85 (s, 1H, H5), 9.65 (d, 1H, 3J = 5.4 Hz, H15), 9.15 (s, 1H, H13), 8.70 (br s, 1H, H1), 8.39–8.32 (m, 2H, H17 + H18), 8.24 (d, 1H, 3J = 7.9 Hz, H7), 7.97 (d, 1H, 3J = 8.7 Hz, H3), 7.94–7.89 (m, 1H, H16), 7.72–7.67 (m, 2H, H4 + H10), 7.44–7.40 (m, 1H, H9), 7.33–7.29 (m, 1H, H8), 6.20 (d, 1H, 3J = 6.1 Hz, Hcy2), 5.91 (d, 1H, 3J = 6.1 Hz, Hcy1), 5.78–5.72 (m, 2H, Hcy1′ + Hcy2′), 2.55–2.47 (m, 1H, Hcy3), 2.24 (s, 3H, Hcy5), 1.00–0.95 (m, 6H, Hcy4 + Hcy4′) ppm. 13C NMR (125 MHz, DMSO-d6): δ 167.3 (CH, C13), 160.3 (Cq, C6), 156.7 (CH, C15), 155.0 (Cq, C13a), 146.1 (Cq, C2), 140.6 (CH, C17), 140.5 (Cq, C11a), 139.1 (Cq, C4a), 138.4 (Cq, C10a), 130.3 (CH, C18), 129.4 (CH, C16), 125.1 (CH, C3), 125.0 (CH, C9), 124.6 (Cq, C6b), 121.8 (CH, C8), 121.4 (CH, C7), 117.5 (CH, C4), 116.0 (CH, C1), 112.6 (Cq, C11b), 112.4 (CH, C10), 107.6 (Cq, C6a), 105.4 (Cq, Ccy2a), 104.9 (Cq, Ccy1a), 87.6 (CH, Ccy2′), 87.2 (CH, Ccy2), 85.4 (CH, Ccy1), 85.0 (CH, Ccy1′), 31.0 (CH, Ccy3), 22.3 (CH3, Ccy4 or Ccy4′), 22.1 (CH3, Ccy4 or Ccy4′), 19.0 (CH3, Ccy5) ppm.
(η6-p-Cymene)[(5,11-dihydroindolo[3,2-c]quinolin-6-on-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoosmium(II) Chloride (1b)
2-Amino-5,11-dihydroindolo[3,2-c]quinolin-6-one (0.12 g, 0.48 mmol), 2-pyridinecarboxaldehyde (51 μL, 0.53 mmol), [Os(p-cymene)Cl2]2 (0.19 g, 0.24 mmol), and EtOH (5 mL). Yield: 0.24 g, 68% (orange-red powder). Anal. Calcd for C31H28N4Cl2OOs·1.5H2O (Mr 760.74): C, 48.94; H, 4.11; N, 7.36. Found: C, 48.87; H, 4.01; N, 7.40. ESI-MS (methanol), positive: m/z 699 [M – Cl]+, 663 [M – Cl – HCl]+. FT-IR (ATR, selected bands, λmax): 3369, 3099, 3065, 3040, 2879, 1643, 1619, 1589, 1475, 1454, 1434, 1392, 1336, 785, 750, 660, 611 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.10 (s, 1H, H11), 11.85 (s, 1H, H5), 9.59 (d, 1H, 3J = 5.4 Hz, H15), 9.56 (s, 1H, H13), 8.63 (br s, 1H, H1), 8.49 (d, 1H, 3J = 7.7 Hz, H18), 8.34–8.29 (m, 1H, H17), 8.24 (d, 1H, 3J = 7.7 Hz, H7), 7.90–7.85 (m, 2H, H3 + H16), 7.70 (d, 1H, 3J = 8.2 Hz, H10), 7.67 (d, 1H, 3J = 8.8 Hz, H4), 7.45–7.40 (m, 1H, H9), 7.33–7.29 (m, 1H, H8), 6.48 (d, 1H, 3J = 5.5 Hz, Hcy2), 6.12 (d, 1H, 3J = 5.5 Hz, Hcy1), 5.97 (d, 1H, 3J = 5.5 Hz, Hcy2′), 5.88 (d, 1H, 3J = 5.5 Hz, Hcy1′), 2.43–2.36 (m, 1H, Hcy3), 2.32 (s, 3H, Hcy5), 0.92 (d, 3H, 3J = 6.9 Hz, Hcy4′), 0.90 (d, 3H, 3J = 6.8 Hz, Hcy4) ppm. 13C NMR (125 MHz, DMSO-d6): δ 168.1 (CH, C13), 160.3 (Cq, C6), 156.5 (Cq, C13a), 156.4 (CH, C15), 146.2 (Cq, C2), 140.7 (CH, C17), 140.5 (Cq, C11a), 139.3 (Cq, C4a), 138.4 (Cq, C10a), 130.3 (CH, C16), 130.2 (CH, C18), 125.4 (CH, C3), 125.0 (CH, C9), 124.6 (Cq, C6b), 121.8 (CH, C8), 121.4 (CH, C7), 117.5 (CH, C4), 116.5 (CH, C1), 112.6 (Cq, C11b), 112.4 (CH, C10), 107.6 (Cq, C6a), 99.0 (Cq, Ccy1a), 97.2 (Cq, Ccy2a), 79.8 (CH, Ccy2′), 79.3 (CH, Ccy2), 76.1 (CH, Ccy1), 75.4 (CH, Ccy1′), 31.3 (CH, Ccy3), 22.7 (CH3, Ccy4′), 22.3 (CH3, Ccy4), 18.9 (CH3, Ccy5) ppm.
(η6-p-Cymene)[(11H-indolo[3,2-c]quinolin-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoruthenium(II) Chloride (2a)
2-Amino-11H-indolo[3,2-c]quinoline (0.15 g, 0.64 mmol), 2-pyridinecarboxaldehyde (67 μL, 0.71 mmol), [Ru(p-cymene)Cl2]2 (0.20 g, 0.32 mmol), and EtOH (5 mL). Yield: 0.27 g, 66% (ochre powder). Anal. Calcd for C31H28N4Cl2Ru·2H2O (Mr 664.59): C, 56.02; H, 4.85; N, 8.43. Found: C, 55.76; H, 4.93; N, 8.48. ESI-MS (methanol), positive: m/z 593 [M – Cl]+. FT-IR (ATR, selected bands, λmax): 3418, 3096, 3068, 3009, 2970, 1597, 1511, 1464, 1366, 1346, 1240, 836, 788, 752 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.54 (s, 1H, H11), 9.79 (s, 1H, H6), 9.70 (d, 1H, 3J = 5.6 Hz, H15), 9.27 (s, 1H, H13), 9.02 (br s, 1H, H1), 8.45–8.36 (m, 4H, H4 + H7 + H17 + H18), 8.19 (dd, 1H, 3J = 9.0 Hz, 4J = 2.3 Hz, H3), 7.98–7.94 (m, 1H, H16), 7.82 (d, 1H, 3J = 8.1 Hz, H10), 7.60–7.56 (m, 1H, H9), 7.44–7.40 (m, 1H, H8), 6.21 (d, 1H, 3J = 6.1 Hz, Hcy2), 5.94 (d, 1H, 3J = 6.1 Hz, Hcy1), 5.88 (d, 1H, 3J = 6.1 Hz, Hcy2′), 5.71 (d, 1H, 3J = 6.1 Hz, Hcy1′), 2.59–2.49 (m, 1H, Hcy3), 2.24 (s, 3H, Hcy5), 1.00–0.97 (m, 6H, Hcy4 + Hcy4′) ppm. 13C NMR (125 MHz, DMSO-d6): δ 168.8 (CH, C13), 156.8 (CH, C15), 154.9 (Cq, C13a), 149.5 (Cq, C2), 145.6 (CH, C6), 144.8 (Cq, C4a), 140.9 (Cq, C11a), 140.7 (CH, C17), 139.6 (Cq, C10a), 130.8 (2CH, C4 + C18), 129.7 (CH, C16), 126.8 (CH, C9), 124.2 (CH, C3), 122.2 (Cq, C6b), 121.7 (CH, C8), 121.0 (CH, C7), 117.4 (Cq, C11b), 115.8 (CH, C1), 115.6 (Cq, C6a), 112.7 (CH, C10), 105.6 (Cq, Ccy2a), 104.6 (Cq, Ccy1a), 87.3 (2CH, Ccy2 + Ccy2′), 85.6 (CH, Ccy1), 85.3 (CH, Ccy1′), 31.0 (CH, Ccy3), 22.3 (CH3, Ccy4 or Ccy4′), 22.2 (CH3, Ccy4 or Ccy4′), 18.9 (CH3, Ccy5) ppm.
(η6-p-Cymene)[(11H-indolo[3,2-c]quinolin-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoosmium(II) Chloride (2b)
2-Amino-11H-indolo[3,2-c]quinoline (0.12 g, 0.51 mmol), 2-pyridinecarboxaldehyde (54 μL, 0.57 mmol), [Os(p-cymene)Cl2]2 (0.20 g, 0.26 mmol), and EtOH (5 mL). Yield: 0.24 g, 65% (red-brown powder). Anal. Calcd for C31H28N4Cl2Os·3H2O (Mr 771.76): C, 48.24; H, 4.44; N, 7.26. Found: C, 48.30; H, 4.20; N, 7.18. ESI-MS (methanol), positive: m/z 683 [M – Cl]+. FT-IR (ATR, selected bands, λmax): 3367, 3063, 3002, 2973, 1598, 1512, 1464, 1367, 1345, 1240, 836, 787, 751 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.53 (s, 1H, H11), 9.80 (s, 1H, H6), 9.68 (s, 1H, H13), 9.64 (d, 1H, 3J = 5.6 Hz, H15), 8.95 (br s, 1H, H1), 8.55 (d, 1H, 3J = 7.6 Hz, H18), 8.42 (d, 1H, 3J = 7.8 Hz, H7), 8.39 (d, 1H, 3J = 8.9 Hz, H4), 8.37–8.33 (m, 1H, H17), 8.10 (dd, 1H, 3J = 8.9 Hz, 4J = 2.0 Hz, H3), 7.93–7.89 (m, 1H, H16), 7.82 (d, 1H, 3J = 8.1 Hz, H10), 7.61–7.56 (m, 1H, H9), 7.45–7.41 (m, 1H, H8), 6.48 (d, 1H, 3J = 5.8 Hz, Hcy2), 6.14 (d, 1H, 3J = 5.8 Hz, Hcy1), 5.98 (d, 1H, 3J = 5.8 Hz, Hcy2′), 5.86 (d, 1H, 3J = 5.8 Hz, Hcy1′), 2.47–2.38 (m, 1H, Hcy3), 2.32 (s, 3H, Hcy5), 0.94–0.90 (m, 6H, Hcy4 + Hcy4′) ppm. 13C NMR (125 MHz, DMSO-d6): δ 169.7 (CH, C13), 156.5 (CH, C15), 156.3 (Cq, C13a), 149.5 (Cq, C2), 145.6 (CH, C6), 144.7 (Cq, C4a), 141.0 (Cq, C11a), 140.8 (CH, C17), 139.6 (Cq, C10a), 130.6 (3CH, C4 + C16 + C18), 126.9 (CH, C9), 124.6 (CH, C3), 122.2 (Cq, C6b), 121.8 (CH, C8), 121.1 (CH, C7), 117.3 (Cq, C11b), 116.4 (CH, C1), 115.5 (Cq, C6a), 112.8 (CH, C10), 98.6 (Cq, Ccy1a), 97.4 (Cq, Ccy2a), 79.5 (2CH, Ccy2 + Ccy2′), 76.3 (CH, Ccy1), 75.9 (CH, Ccy1′), 31.2 (CH, Ccy3), 22.6 (CH3, Ccy4 or Ccy4′), 22.3 (CH3, Ccy4 or Ccy4′), 18.9 (CH3, Ccy5) ppm.
(η6-p-Cymene)[(6-chloro-11H-indolo[3,2-c]quinolin-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoruthenium(II) Chloride (3a)
2-Amino-6-chloro-11H-indolo[3,2-c]quinoline (0.15 g, 0.56 mmol), 2-pyridinecarboxaldehyde (60 μL, 0.62 mmol), [Ru(p-cymene)Cl2]2 (0.17 g, 0.27 mmol), and EtOH (3 mL). Yield: 0.20 g, 55% (yellow powder). Anal. Calcd for C31H27N4Cl3Ru·2.5H2O (Mr 708.04): C, 52.59; H, 4.56; N, 7.91. Found: C, 52.50; H, 4.16; N, 7.93. ESI-MS (methanol), positive: m/z 627 [M – Cl]+, 609 [M – Cl – HCl + H2O]+, 591 [M – Cl – HCl]+. FT-IR (ATR, selected bands, λmax): 3334, 3063, 2979, 1596, 1504, 1358, 1243, 1173, 962, 832, 744, 651, 614 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.72 (s, 1H, H11), 9.68 (d, 1H, 3J = 5.4 Hz, H15), 9.26 (s, 1H, H13), 8.96 (d, 1H, 4J = 2.2 Hz, H1), 8.51 (d, 1H, 3J = 8.0 Hz, H7), 8.43–8.36 (m, 2H, H17 + H18), 8.30 (d, 1H, 3J = 8.9 Hz, H4), 8.20 (dd, 1H, 3J = 8.9 Hz, 4J = 2.2 Hz, H3), 7.98–7.94 (m, 1H, H16), 7.88 (d, 1H, 3J = 8.2 Hz, H10), 7.66–7.62 (m, 1H, H9), 7.51–7.47 (m, 1H, H8), 6.20 (d, 1H, 3J = 6.2 Hz, Hcy2), 5.93 (d, 1H, 3J = 6.2 Hz, Hcy1), 5.79 (d, 1H, 3J = 6.1 Hz, Hcy2′), 5.70 (d, 1H, 3J = 6.1 Hz, Hcy1′), 2.57–2.48 (m, 1H, Hcy3), 2.24 (s, 3H, Hcy5), 1.00–0.96 (m, 6H, Hcy4 + Hcy4′) ppm. 13C NMR (125 MHz, DMSO-d6): δ 169.0 (CH, C13), 156.8 (CH, C15), 154.9 (Cq, C13a), 149.7 (Cq, C2), 146.1 (Cq, C6), 144.8 (Cq, C4a), 142.6 (Cq, C11a), 140.7 (CH, C17), 139.5 (Cq, C10a), 130.8 (CH, C18), 130.4 (CH, C4), 129.7 (CH, C16), 127.1 (CH, C9), 124.9 (CH, C3), 122.1 (CH, C8), 122.0 (CH, C7), 121.1 (Cq, C6b), 117.0 (Cq, C11b), 115.8 (CH, C1), 112.9 (CH, C10), 112.7 (Cq, C6a), 105.7 (Cq, Ccy2a), 104.6 (Cq, Ccy1a), 87.2 (2CH, Ccy2 + Ccy2′), 85.6 (CH, Ccy1), 85.3 (CH, Ccy1′), 31.0 (CH, Ccy3), 22.3 (CH3, Ccy4 or Ccy4′), 22.1 (CH3, Ccy4 or Ccy4′), 18.9 (CH3, Ccy5) ppm.
(η6-p-Cymene)[(6-chloro-11H-indolo[3,2-c]quinolin-2-yl)(1-κN-pyridin-2-ylmethylidene)-κN-amine]chloridoosmium(II) Chloride (3b)
2-Amino-6-chloro-11H-indolo[3,2-c]quinoline (0.20 g, 0.75 mmol), 2-pyridinecarboxaldehyde (79 μL, 0.82 mmol), [Os(p-cymene)Cl2]2 (0.30 g, 0.37 mmol), and EtOH (25 mL). Yield: 0.28 g, 48% (red-brown powder). Anal. Calcd for C31H27N4Cl3Os·2H2O (Mr 788.19): C, 47.37; H, 3.96; N, 7.11. Found: C, 47.03; H, 3.77; N, 6.97. ESI-MS (methanol), positive: m/z 717 [M – Cl]+, 681 [M – Cl – HCl]+. FT-IR (ATR, selected bands, λmax): 3353, 3057, 2977, 1595, 1505, 1356, 1243, 1175, 960, 836, 740, 657, 616, 590 cm–1. 1H NMR (500 MHz, DMSO-d6): δ 13.70 (s, 1H, H11), 9.67 (s, 1H, H13), 9.63 (d, 1H, 3J = 5.8 Hz, H15), 8.90 (d, 1H, 4J = 2.3 Hz, H1), 8.54 (d, 1H, 3J = 7.7 Hz, H18), 8.51 (d, 1H, 3J = 8.0 Hz, H7), 8.37–8.32 (m, 1H, H17), 8.28 (d, 1H, 3J = 8.9 Hz, H4), 8.11 (dd, 1H, 3J = 8.9 Hz, 4J = 2.3 Hz, H3), 7.94–7.89 (m, 1H, H16), 7.87 (d, 1H, 3J = 8.1 Hz, H10), 7.66–7.62 (m, 1H, H9), 7.51–7.47 (m, 1H, H8), 6.48 (d, 1H, 3J = 5.8 Hz, Hcy2), 6.13 (d, 1H, 3J = 5.8 Hz, Hcy1), 5.98 (d, 1H, 3J = 5.8 Hz, Hcy2′), 5.85 (d, 1H, 3J = 5.8 Hz, Hcy1′), 2.46–2.39 (m, 1H, Hcy3), 2.32 (s, 3H, Hcy5), 0.94–0.90 (m, 6H, Hcy4 + Hcy4′) ppm. 13C NMR (125 MHz, DMSO-d6): δ 169.8 (CH, C13), 156.5 (CH, C15), 156.3 (Cq, C13a), 149.7 (Cq, C2), 146.3 (Cq, C6), 144.9 (Cq, C4a), 142.6 (Cq, C11a), 140.8 (CH, C17), 139.5 (Cq, C10a), 130.6 (CH, C16), 130.6 (CH, C18), 130.4 (CH, C4), 127.1 (CH, C9), 125.2 (CH, C3), 122.2 (CH, C8), 122.0 (CH, C7), 121.1 (Cq, C6b), 117.0 (Cq, C11b), 116.3 (CH, C1), 112.9 (CH, C10), 112.7 (Cq, C6a), 98.5 (Cq, Ccy1a), 97.5 (Cq, Ccy2a), 79.5 (CH, Ccy2), 79.5 (CH, Ccy2′), 76.4 (CH, Ccy1), 76.0 (CH, Ccy1′), 31.2 (CH, Ccy3), 22.6 (CH3, Ccy4 or Ccy4′), 22.3 (CH3, Ccy4 or Ccy4′), 18.9 (CH3, Ccy5) ppm.
Crystallographic Structure Determination
X-ray diffraction measurements were performed on a Bruker X8 APEXII CCD diffractometer. Single crystals were positioned at 35, 35, 40, and 35 mm from the detector, and 623, 883, 606, and 1932 frames were measured, each for 30, 80, 30, and 40 s over 1° scan width for 1a,1b, 2a, and 3a, respectively. The data were processed using SAINT software.46 Crystal data, data collection parameters, and structure refinement details are given in Table S1 (Supporting Information). The structures were solved by direct methods and refined by full-matrix least-squares techniques. Non-H atoms were refined with anisotropic displacement parameters. H atoms were inserted in calculated positions and refined with a riding model. The arene ligand in 2a·C2H5OH·H2O was found to be disordered over two positions with sof 0.53:0.43. The following software programs and computer were used: structure solution and refinement, SHELXS-97 and SHELXL-97;47 molecular diagrams, ORTEP-3;48 computer, Intel CoreDuo.
Cell Lines and Cell Culture Conditions
For cytotoxicity determination, three cell lines were used: A549, a human non-small cell lung cancer cell line, and SW480, a human colon carcinoma cell line (both purchased from American Type Culture Collection), as well as CH1, a human ovarian carcinoma cell line (kindly provided by Lloyd R. Kelland, CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, U.K.). Cells were grown as adherent monolayer cultures in 75 cm2 culture flasks (Iwaki/Asahi Technoglass) in complete culture medium: i.e., Minimal Essential Medium supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 1% v/v nonessential amino acids (from 100× ready-to-use stock) and 4 mM l-glutamine but without antibiotics at 37 °C under a moist atmosphere containing 5% CO2 and 95% air. For in vivo experiments, the murine colon cancer cell model CT-26 (purchased from American Type Culture Collection, Manassas, VA) was used. Cells were grown in DMEM/F12 supplemented with 10% FCS. All cell culture media and reagents were purchased from Sigma-Aldrich Austria.
Cytotoxicity Assay
Cytotoxicity was determined by the colorimetric MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) as described previously.49 Briefly, cells were harvested by trypsinization and seeded into 96-well plates in volumes of 100 μL/well. Depending on the cell line, different cell densities were used to ensure exponential growth of the untreated controls during the experiment: 1.0 × 103 (CH1), 2.0 × 103 (SW480), 3.0 × 103 (A549). In the first 24 h, the cells were allowed to settle and resume exponential growth. Then the test compounds were dissolved in DMSO, serially diluted in complete culture medium, and added to the plates in volumes of 100 μL/well such that the DMSO content did not exceed 1% v/v. Due to the limited solubility of some compounds, the medium in the wells used for the highest concentrations was removed and 200 μL/well of the substance dilution was added. After continuous exposure for 96 h (in the incubator at 37 °C and under 5% CO2), the medium was replaced by 100 μL/well RPMI 1640 medium (supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine) and 20 μL/well MTT solution (MTT reagent in phosphate-buffered saline, 5 mg/mL), and plates were incubated for a further 4 h. Then the medium/MTT mixture was removed, and the formazan that formed was dissolved in DMSO (150 μL/well). Optical densities at 550 nm were measured (reference wavelength 690 nm) with a microplate reader (Tecan Spectra Classic). The quantity of viable cells was expressed as a percentage of untreated controls, and 50% inhibitory concentrations (IC50) were calculated from concentration-effect curves by interpolation. Every test was repeated in at least three independent experiments, each consisting of three replicates per concentration level.
Cell Cycle Studies
For this assay, CH1 cells were harvested by trypsinization and 8 × 104 cells in 1.5 mL/well were seeded into 12-well plates. In the first 24 h, the cells were allowed to settle and resume exponential growth. Thereafter, stocks of the test compounds in DMSO were diluted in complete culture medium and 1.5 mL/well was added to the plate such that the DMSO content did not exceed 0.5% v/v. For concentrations ≥50 μM, the medium was removed from the wells before the dissolved complexes were added. After continuous exposure for 24 h (in the incubator at 37 °C and under 5% CO2), the cells were washed with PBS and trypsinized. Trypsinization was stopped with MEM, the cells were centrifuged (300g, 3 min), and the supernatant was discarded. Then the cells were washed with PBS once more and resuspended in 600 μL PI/HSF buffer (0.1% v/v Triton X-100, 0.1% w/v sodium citrate, in PBS) containing 50 μg/mL propidium iodide (PI). After incubation overnight at 4 °C in the dark, 5 × 103 cells were measured by flow cytometry with a Millipore guava easyCyte 8HT instrument. Data were evaluated by FlowJo software (Tree Star) using Dean Jett Fox algorithms.
Cellular Accumulation
Studies on cellular accumulation were performed according to the method described previously.50 Briefly, SW480 cells were seeded in 6-well plates in densities of 3 × 105 cells per well in aliquots of 2.5 mL complete culture medium. Samples and corresponding adsorption/desorption controls were located on the same plate. For determination of the cell number, three wells of a separate plate were seeded in the same manner. Plates were kept at 37 °C for 24 h. After removal of the medium used for settlement of the cells, 2.5 mL of the solution containing the test compounds (from DMSO stocks diluted with complete culture medium, yielding a final DMSO concentration below 0.5% v/v) was added to the wells. During exposure (2 h at 37 °C), the cell number in three wells was determined by counting in a hemocytometer upon trypsinization. After exposure, the medium was removed, cells were washed three times with PBS and lysed with 0.5 mL sub-boiled HNO3 per well for 1 h at room temperature, and Ru was quantified by ICP-MS in aliquots of 400 μL diluted to a total volume of 8 mL and internally standardized with indium (0.5 ppb). The amount of adsorbed/desorbed ruthenium was subtracted from the corresponding uptake sample, and the resulting cell-associated amount was divided by the average cell number. Results are based on at least four independent experiments, each consisting of triplicates.
Metal concentrations were determined by an ICP-MS instrument (Agilent 7500ce, Waldbronn, Germany), equipped with a CETAC ASX-520 autosampler and a MicroMist nebulizer, at a sample uptake rate of approximately 0.25 mL/min. Indium and ruthenium standards were obtained from CPI International (Amsterdam, The Netherlands). Standards were freshly prepared for each analysis in matrices matching the sample matrix with regard to internal standard and concentration of the acid. Nitric acid (p.a.) was purchased from Fluka (Buchs, Switzerland) and further purified in a quartz sub-boiling point distillation unit. All samples and dilutions were prepared with Milli-Q water (18.2 MΩ cm). In order to monitor plasma stability and allow manual linear drift correction, a ruthenium standard (approximately 1 ppb) was measured every 12–18 samples. At the end of the sequence, the precision of the measurement was checked by rerunning approximately 5% of randomly selected samples covering the whole measurement time. The accuracy was checked by rerunning samples, whose concentrations were determined independently during a previous sequence. Data analysis is based on the isotopes 115In, 101Ru, and 102Ru.
Animals
Six- to eight-week-old female Balb/c mice were purchased from Harlan Laboratories (San Pietro al Natisone, Italy). The animals were kept in a pathogen-free environment, and every procedure was done in a laminar airflow cabinet. Experiments were carried out according to the Austrian and FELASA guidelines for animal care and protection.
In Vivo Experiments
CT-26 cells (106 cells in serum-free DMEM medium) were injected subcutaneously into the right flank. Therapy was started when tumor nodules were palpable. Animals were treated with 2a (25.9 mg/kg i.p. and 51.7 mg/kg p.o. dissolved in 10% DMSO before administration) and 2b (30 mg/kg i.p. and 60 mg/kg p.o dissolved in 10% DMSO before administration), each for 5 subsequent days. Animals were controlled for distress development every day, and tumor size was assessed regularly by caliper measurement. Tumor volume was calculated using the formula (length × (width)2)/2.
Results and Discussion
Synthesis and Characterization of the Compounds
2-Nitro-5,11-dihydroindolo[3,2-c]quinolin-6-one and 6-chloro-2-nitro-11H-indolo[3,2-c]quinoline (A and C, respectively, in Scheme 1) were prepared according to the literature protocols.51 The nitro group in C was reduced with iron powder under sonication with formation of species E, in analogy to a procedure published previously.45 Because of its scarce solubility in common solvents, A could not be reduced by using this procedure. Reduction was therefore carried out by stirring the compound in neat hydrazine hydrate at 118 °C under an argon atmosphere for 135 h with formation of B. 2-Amino-11H-indolo[3,2-c]quinoline (D) was easily obtained by stirring 6-chloro-2-nitro-11H-indolo[3,2-c]quinoline in neat hydrazine hydrate at 118 °C under Ar for 27 h. Unlike other substituents in position 2 of the indoloquinoline backbone, the amino group seems to facilitate reduction of the imidoyl chloride, yielding the quinoline with hydrogen in position 6. Substitution of the chloride with hydrazine, as reported in all other cases,43,44,49 has not been observed.
Condensation of the aromatic amines with 2-formylpyridine yielded ligands that showed poor stability in wet organic solvents, hydrolyzing back with formation of the starting materials. Therefore, the complexes were generated in situ via one-pot three-component synthesis from the corresponding 2-aminoindoloquinoline derivative, 2-formylpyridine, and the appropriate metal–arene dimer [M(p-cymene)Cl2]2, where M = Ru, Os (see the Experimental Section for details). All complexes were characterized by ESI-MS in methanol, showing peaks at m/z 609, 699, 593, 693, 627, and 717, respectively, for 1a,b–3a,b due to [M – Cl]+ ions. Additional peaks attributed to [M – Cl – HCl]+ at m/z 663, 591, and 681 were detected in the case of 1b and 3a,b. Peaks that could be assigned to free ligand or formation of [M2(p-cymene)2(μ-OMe)3]+, as commonly noted for more labile complexes, have not been detected. The stability of the complexes in 1% DMSO/water solution was further investigated by UV–vis measurements (Figure S1, Supporting Information). In the case of 3a,b, no change in the absorption spectra over 24 h was observed. The complexes remained intact in aqueous media, as was also confirmed by ESI mass spectrometry. Whereas 1b and 2b showed only minor changes over 24 h, the ruthenium complexes 1a and 2a seemed to hydrolyze slowly. An isosbestic point at about 450 nm is clearly seen. The greater inertness of osmium complexes is well documented in the literature.41,42 The effect of MEM on the complexes was also investigated to probe their resistance to chemical environment on application in vitro. MEM consists mainly of amino acids, salts (Na+, K+, Mg2+, Ca2+), vitamins, and glucose. It is used in cell culture experiments. With the exception of 3b, where precipitation led to decreasing absorbance, the spectra especially of the ruthenium compounds showed smaller changes in comparison to those obtained in 1% DMSO/water (Figure S2, Supporting Information), which may be due to the higher chloride concentration and partially suppressed hydrolysis. The stability of complexes 2a,b, which were tested in vivo, was also investigated in a solution of 1% DMSO/water at pH 3.4. The pH was adjusted by the addition of concentrated HCl to create conditions similar to those inside the stomach of mice.52 The spectra of both complexes remained unchanged over 24 h (Figure S3, Supporting Information), indicating that they were stable at such a pH and, furthermore, that the hydrolysis of 2a can be suppressed by higher chloride concentrations.
Crystal Structures
The results of the X-ray diffraction studies of [(η6-p-cymene)Ru(L1)Cl]Cl·C2H5OH·H2O (1a·C2H5OH·H2O), [(η6-p-cymene)Os(L1)Cl]Cl·C2H5OH·H2O (1b·C2H5OH·H2O), [(η6-p-cymene)Ru(L2)Cl]Cl·C2H5OH·H2O (2a·C2H5OH·H2O), [(η6-p-cymene)Ru(L3)Cl]Cl·4H2O (3a·4H2O) are shown in Figures 1–3. All complexes have a typical “three-leg piano-stool” geometry of ruthenium(II) and osmium(II) arene complexes,53−56 with an η6-π-bound p-cymene ring forming the seat and three other donor atoms (two nitrogens N12 and N15 of indolo[3,2-c]quinoline and one chlorido ligand) as the legs of the stool. Selected bond distances and angles are given in Table 1. All complexes crystallize as racemates, owing to the presence of the stereogenic metal center.
Figure 1.

ORTEP view of the cations in 1a (left) and 1b (right) with thermal ellipsoids drawn at the 50% probability level.
Figure 3.
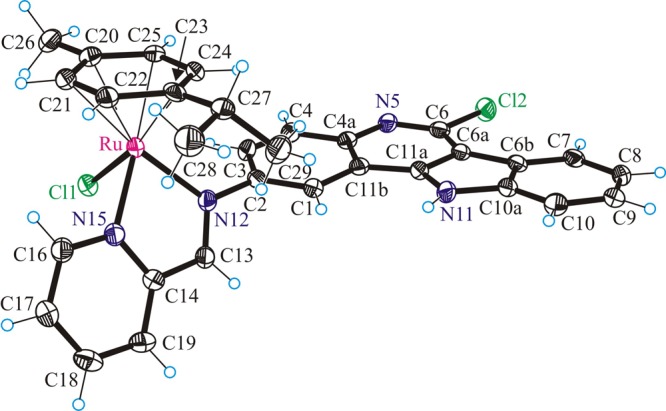
ORTEP view of the cation in 3a with thermal ellipsoids drawn at the 50% probability level.
Table 1. Selected Bond Distances (Å) and Angles (deg) for Complexes 1a·C2H5OH·H2O, 1b·C2H5OH·H2O, 2a·CH3OH·H2O,a and 3a·4H2O.
| 1a·C2H5OH·H2O | 1b·C2H5OH·H2O | 2a·CH3OH·H2O | 3a·4H2O | |
|---|---|---|---|---|
| M–Cl | 2.3819(10) | 2.3920(16) | 2.3941(15) | 2.4024(11) |
| M–N12 | 2.077(3) | 2.081(5) | 2.079(5) | 2.098(3) |
| M–N15 | 2.071(3) | 2.080(5) | 2.090(5) | 2.098(3) |
| M–Carene(av) | 2.198(2) | 2.206(3) | 2.219(3) | |
| Carene–Carene(av) | 1.410(1) | 1.417(1) | 1.425(1) | |
| N12–M–N15 | 76.30(11) | 75.83(18) | 76.0(2) | 76.96(13) |
| N12–M–Cl | 87.56(8) | 86.98(13) | 85.58(9) | 84.16(9) |
| N15–M–Cl | 84.33(9) | 83.79(14) | 85.22(9) | 84.12(9) |
M–Carene(av) and Carene–Carene(av) have not been quoted because of the disorder observed for the arene ligand in 2a.
Figure 2.
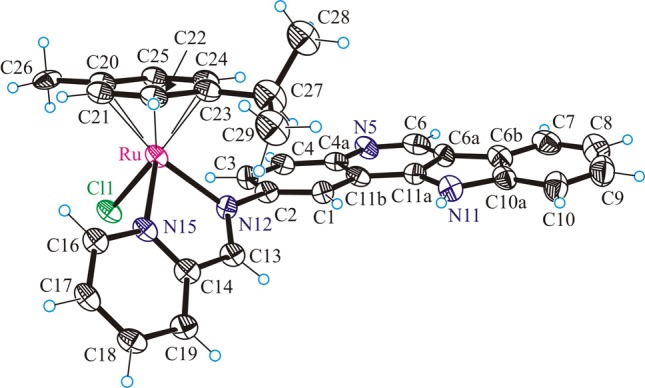
ORTEP view of the cation in 2a with thermal ellipsoids drawn at the 50% probability level. Only one position for the disordered arene ligand is shown for clarity.
Upon binding to ruthenium(II) or osmium(II), the ligands L1–L3 form the five-membered chelate ring N12C13C14N15M (M = Ru, Os). The torsion angles θN12–C13–C14–N15, which serves as measures of the distortion of the chelate ring from planarity, are −0.7(5), 0.1(8), −1.2(8), and −2.7(5)° for 1a,b, 2a, and 3a, respectively. This almost perfectly planar chelate ring forms a dihedral angle with the flat indoloquinoline backbone, which can be described by the torsion angles θC1–C2–N12–C13 of 50.8(4), 50.6(8), 52.8(8), and 57.2(5)° in 1a,b, 2a, and 3a, respectively.
The lactam unit in 1a,b is involved in complex pairing through strong intermolecular hydrogen bonding, as shown in Figure 4 for 1a. The atom N5 acts as a proton donor, while O6 acts as a proton acceptor.
Figure 4.
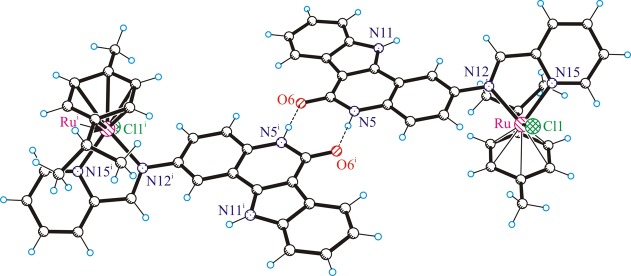
Centrosymmetric dimeric associates of the cations of 1a stabilized by hydrogen-bonding interactions involving the lactam unit: i.e., N5–H···O6i (N5–H = 0.88 Å, H···O6i = 1.914 Å, N5···O6i = 2.778 Å, N5–H···O6i = 167.3°). Atoms marked with i are generated via the symmetry transformation −x + 1, −y + 1, −z + 1.
Such complex pairing cannot occur in 2a and 3a. Instead the presence of isolated pairs of complex cations with offset parallel arrangement stabilized by π-stacking interactions has been observed, as shown in Figure 5. For 3a, the interplanar separation of the indoloquinoline backbones in the pairs is 3.485 Å.
Figure 5.
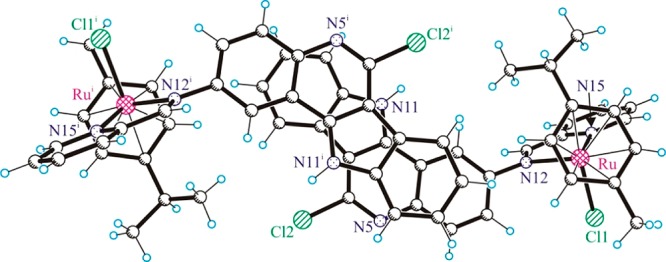
Centrosymmetric dimeric associates of the cations of 3a stabilized by π–π stacking interactions.
Cytotoxicity in Cancer Cells
The cytotoxicity of ruthenium(II) and osmium(II) complexes was determined by an MTT assay in three human cancer cell lines, namely, A549 (non-small cell lung carcinoma), CH1 (ovarian carcinoma), and SW480 (colon adenocarcinoma), mostly yielding IC50 values in the micromolar concentration range. The corresponding metal-free ligands could not be tested because of insufficient stability. A549 was the least sensitive cell line to all tested compounds, with IC50 values >80 μM in the case of compounds 1a,b and 3b (a further increase of concentrations was not possible because of the low solubility of the compounds). IC50 values in SW480 cells are up to 4 times and those in CH1 cells at least 4 up to 40 times lower than in A549 cells, as can be seen in Table 2 and Figure 6.
Table 2. Cytotoxicity of Ruthenium and Osmium Complexes with Indoloquinoline-Based Ligands in Three Human Cancer Cell Lines.
| IC50 (μM), 96 ha |
|||
|---|---|---|---|
| A549 | SW480 | CH1 | |
| 1a | >80 | >80 | 20 ± 2 |
| 1b | >80 | 48 ± 5 | 7.9 ± 0.9 |
| 2a | 27 ± 3 | 7.0 ± 1.7 | 3.2 ± 0.3 |
| 2b | 53 ± 9 | 28 ± 6 | 9.9 ± 2.1 |
| 3a | 51 ± 2 | 17 ± 3 | 1.3 ± 0.5 |
| 3b | >80 | 25 ± 2 | 3.1 ± 0.8 |
50% inhibitory concentrations (means ± standard deviations from at least three independent experiments), as obtained by the MTT assay using exposure times of 96 h.
Figure 6.

Concentration-effect curves of ruthenium– and osmium–indoloquinoline complexes 1a,b–3a,b in the human cancer cell lines A549 (A), CH1 (B), and SW480 (C), determined by the MTT assay using continuous exposure for 96 h.
Comparison of ruthenium(II) with osmium(II) complexes revealed the following relationships: ruthenium complexes (2a, 3a) are at least 1.5 and up to 4 times more potent than osmium complexes (2b, 3b) in all three cell lines, except for the pair 1a,b, of which the osmium complex is more active (Table 2, Figure 6). This deviation may be associated with the presence of a lactam unit in the indoloquinoline-based ligand. The ruthenium compound 1a is the least active of all tested compounds. No systematic structure–activity relationships could be observed for the presence vs absence of a chloro substituent at position 6 of the indoloquinoline. In contrast to previously reported ruthenium(II) and osmium(II) complexes with modified indoloquinoline ligands,40,43,44 the expected high cytotoxicities were not found. In the case of complexes with the binding moiety attached in position 6 of the indoloquinoline backbone,40,44 the osmium(II)-based complexes are more active than or equal to their ruthenium(II) analogues, whereas the compounds reported here are up to 5 times less cytotoxic in the case of ruthenium compounds and up to 100-fold less in the case of osmium complexes. One of the reasons may be the difference in the structure of side chain, but the different substituent pattern (coordination via position 2 of the indoloquinoline backbone) is more likely to account for this finding.
Effects on Cell Cycle Distribution
As reported previously, indoloquinolines affect cell cycle progression,40 and a free lactam unit is known to be favorable for cdk inhibition in the case of related metal-free indolobenzazepines.19 Due to these facts, the effects of the new ruthenium and osmium complexes on the cell cycle were studied. For this purpose, exponentially growing CH1 cells were treated for 24 h with different concentrations of the compounds, stained with propidium iodide, and the amount of DNA was analyzed by flow cytometry. As illustrated in Figure 7, the effects of osmium complexes on the cell cycle are negligible, whereas those of their ruthenium analogues are more pronounced. The strongest effects were observed in the case of 3a, where a 20% increase of the G2/M phase fraction and a ∼ 25% decrease of the G0/G1 phase fraction were found at a concentration of 10 μM. At the highest concentration, the G2/M arrest changes into an apparent S phase arrest, which might partially result from a loss of G2/M arrested cells due to cell death. 1a is less active than 3a; still, a ∼20% increase of the G2/M fraction at a concentration of 40 μM was found (Figures 7 and 8). A positive influence of the lactam unit, which might be expected on the basis of structure–activity relationships of metal-free indolobenzazepines reported by other authors,19 was not observed in the case of 1b, however. These results lead to the conclusion that the metal center plays an important role in the activity of the complexes on cell cycle distribution, whereas the effect of ligand variation is less pronounced.
Figure 7.

Concentration-dependent impact of complexes 1a,b–3a,b on the cell cycle distribution of CH1 cells after 24 h continuous exposure.
Figure 8.

Cell cycle analysis of CH1 cells after 24 h treatment. A selection of histograms shows the strong effects of 1a (bottom right) and 3a (bottom left) on cell cycle distribution. The DNA content of cells was analyzed by flow cytometry upon staining with propidium iodide and evaluation with FlowJo. Gemcitabine (top right) was used as a positive control. The black line shows the measured curve and the violet line the calculated cell cycle distribution, as obtained by the FlowJo cell cycle tool.
Cellular Accumulation
As CH1 cells are known to detach easily during the washing steps required in the cellular uptake experimental procedure, and A549 cells were the least sensitive ones, cell-associated concentrations of ruthenium were determined in SW480 cells upon exposure to 1a–3a. The concentration of the compounds during exposure was chosen to correspond roughly to an average IC50 in SW480 cells (10 μM). The results (Figure 9) show that 1a and 2a accumulate in concentrations of ∼5 fg/cell, while 3a accumulated much better (∼30 fg/cell). A comparison of these results with the cytotoxicity data shows that complex 1a is the least active (IC50 > 80 μM), in line with the lowest accumulation. On the other hand, the most potent complex (2a, IC50 = 7.0 μM) accumulates only to a slightly higher extent than 1a, whereas 3a, which accumulates to the highest degree in 2 h, is less cytotoxic than 2a after 96 h in the MTT assay (IC50 = 17 μM).
Figure 9.
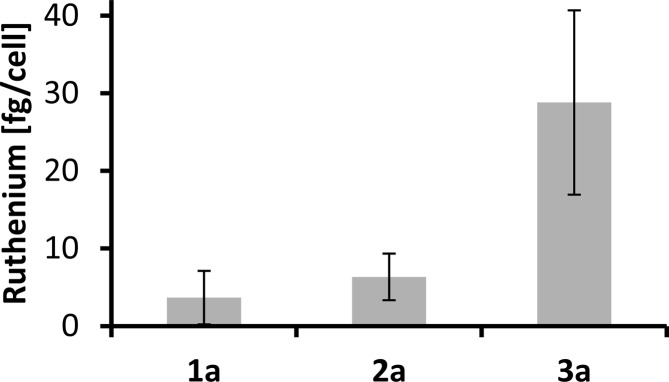
Cellular ruthenium accumulation in SW480 cells, treated for 2 h with 10 μM solutions of 1a (n = 5), 2a (n = 4), and 3a (n = 7). Values are means ± standard deviations.
In contrast to the case of the ruthenium compounds, determination of the cellular accumulation of the analogous osmium compounds led to implausible results: the measured concentration of Os obtained from cell-free adsorption blanks was higher than that from wells containing cells, resulting in apparently negative cellular accumulations. In order to rule out an unexpected impact of the polystyrene material, experiments with 1b were repeated using glass dishes. In addition, formation of volatile OsO4 in the presence of oxidizing agents (such as nitric acid during cell lysis) was taken into consideration. Cells were lysed under basic conditions by tetramethylammonium hydroxide followed by acidification with hydrochloric acid. However, strongly fluctuating, partially negative values persisted in both studies, indicating an uncontrollable process rather than a systematic error. As reported in the literature, quantification of osmium by ICP-MS is problematic and error-prone. Osmium-containing samples are routinely digested under oxidizing conditions in tightly sealed vessels, and the resulting volatile OsO4 is directly transferred to the ICP-MS. Hence, it is typically only applied for determination of isotopic ratios in geochronologic applications.57−59
Anticancer Activity in Vivo
The anticancer efficacy of compound 2b in comparison to its ruthenium analogue 2a was investigated in the murine colon cancer model CT-26. As shown in Figure 10, 2b displayed significant growth-inhibitory potential (P < 0.01 for both administration routes assessed by two-way ANOVA with Bonferroni post-test) not only when applied intraperitoneally but also when given orally. In contrast, 2a at equimolar concentrations had no anticancer activity in this mouse model (Figure S4, Supporting Information). 2a,b were both well tolerated, as can be seen from Figure S5 (Supporting Information), showing that the body weight remained almost unaffected during treatment.
Figure 10.

Anticancer activity of 2b in vivo. CT-26 cells were injected subcutaneously in the right flank of BALB/c mice. After the tumor was palpable, mice were treated for 5 days (day 5–9) with 30 mg/kg (i.p.) and 60 mg/kg (p.o.) of 2b, respectively. Tumor volumes were calculated as described in the Experimental Section. Each experimental group contained four animals. Data are means ± SD.
To the best of our knowledge, there is only one other example of an osmium–arene complex with in vivo tumor growth-inhibitory properties.60 This complex, [M(η6-p-cymene)(azpy)I]PF6, where azpy is a chelating N,N-dimethyl-4-(pyridine-2-yldiazenyl)aniline ligand, was proven to be highly active in vitro,61 and it retarded tumor growth after a single-dose 40 mg/kg subcutaneous injection in a HCT116 human colorectal cancer xenograft model. However, in vivo data of its ruthenium congener62 are not available. For the only other Ru/Os–arene couple with in vivo data available, namely [M(η6-biphenyl)(κN,κN′-ethylenediamine)Cl]+, experiments in a MCa (murine mammary carcinoma) yielded reverse results. The ruthenium complex led to reduction of primary tumor volume and decreased lung metastasis formation, whereas the osmium congener did not show any activity.63 For either of the complexes described in the literature, in vivo data for oral application are not available.
Final Remarks
Six novel ruthenium– and osmium–arene complexes with indolo[3,2-c]quinoline-based ligands have been synthesized and characterized. These complexes contain an intact lactam unit, allowing for the first time the determination of its effect on antiproliferative activity. The complexes were tested in three different human cancer cell lines (A549, SW480, and CH1) and exhibited IC50 values between 1.3 and >80 μM. Cellular accumulation studies revealed no direct correlation between cytotoxic activity and cellular accumulation in SW480 cells, as the most active compound 2a showed lower accumulation than 3a. 3a, the most active complex in CH1 cells, also showed the most pronounced effect on the cell cycle distribution in this cell line. In vivo experiments using a murine tumor model indicated a significant tumor growth inhibitory potential of the osmium compound 2b but not of its ruthenium counterpart, 2a. These findings will serve as a basis for further improvement and development of orally applicable anticancer drug candidates with indoloquinoline-based ligands, particularly as 2b showed distinct in vivo tumor growth inhibition both after intraperitoneal and oral application. Furthermore, these results clearly show the need for additional in-depth studies concerning the influence of the metal center in metallorganic drug research, as from comparison with the very limited data available,60,63 no trend can be inferred regarding the biological consequences of replacing ruthenium by osmium in an otherwise identical complex.
Acknowledgments
We thank Prof. M. Galanski for recording the 2D NMR data, A. Luganschi and A. Dobrov for measuring the ESI mass spectra, and A. Roller for the collection of the X-ray data. L.K.F. greatly acknowledges the contribution of R. Bugl to the synthesis of some compounds. The financial support of the FWF (Austrian Science Fund, Project Number P22339-N19) is kindly acknowledged.
Supporting Information Available
Figures, a table, and CIF files giving the numbering used for NMR assignment, X-ray data collection details and crystallographic data, UV–vis spectra, in vivo data for 2a, and body weight data for the in vivo experiments of 2a,b. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Hartinger C. G.; Dyson P. J. Chem. Soc. Rev. 2009, 38, 391–401. [DOI] [PubMed] [Google Scholar]
- Chavain N.; Biot C. Curr. Med. Chem. 2010, 17, 2729–2745. [DOI] [PubMed] [Google Scholar]
- Gasser G.; Metzler-Nolte N. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [DOI] [PubMed] [Google Scholar]
- Patra M.; Gasser G.; Metzler-Nolte N. Dalton Trans. 2012, 41, 6350–6358. [DOI] [PubMed] [Google Scholar]
- Ornelas C. New J. Chem. 2011, 35, 1973–1985. [Google Scholar]
- Nguyen A.; Vessieres A.; Hillard E. A.; Top S.; Pigeon P.; Jaouen G. Chimia 2007, 61, 716–724. [Google Scholar]
- Pigeon P.; Top S.; Vessieres A.; Huche M.; Goermen M.; El Arbi M.; Plamont M.-A.; McGlinchey M. J.; Jaouen G. New J. Chem. 2011, 35, 2212–2218. [Google Scholar]
- Kasiotis K. M.; Haroutounian S. A. Curr. Org. Chem. 2012, 16, 335–352. [Google Scholar]
- Polyak K.; Vogt P. K. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2715–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dive D.; Biot C. ChemMedChem 2008, 3, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A.; Rajapakse C. S. K.; Sanchez-Delgado R. A.; Varela-Ramirez A.; Lema C.; Aguilera R. J. J. Inorg. Biochem. 2010, 104, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C.; Salas P. F.; Patrick B. O.; Kock C. de; Smith P. J.; Adam M. J.; Orvig C. Organometallics 2012, 31, 5748–5759. [Google Scholar]
- Mombo-Ngoma G.; Supan C.; Dal-Bianco M. P.; Missinou M. A.; Matsiegui P.-B.; Salazar C. L. O.; Issifou S.; Ter-Minassian D.; Ramharter M.; Kombila M.; Kremsner P. G.; Lell B. Malar. J. 2011, 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supan C.; Mombo-Ngoma G.; Dal-Bianco M. P.; Salazar C. L. O.; Issifou S.; Mazuir F.; Filali-Ansary A.; Biot C.; Ter-Minassian D.; Ramharter M.; Kremsner P. G.; Lell B. Antimicrob. Agents Chemother. 2012, 56, 3165–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gani O. A. B. S. M.; Engh R. A. Nat. Prod. Rep. 2010, 27, 489–498. [DOI] [PubMed] [Google Scholar]
- Meggers E.; Atilla-Gokcumen G.; Bregman H.; Maksimoska J.; Mulcahy S.; Pagano N.; Williams D. Synlett 2007, 1177–1189. [Google Scholar]
- Paull K. D.; Hamel E.; Malspeis L. In Cancer Chemotherapeutic Agents; Foye W. O., Ed.; American Chemical Society: Washington DC, 1995; pp 9–45. [Google Scholar]
- Zaharevitz D. W.; Gussio R.; Leost M.; Senderowicz A. M.; Lahusen T.; Kunick C.; Meijer L.; Sausville E. A. Cancer Res. 1999, 59, 2566–2569. [PubMed] [Google Scholar]
- Schultz C.; Link A.; Leost M.; Zaharevitz D. W.; Gussio R.; Sausville E. A.; Meijer L.; Kunick C. J. Med. Chem. 1999, 42, 2909–2919. [DOI] [PubMed] [Google Scholar]
- Stukenbrock H.; Mussmann R.; Geese M.; Ferandin Y.; Lozach O.; Lemcke T.; Kegel S.; Lomow A.; Burk U.; Dohrmann C.; Meijer L.; Austen M.; Kunick C. J. Med. Chem. 2008, 51, 2196–2207. [DOI] [PubMed] [Google Scholar]
- Knockaert M.; Wieking K.; Schmitt S.; Leost M.; Grant K. M.; Mottram J. C.; Kunick C.; Meijer L. J. Biol. Chem. 2002, 277, 25493–25501. [DOI] [PubMed] [Google Scholar]
- Kunick C.; Zeng Z.; Gussio R.; Zaharevitz D.; Leost M.; Totzke F.; Schaechtele C.; Kubbutat M. H. G.; Meijer L.; Lemcke T. ChemBioChem 2005, 6, 541–549. [DOI] [PubMed] [Google Scholar]
- Pies T.; Schaper K.-J.; Leost M.; Zaharevitz D. W.; Gussio R.; Meijer L.; Kunick C. Arch. Pharm. 2004, 337, 486–492. [DOI] [PubMed] [Google Scholar]
- Becker A.; Kohfeld S.; Lader A.; Preu L.; Pies T.; Wieking K.; Ferandin Y.; Knockaert M.; Meijer L.; Kunick C. Eur. J. Med. Chem. 2010, 45, 335–342. [DOI] [PubMed] [Google Scholar]
- Tolle N.; Kunick C. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2011, 11, 1320–1332. [DOI] [PubMed] [Google Scholar]
- Lahusen T.; De Siervi A.; Kunick C.; Senderowicz A. M. Mol. Carcinog. 2003, 36, 183–194. [DOI] [PubMed] [Google Scholar]
- Dobrov A.; Arion V. B.; Kandler N.; Ginzinger W.; Jakupec M. A.; Rufinska A.; Graf von Keyserlingk N.; Galanski M.; Kowol C.; Keppler B. K. Inorg. Chem. 2006, 45, 1945–1950. [DOI] [PubMed] [Google Scholar]
- Ginzinger W.; Arion V. B.; Giester G.; Galanski M.; Keppler B. K. Cent. Eur. J. Chem. 2008, 6, 340–346. [Google Scholar]
- Schmid W. F.; Zorbas-Seifried S.; John R. O.; Arion V. B.; Jakupec M. A.; Roller A.; Galanski M.; Chiorescu I.; Zorbas H.; Keppler B. K. Inorg. Chem. 2007, 46, 3645–3656. [DOI] [PubMed] [Google Scholar]
- Schmid W. F.; John R. O.; Mühlgassner G.; Heffeter P.; Jakupec M. A.; Galanski M.; Berger W.; Arion V. B.; Keppler B. K. J. Med. Chem. 2007, 50, 6343–6355. [DOI] [PubMed] [Google Scholar]
- Schmid W. F.; John R. O.; Arion V. B.; Jakupec M. A.; Keppler B. K. Organometallics 2007, 26, 6643–6652. [Google Scholar]
- Primik M. F.; Mühlgassner G.; Jakupec M. A.; Zava O.; Dyson P. J.; Arion V. B.; Keppler B. K. Inorg. Chem. 2010, 49, 302–311. [DOI] [PubMed] [Google Scholar]
- Mühlgassner G.; Bartel C.; Schmid W. F.; Jakupec M. A.; Arion V. B.; Keppler B. K. J. Inorg. Biochem. 2012, 116, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion V. B.; Dobrov A.; Goeschl S.; Jakupec M. A.; Keppler B. K.; Rapta P. Chem. Commun. 2012, 48, 8559–8561. [DOI] [PubMed] [Google Scholar]
- Lavrado J.; Moreira R.; Paulo A. Curr. Med. Chem. 2010, 17, 2348–2370. [DOI] [PubMed] [Google Scholar]
- Hu X.-W.; Chien C.-M.; Yang S.-H.; Lin Y.-H.; Lu C.-M.; Chen Y.-L.; Lin S.-R. Cell Biol. Toxicol. 2006, 22, 417–427. [DOI] [PubMed] [Google Scholar]
- Yang S.-H.; Chien C.-M.; Lu C.-M.; Chen Y.-L.; Chang L.-S.; Lin S.-R. Leuk. Res. 2007, 31, 1413–1420. [DOI] [PubMed] [Google Scholar]
- Chien C.-M.; Yang S.-H.; Lin K.-L.; Chen Y.-L.; Chang L.-S.; Lin S.-R. Chem.-Biol. Interact. 2008, 176, 40–47. [DOI] [PubMed] [Google Scholar]
- Tzeng C.-C.; Chen Y.-L.; Lin J.-J.; Lu C.-M.. Indolo[3,2-c]quinoline derivatives as inhibitors of DNA replication and transcription and their preparation, pharmaceutical compositions and use in the treatment of cancer. U.S. Pat. Appl. US2009/0298846, 2009.
- Filak L. K.; Mühlgassner G.; Jakupec M. A.; Heffeter P.; Berger W.; Arion V. B.; Keppler B. K. J. Biol. Inorg. Chem. 2010, 15, 903–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock A. F. A.; Habtemariam A.; Fernandez R.; Walland V.; Fabbiani F. P. A.; Parsons S.; Aird R. E.; Jodrell D. I.; Sadler P. J. J. Am. Chem. Soc. 2006, 128, 1739–1748. [DOI] [PubMed] [Google Scholar]
- Ginzinger W.; Mühlgassner G.; Arion V. B.; Jakupec M. A.; Roller A.; Galanski M.; Reithofer M.; Berger W.; Keppler B. K. J. Med. Chem. 2012, 55, 3398–3413. [DOI] [PubMed] [Google Scholar]
- Filak L. K.; Mühlgassner G.; Bacher F.; Roller A.; Galanski M.; Jakupec M. A.; Keppler B. K.; Arion V. B. Organometallics 2011, 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filak L. K.; Göschl S.; Hackl S.; Jakupec M. A.; Arion V. B. Inorg. Chim. Acta 2012, 393, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble A. B.; Garner J.; Gordon C. P.; O’Conner S. M. J.; Keller P. A. Synth. Commun. 2007, 37, 2777–2786. [Google Scholar]
- Saint-Plus, version 7.06a and APEX2; Bruker-Nonius AXS Inc., Madison, WI, 2004.
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Johnson G. K.Report ORNL-5138; Oak Ridge National Laboratory, Oak Ridge, TN, 1976.
- Primik M. F.; Göschl S.; Jakupec M. A.; Roller A.; Keppler B. K.; Arion V. B. Inorg. Chem. 2010, 49, 11084–11095. [DOI] [PubMed] [Google Scholar]
- Egger A. E.; Rappel C.; Jakupec M. A.; Hartinger C. G.; Heffeter P.; Keppler B. K. J. Anal. At. Spectrom. 2009, 24, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J.; Engqvist R.; Stålhandske C.; Wallberg H. Tetrahedron 2003, 59, 1033–1048. [Google Scholar]
- McConnell E. L.; Basit A. W.; Murdan S. J. Pharm. Pharmacol. 2008, 60, 63–70. [DOI] [PubMed] [Google Scholar]
- El-khateeb M.; Damer K.; Görls H.; Weigand W. J. Organomet. Chem. 2007, 692, 2227–2233. [Google Scholar]
- Kandioller W.; Hartinger C. G.; Nazarov A. A.; Kuznetsov M. L.; John R. O.; Bartel C.; Jakupec M. A.; Arion V. B.; Keppler B. K. Organometallics 2009, 28, 4249–4251. [Google Scholar]
- Zagermann J.; Kuchta M. C.; Merz K.; Metzler-Nolte N. J. Organomet. Chem. 2009, 694, 862–867. [Google Scholar]
- Albrecht C.; Gauthier S.; Wolf J.; Scopelliti R.; Severin K. Eur. J. Inorg. Chem. 2009, 1003–1010. [Google Scholar]
- Gregoire D. C. Anal. Chem. 1990, 62, 141–146. [Google Scholar]
- Nam K. H.; Isensee R.; Infantino G.; Putyera K.; Wang X. Spectroscopy (Duluth, MN, U.S.) 2011, 26, 36–41. [Google Scholar]
- Jin X.-D.; Du A.-D.; Li W.-J.; Xiang P.; Sakyi P. A.; Zhang L.-C. J. Anal. At. Spectrom. 2011, 26, 1245–1252. [Google Scholar]
- Shnyder S. D.; Fu Y.; Habtemariam A.; van Rijt S. H.; Cooper P. A.; Loadman P. M.; Sadler P. J. MedChemComm 2011, 2, 666–668. [Google Scholar]
- Fu Y.; Habtemariam A.; Pizarro A. M.; van Rijt S. H.; Healey D. J.; Cooper P. A.; Shnyder S. D.; Clarkson G. J.; Sadler P. J. J. Med. Chem. 2010, 53, 8192–8196. [DOI] [PubMed] [Google Scholar]
- Dougan S. J.; Habtemariam A.; McHale S. E.; Parsons S.; Sadler P. J. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11628–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamo A.; Masi A.; Peacock A. F. A.; Habtemariam A.; Sadler P. J.; Sava G. J. Inorg. Biochem. 2010, 104, 79–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.