

Abstract
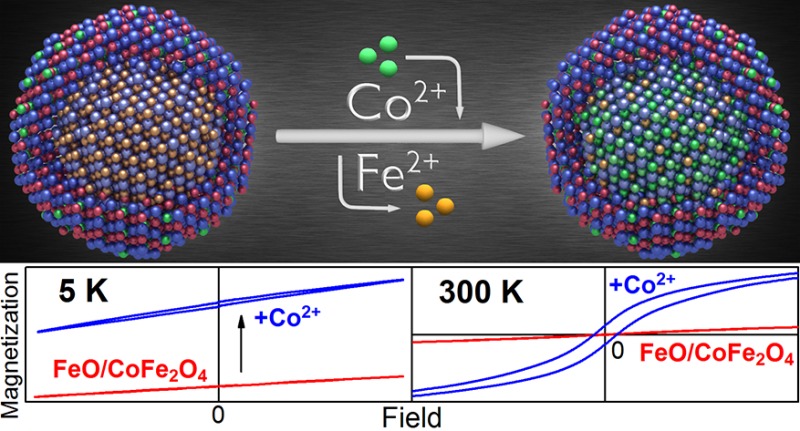
For three types of colloidal magnetic nanocrystals, we demonstrate that postsynthetic cation exchange enables tuning of the nanocrystal’s magnetic properties and achieving characteristics not obtainable by conventional synthetic routes. While the cation exchange procedure, performed in solution phase approach, was restricted so far to chalcogenide based semiconductor nanocrystals, here ferrite-based nanocrystals were subjected to a Fe2+ to Co2+ cation exchange procedure. This allows tracing of the compositional modifications by systematic and detailed magnetic characterization. In homogeneous magnetite nanocrystals and in gold/magnetite core shell nanocrystals the cation exchange increases the coercivity field, the remanence magnetization, as well as the superparamagnetic blocking temperature. For core/shell nanoheterostructures a selective doping of either the shell or predominantly of the core with Co2+ is demonstrated. By applying the cation exchange to FeO/CoFe2O4 core/shell nanocrystals the Neél temperature of the core material is increased and exchange-bias effects are enhanced so that vertical shifts of the hysteresis loops are obtained which are superior to those in any other system.
Keywords: magnetic nanocrystals, cation exchange, exchange bias, nanoheterostructures, ferrites
Postsynthetic substitution reactions, including galvanic replacement and cation exchange, applied to colloidal nanocrystals,1−14 represent a simple and versatile tool to achieve nanoarchitectures not readily accomplishable by other methods. The galvanic replacement is predominantly applied to form noble metal nanocrystals, such as Au nanocages and nanoboxes, by reacting solutions of appropriate salts with nanocrystals as “nanotemplates”.1−7 The cation exchange is a similar process, however, usually applied to compound semiconductors.8−14 By cation exchange procedures, e.g., nanorod superlattices of regularly spaced Ag2S quantum dots in CdS colloidal quantum rods,9 nonepitaxial hybrid nanostructures with gold nanoparticle cores and CdS shells,15 or branched nanocrystals with either CdSe or Cu2-xSe central cores and Cu2S pods,16 have been demonstrated. In contrast to the galvanic replacement where often substantial morphology changes are observed (e.g., hollow structures are obtained from solid nanoparticles)3,4 during cation exchange the nanocrystal shape is nearly preserved.12,14,16 The latter is caused by an anionic framework conservation, which might result from the larger ionic size of the anions in the lattice, causing their lower diffusion velocity as compared to that of the cations.17 As will be discussed below, also the coordinating solvent applied during cation exchange might contribute to the anionic framework conservation. Cation exchange reactions have been applied also to improve the properties of semiconductor nanocrystals. Treating PbX (X = S, Se, Te) with Cd2+ ions results, e.g., in the formation of protective CdX shells, causing a significant increase of the photoluminescence quantum yield of these materials.18,19
While the high potential for tailoring of material properties by the cation exchange treatment has been demonstrated for ionic semiconductors (primarily chalcogenides),8−20 here it is applied to ionic magnetic oxide materials. In particular, ferrite-based single component nanocrystals and multicomponent core/shell nanocrystal hererostructures were chosen as starting materials for the cation exchange treatments (Figure 1), because of their high potential in biomedical sciences21−31 and their highly reproducible synthesis by facile solution-phase thermolysis routes.32−35 Synthesizing them as nanoheterostructures allows obtaining physical properties which are unreachable in bulk materials.36 Two examples for such nanoheterostructures with properties not reachable in bulk are presented in the following to demonstrate the cation exchange procedure. The first example are nanoheterostructures combining ferrites with coinage metals (Figure 1b) exhibiting combined plasmonic and magnetic properties.37−39 The second example are core/shell nanocrystals combining magnetic materials of different magnetic phases (Figure 1c), showing pronounced exchange bias effects.40−43 The latter can be used to overcome some fundamental limitations of nanomaterials such as the superparamagnetic limit for a given nanocrystal size41 and are essential for the operation of magneto-electronic devices such as spin valves and magnetic tunnel junctions.44
Figure 1.

Three classes of magnetic nanocrystals are investigated: (a, e) homogeneous spherical magnetite (Fe3O4) nanocrystals, (b, f) gold/magnetite core/shell nanocrystals, and (c, g) Wüstite (FeO)/magnetite core/shell nanocrystals. (a–c) TEM images of the starting materials. (d) Scheme of the cation exchange procedure in the three different nanocrystal types with inverse spinel or rock-salt crystal structure, (blue balls represent oxygen atoms; red balls, Fe3+; orange, Fe2+; green, Co2+; gold, Au atoms). (e, f) are high-angle annular dark field STEM images of nanocrystals after the Fe2+ to Co2+ cation exchange procedure is performed. The ion concentration distributions for Fe (red) and Co (green), and Au (golden) across the nanocrystals, shown by the EDX line scans (obtained for transitions from the K or M shell respectively), evidence the Co doping after the cation exchange treatment.
The structural similarity between Fe3O4, more precisely written as FeIIFeIII2O4, to metal-doped spinel-type ferrites, MIIFeIII2O4 (M abbreviates the transition metals Co, Ni, or Mn), allows the magnetic engineering of iron oxide nanocrystals by replacing the Fe2+ by M2+.26,32,45 While uniform MIIFeIII2O4 nanocrystals are usually synthesized from mixtures of appropriate precursors,26,32,45 synthesizing core/shell structures with MIIFeIII2O4 shells covering either noble metal seeds or cores of other magnetic metal oxides is hardly achievable by conventional seeded-growth methods. For such cases the cation exchange procedure offers an elegant way to obtain the desired morphologies and magnetic doping levels by a simple post synthetic treatment, as is shown below. Applied to Fe3O4, the cation exchange is especially attractive when it is performed with Co2+, because even though the mass magnetization of CoFe2O4 is almost identical to that of pure magnetite, several other important parameters such as the coercivity and the magnetic blocking temperature are significantly increased, due to the higher spin–orbit coupling.46 Thus, even small compositional modifications due to the cation exchange can be sensitively traced by inspecting the magnetic properties of the nanocrystals. While here the Fe2+ is replaced by Co2+ the same procedure can be easily expanded to further transition metals, in order to fine-tune the nanocrystals magnetic properties, and optimize them for different requirements of potential applications, including magnetic hyperthermia,27 or spin-electronic device developments,44 or for the rapidly increasing research field of magnonics.47
To perform the Fe2+ to Co2+ cation exchange the nanocrystals, stabilized by oleic acid (OA), are treated by the following procedure: 1.2 g of CoCl2 are dispersed in 28 mL of oleylamine. This mixture is degassed under vacuum and heated to 120 °C for 20 min to completely dissolve the CoCl2. After cooling the solution down to room temperature in an argon atmosphere, 20 mg of nanocrystals dispersed in 0.5 mL of chloroform are added to the mixture, which is then degassed under vacuum again and heated for 20 min to 100 °C to remove the chloroform, before 1.5 mL trioctylphosphine (TOP) is injected under Ar atmosphere. The reaction is taking place at temperatures between 200 and 220 °C, and the Co content in the final nanocrystals can be controlled by both, reaction temperature and time, which was varied between 5 and 80 min. This procedure results in a cation exchange reaction which greatly preserves the nanocrystals core/shell structures, sizes and shapes (Figure S1–S3, Supporting Information show examples of Fe3O4 nanocrystals after the cation exchange process). Significant morphological changes are observed, however, when the reaction temperature increases above 220 °C, because the nanocrystals start to dissolve and to form aggregates of much smaller nanocrystals, or when the treatment time exceeds 80 min (Figures S3, Supporting Information). Also the addition of TOP was found to be of crucial importance, because in its absence at the reaction temperature the nanocrystals undergo fast digestive ripening leading to amorphous and small polydisperse nanoparticles (Figure S4, Supporting Information). Because of the high affinity of Co2+ and Fe2+ to oleylamine48 not only the soluble salts are nicely dissolved but also insoluble compounds like iron oxide can be etched by solvation of Fe2+ with oleylamine at high temperature. In contrast, TOP has a significantly lower affinity to Fe2+ and Co2+.48 Thus, addition of TOP ligands to the oleylamine dispersion influences the equilibrium between solvated Fe2+ or Co2+ ions and cobalt/iron in the lattice. As was shown before for the case of cation exchange in CdSe nanocrystals treated by Pd2+, and Pt4+ also in the present case by changing the solvent environment the thermodynamics of cation solvation is modified.49 Another plausible effect of TOP may be related to its strongly reductive power and the ability to scavenge the traces of oxygen, thus protecting the Fe2+ ions in the lattice. It should be noted that in contrast to the cations Co2+ and Fe2+ the anion O2- does not react with oleylamine, because both, the O2- and the olylamine represent bases according to the theory of Pearson.50 This might be another important reason why the anion framework is preserved during the cation exchange process.
The cation exchange is stopped by cooling down the nanocrystal solution. Chloroform is added to the solution and the nanocrystals are aggregated by ethanol together with acetone for the washing procedure. Few drops of OA are added to improve the colloidal stability in the chloroform solvent. The whole washing procedure is repeated 6 times and finished by filtration. This procedure removes the CoCl2 and aggregates of other byproducts from the nanocrystal solution, to allow accurate determination of the Co content in the nanocrystals, probed by (i) EDX on relatively thick nanocrystal films, (ii) by Rutherford backsattering spectrometry (RBS) performed on nanocrystal (sub)monolayers, and (iii) by atomic absorption spectroscopy (AAS) performed from nanocrystals which are completely dissolved in acidic solutions. The Co concentrations are deduced from all three methods, and for the presented samples the scatter between the data is less than ±1%.
To facilitate a comparison between the results of the three nanocrystal systems under investigation (Figures 1 and S1 (Supporting Information)), we have chosen in all cases spherically shaped nanocrystals with mean diameters close to 20 nm. The first system represents homogeneous magnetite nanocrystals with a mean diameter of 21 nm (Figures 1a, S1a, S2, and S3 (Supporting Information)), synthesized by thermal decomposition of iron-oleate in the presence of oleic acid.33 In this case the cation exchange results in a rather homogeneous Co alloying of the nanocrystals, by replacing a part of the Fe2+, residing on the octahedral sites of the inverse spinel structure of magnetite, by Co2+ (sketched in Figure 1d). This scenario is confirmed by the energy dispersive X-ray (EDX) analysis, performed after cation exchange by scanning transmission electron microscopy (STEM). The EDX line profile in Figure 1e shows an almost as smooth Co distribution across the nanocrystal as is also found for Fe. After 30 min of reaction with CoCl2 at 220 °C in this sample a Co content of 8% is detected, e.g., in the RBS spectrum in Figure 2a by the appearance of a distinct Co peak on the right-hand side of the Fe peak. TEM images confirm that the size and shape of the nanocrystals is almost preserved (Figure 2b,c; the inset in Figure 2d shows the nanocrystals after Co exchange and a direct comparison of the nanocrystals before and after treatment is given in Figure S2, Supporting Information). While the Co alloying causes only subtle effects with respect to nanocrystal morphology, it is greatly affecting their magnetic properties. The magnetic response changes because of strong spin–orbit couplings at the Co2+ sites and the concomitant increase of the magnetic anisotropy. The latter has a direct effect on the temperature dependence of the zero-field-cooling magnetization, showing the typical behavior of magnetic nanocrystals (Figure 2d). At low temperature the magnetic moments of the individual nanocrystals, resulting from the ferromagnetically coupled M2+ ion spins (the magnetic moments of the Fe3+ ions are antiferromagnetically coupled in ferrites and thus do not contribute to the magnetization), are blocked and are pointing randomly into all directions, so that the total magnetization of the sample is zero. With increasing temperature the sample magnetization, given here per weight of the magnetic ions (Fe and Co), increases and at high temperatures the thermal energy of the nanocrystals overcomes the magnetic anisotropic energy barrier between different magnetic easy axes and between the spin up and spin down direction within the same easy axis, respectively. As a consequence the magnetization typically shows a superparamagnetic behavior with a 1/T dependence of the sample magnetization (for the present sample the 1/T dependence can be expected to occur well above 300 K, whereas for the nanocrystals described in the next paragraph it is clearly displayed in Figure 3a). Thus, the magnetization as a function of temperature exhibits a maximum at the so defined blocking temperature TB. For the Fe3O4 nanocrystals with a size of 21 nm this blocking temperature is found at 250 K, whereas the Co treatment increases TB by 60 to 310 K. This causes also an opening of the hysteresis loop close to room temperature, which is observed after cation exchange but is completely absent for the initial Fe3O4 nanocrystals (inset in Figure 2e). From the opening of the hysteresis loops the values of the coercivity field, HC, and remanent magnetization, MR, are measured at zero magnetization and zero external field, respectively (indicated in the inset of Figure 2e). Both values are strongly temperature dependent, and at all temperatures, HC is at least twice as high after the Co treatment (Figure 2e, the corresponding hysteresis data before and after Co2+ treatment are shown in Figure S5, Supporting Information). Thus, the Co treatment results in an overall increase of coercivity, remanence and blocking temperature, due to an increased magneto-crystalline anisotropy. The latter is directly probed by measuring the relaxation of the nanocrystals magnetic moment, after aligning them by an external magnetic field.51 If the nanoparticles are immobilized, magnetorelaxometry experiments provide a purely Neél relaxation time τN due to flipping of the magnetic moments within the nanocrystals caused by thermal activation. The flipping time is exponentially proportional to the magnetocrystalline anisotropy energy.51 After Co2+ treatment of the nanocrystals, room temperature flux gate magnetometry detects an increase in τN (Figure S6, Supporting Information). An even more significant increase of τN due to the Co2+ treatment of magnetite nanocrystals is observed for smaller nanocrystal sizes (Figure S6b, Supporting Information). In Figure S7 (Supporting Information), the results for 9 nm large nanocrystals are summarized, showing also an increase of TB from 150 to 206 K and an increase of the coercivity by a factor of 5.7. It should be noted that these observed modifications of the magnetic properties are solely due to the exchange of the Fe2+ to the Co2+ ions and not due to different states of nanocrystal aggregation. The latter is ruled out by concentration dependent magnetization measurements, showing almost no dependence of the normalized magnetization versus temperature, at least when the dilution of nanocrystals is performed in a good solvent (Figure S8, Supporting Information).
Figure 2.
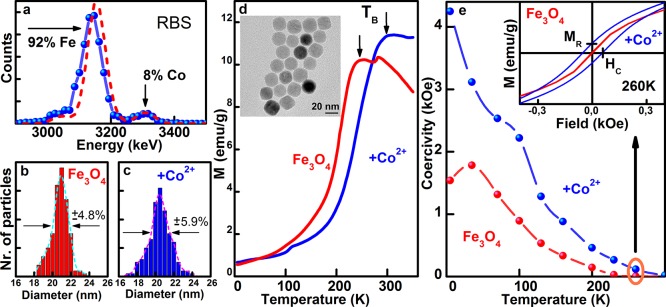
Co2+-treated magnetite (Fe3O4) nanocrystals. (a) Rutherford backscattering provides a Co/Fe ration of 8% to 92% after the Co2+ treatment is performed at 220 °C for 30 min. The dashed line is a model calculation. The mean sizes and size distribution, extracted from TEM images (Figure S2, Supporting Information), before (b) and after Co treatment (c), are almost identical. (d) Zero field cooled magnetization data, measured at an applied field of 100 Oe, evidence an increase of the blocking temperature TB upon Co2+ treatment, as is indicated by the arrows. Inset: TEM image of the nanocrystals after Co2+ treatment. (e) Temperature dependent coercivity field, extracted from the data shown in Figure S5, Supporting Information. The inset shows exemplarily the 260 K hysteresis loops, exhibiting an opening after Co2+ treatment while being absent before the treatment. MR and HC are indicated for the hysteresis loop obtained after Co treatment.
Figure 3.

Co2+ treated gold/Fe3O4 core/shell nanocrystals. (a) Zero-field cooled magnetization curves, measured at an applied field of 100 Oe, evidence an increased blocking temperature after Co2+ treatment. The inset shows the effect of the Co2+ treatment on low temperature hysteresis loops. (b) Normalized optical absorbance spectra exhibit plasmon peaks close to 600 nm wavelength. (c) TEM overview (bright field) of the nanocrystals after Co2+ treatment. The inset shows a dark field image of an individual nanocrystal, highlighting the defect structure (polycrystallinity) of the shell.
The second system investigated here represents a nanoheterostructure, formed by a gold core and a relatively uniform magnetite shell (Figures 1b, S1b, and S9 (Supporting Information)). Such magnetic–plasmonic core shell nanoparticles have attracted great interest because the localized surface plasmon of the gold cores provide an optical addressability for tracking or monitoring of the magnetic nanoparticles.38 The gold/Fe3O4 core/shell nanocrystals are grown by decomposition of iron pentacarbonyl on gold seed nanocrystals, and subsequent in situ oxidation of the iron shell into Fe3O4.39 The shortcoming of these core/shell nanocrystals is the relatively small magnetic moment observed at room temperature due to their limited shell thickness and their small magnetic blocking temperature.39 Applying the Fe2+ to Co2+ cation exchange to these nanocrystals improves both of these parameters, TB from 68 K to about 110 K and the room temperature magnetization from 0.62 emu/g to 1.08 emu/g (at a field of 100 Oe, Figure 3a). These improvements, together with an increase of the coercivity at low temperatures (inset in Figure 3a), is obtained here by doping of the nanocrystal shell by 11% of Co, as is deduced also by RBS (Figure S9, Supporting Information). Because of the high chemical stability of the gold core, it is not affected by any treatments with Co2+ precursors (Figure 1d,f). Thus, the optical properties of the nanoheterostrucures, dominated by the plasmonic feature of the gold nanocrystal core, are almost preserved. As shown in Figure 3b, the plasmon resonance found at 553 nm before Co2+ treatment, shifts only by 33 nm due to a change of the dielectric function of the nanocrystal shells. While the inert gold core is unaffected by the Co treatment, the shell becomes more polycrystalline during the cation exchange process (Figures 3c and S9 (Supporting Information)), which is important because defects in the lattice structure facilitate cation diffusion from the nanocrystals surfaces to their interior.20,52 That such a diffusion is indeed taking place is not only evidenced by the EDX line scans shown in Figure 1e,f but is confirmed also by the following experiments performed on FeO/CoFe2O4 core/shell nanocrystals.
The situation is completely different in the third system, which are magnetic heterostructure nanocrystals combining antiferromagnetic (AFM) and ferrimagnetic components. This combination is of special interest because it provides a set of unique magnetic properties by exchange coupling via the heterostructures interface,40−43 These exchange bias effects are important for giant magnetoresistance devices or spin valves,58 which are applied e. g., in magnetic-random access memories (MRAMS) or in the read heads of hard discs. The present samples are spherical core/shell nanocrystals with FeO (Wüstite) in the center and CoFe2O4 shells (Figure 1c).43 These nanocrystals exhibit a cubic NaCl crystal structure in the core, while the shell is formed by an inverse spinel structure. Interestingly, these nanocrystals are grown by thermolysis of a mixture of iron- and cobalt oleate, which initially was expected to result in homogeneously Co-doped magnetite nanocrystals as those shown above, obtained by the cation exchange treatment of Fe3O4 nanocrystals. The hysteresis loops of these FeO/CoFe2O4 exhibit in particular very pronounced effects due to the exchange coupling, primarily anomalously large vertical shifts, VS, but dependent on temperature also increased coercivity (Figure 4a). These VSs are observed, when prior to the measurements all magnetic moments in the samples are aligned at high temperatures by an applied external field, which is maintained during cooling down. After cooling the samples, the hysteresis loops are measured by varying and reversing the external magnetic field. The vertical shifts of the hysteresis loops observed after field cooling indicate that a part of the ferromagnetic spins, which are present in our samples in the CoFe2O4 shell, are pinned by exchange interactions via the interface to the spins of the antiferromagnetic FeO core, which does not reverse under reversal of the external magnetic field.43 VSs are observed when the effective Zeeman energy (difference between Zeemen energy and anisotropy energy) of the ferro- or ferrimagnetic material is smaller than the anisotropy energy of the antiferromagnetic material as well as smaller than the interface exchange energy.53 In the present nanocrystals, this condition is fulfilled because of the large magnetic anisotropy present in CoFe2O4, which reduces the effective Zeeman energy in this material. In the present sample at 5 K, the VS is very pronounced and it amounts to 60% of the maximum magnetization, Mmax, measured at an applied field of 1 T. This value is as high as the highest value reported for specially designed layered heterostructure systems.54
Figure 4.

Hysteresis loops of FeO/CoFe2O4 core/shell nanocrystals before (a) and after (b) Co2+ treatment, measured after field cooling. The averaged Co content is 18% in part a and 27% in part b. The vertical loop shift VS, the maximum magnetization Mmax, and the opening of the hysteresis Ho are exemplarily indicated.
While the exchange bias effects are very pronounced at low temperatures, they are unfortunately limited to temperatures below 200 K, which is the Neél temperature of the FeO core (TN(FeO)), at which the AF to paramagnetic phase transition occurs. To increase the maximum temperature where exchange bias effects can be found, which is mandatory because any application of these materials in devices such as spin valves requires this effect to be present at room temperature, the core material has to be modified. One possibility is given by changing the core from FeO toward Fe1-xCoxO, because CoO has a Neél temperature close to room temperature.55 This modification of the core is again performed by the cation exchange procedure. In this case it hardly affects the CoFe2O4 shell, because it is already free of Fe2+ but predominantly affects the FeO core material. Indeed, after the cation-exchange treatment a rather uniform distribution of Co across the nanocrystal diameter is found (Figure 1g). This indicates that in this case the Co2+ cation exchange converts the FeO to CoxFe1-xO (sketched in Figure 1d).
The Co doping of the Wüstite cores is confirmed by the magnetic properties of the nanocrystals after the Co2+ treatment. The hysteresis loops still show the characteristic features of exchange bias in the form of vertical loop shifts (Figure 4b). At 5 K, the VS even increased to 78% of Mmax., which is the to the best of our knowledge the highest value ever reported for any system.54 The value of the magnetization also increased, and the characteristic exchange bias effects are observed even above the Neél temperature of FeO (TN(FeO)). For the starting material, the latter can be clearly identified by an abrupt increase of the temperature dependent magnetization, measured after sample cooling in zero field and applying during the measurement a small field of 100 Oe (Figure 5a). This abrupt magnetization increase close to 190 K is caused by the AF to paramagnetic phase transition of the core material, since in the AF phase all magnetic moments are canceling each other, whereas in the paramagnetic phase they contribute to the magnetization, because the small external field aligns them into field direction. The maximum of this curve, found close to 240 K corresponds to the superparamagnetic blocking temperature of the CoFe2O4 shell TB(CoFe2O4). While TN of the core and TB of the shell are well resolved in the magnetization data of the initial material, the Co2+ treatment results in a shift of both parameters to higher temperatures. While TN cannot be clearly identified, (green and blue lines in Figure 5a) TB still corresponds to the magnetization maximum. For the sample with the highest averaged Co2+ content of 27% it is found at 323 K, well above room temperature. Thus, the hysteresis loop of this sample shows a finite opening at 300 K, whereas initial nanocrystals show only a superparamagnetic behavior (inset in Figure 5b).
Figure 5.

Effect of the Co2+ treatment on the exchange bias features of FeO/CoFe2O4 nanocrystals. (a) Magnetization after zero field cooling, measured with an applied field of 100 Oe, shows a disappearance of the antiferromagnetic to paramagnetic phase transition upon Co2+ treatment (note that the data for the initial material is magnified by a factor of 10). (b) Opening of the hysteresis loop, measured at their vertically shifted center, is greatly enhanced. The inset shows an open hysteresis loop at 300 K for the Co treated nanocrystals (blue line) whereas the initial material (red line) exhibits a completely closed hysteresis. (c) Vertical loop shifts before (red line) and after (blue line) Co2+ treatment. The inset shows the high temperature region on magnified scale.
To obtain an estimate for the Neél temperature of the core after Co2+ treatment, a closer inspection of the exchange bias effects is required. In the present sample, the exchange bias across the interface affects two parameters, the opening of the hysteresis loop (Ho, indicated in Figure 4b) measured at the center of the loop, (which is eventually shifted in vertical direction), and the vertical shift of the loop (indicated in Figure 4a). Also the hysteresis opening Ho shows a characteristic feature close to the Neél temperature. For the initial material a peak is observed in Ho versus T close to TN indicating an enhancement of the coercivity. This enhancement is again caused by the exchange interaction via the interface, because close to TN the anisotropy energy of the antiferromagnet becomes strongly reduced. As soon as it is smaller than the exchange interaction across the interface, the antiferromagnetically aligned spins of the cores rotate coherently with the ferromagnetically aligned spins of the shells under field reversal,53 thus enhancing the coercivity of the whole particles. Above TN the core is in the paramagnetic state and the hysteresis opening is solely caused by the anisotropy energy of the ferromagnetic shell. After Co2+ treatment the opening of the hysteresis loop is increased at all temperatures, at the maximum of Ho versus temperature the opening amounts 5143 Oe, which is 18.8 times larger than for the initial material. Similarly, the VS of the hysteresis loop increased by the Co2+ treatment at all temperatures. At low temperature it increases from 6.97 to 43.51 emu/g (Figure 5c) and a finite vertical shift of the hysteresis loop is observed even up to room temperature (inset in Figure 5c). Since the VS is a results solely from the exchange interaction between core and shell of different magnetic phases, its observation at room temperature confirms the presence of an antiferromagnetic phase at least for a part of the nanocrystal batch. Thus, the Co2+ treatment shifted the AF phase transition temperature from 200 K, as observed for the initial material, to room temperature, which is only possible if the FeO cores of the nanocrystals have been changed to be rich of CoO. This fact evidence that the Fe2+ to Co2+ exchange can act quite efficiently even through an protective nanocrystal shell. This result is also of interest, because the observation of exchange bias effects is also the precondition for any application of these materials in spin valve based devices.58
Overall, by the Co2+ treatment the Co content in the FeO/CoFe2O4 nanocrystals increased from 18% to 27%, as is evidenced by AAS and RBS (Figure S10, Supporting Information). It changed, however, also the morphology of the nanocrystals. From inspections of TEM images (Figure S11, Supporting Information) the morphological changes are hardly seen, because of the polydispersity of the core/shell dimensions. However, the changes are clearly revealed by synchrotron X-ray diffraction, performed at beamline P02/Hasylab Hamburg with 60 keV X-ray photons. The diffraction patterns shown in Figure 6a can be well fitted by assuming separate phases of CoFe2O4 and of Fe1-xCoxO4, by allowing the lattice parameters to vary with respect to that of bulk. By zooming out the region around a momentum transfer of 2.5 Å–1 the effect of the Co2+ treatment can be monitored (Figure 6b). Here the narrow peak at 2.53 Å–1 corresponds to the FeO core and that at 2.46 5 Å–1 to the CoFe2O4 shell. For the core peak the Co2+ treatment results in a shift to larger diffraction angles, corresponding to a decrease of the lattice parameter, indicating the incorporation of Co into the cubic lattice of FeO (bulk FeO has a lattice parameter of 0.4332 nm while that of CoO is 0.4214 nm).56,57 The second peak shows a much smaller peak shift, but a rather pronounced narrowing of its width, and an increase of its intensity, both indicating an increase of the dimensions of the CoFe2O4 shell. From a fit of the data in Figure 6a the initial nanocrystals exhibit a core volume which is 41% of the nanocrystals total volume, whereas after the Co2+ treatment for 80 min the core volume shrinks to an average value of 8% (aliquots taken for shorter treatment times indicate that this shrink of the core and growth of shell is a continuous process). This increase of the CoFe2O4 phase during Co exchange explains also the observed increase of the magnetic moment of the nanocrystals (Figure 4 and 5) because the antiferromagnetic core does not contribute to the magnetization. All these observations, however, confirm that the Co is able to diffuse into the nanocrystals sufficiently far to change the core composition from FeO toward Fe1-xCoxO, with increased Neél temperature. The growth of the CoFe2O4 shell in expense of the core we attribute to a partial oxidation of the Fe2+ present in the Fe1-xCoxO core after cation exchange. This process requires oxygen to diffuse through the shell to the core, which is facilitated by the cation exchange, producing defects in the shell acting as diffusion channels.20,52 Thus, the oxidation takes place after the samples are exposed to the ambient after the cation exchange is finished. Before the cation exchange process, these defect related diffusion channels were obviously absent, because the Fe3O4/FeO core/shell nanocrystals we found to have a stable morphology and composition for several years.
Figure 6.
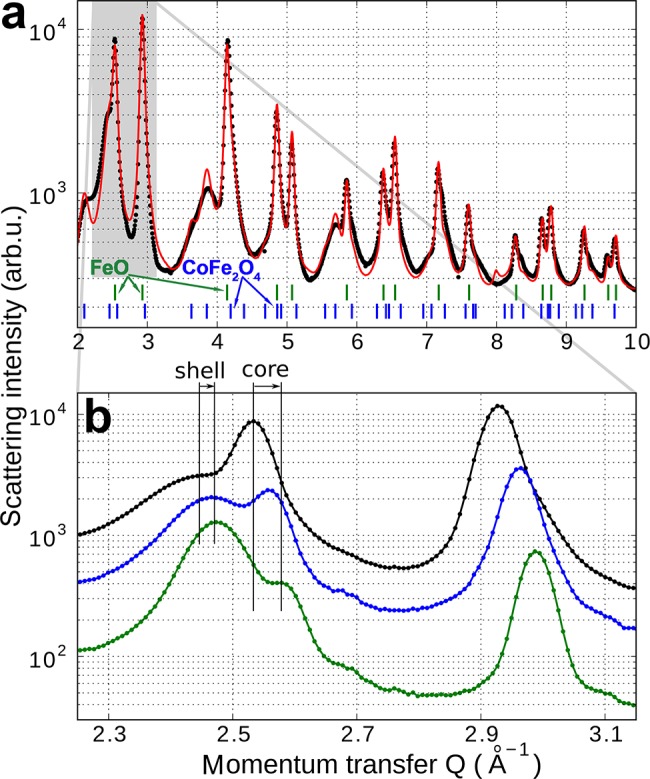
Change of the core and shell morphology during Co2+ treatment. (a) Synchrotron X-ray powder diffraction datat from FeO/CoFe2O4 nanocrystals. The fit of the data (red line) is used to extract the ratio of core to shell volume. The green and blue dashes on the bottom indicate the peak positions of bulk CoFe2O4 and FeO, respectively. (b) Enlarged scale allows the observation of peak shifts and intensity changes upon Co2+ treatment.
In conclusion, the cation exchange process is demonstrated here to be a valuable tool for tuning and improving the magnetic properties of different metal oxide based colloidal nanocrystals. Demonstrated is an increase of the superparamagnetic blocking temperature, of magnetic remanence and coercivity as well as of various exchange bias effects, by replacing Fe2+ by Co2+, due to an enhancement of the magnetocrystalline anisotropy. Outstanding are the demonstrated vertical shifts of the hysteresis loops in Co2+ treated core/shell nanocrystals, which amounts up to 78% of the saturation magnetization at low temperature, representing a substantially higher value than in any other system. The sizes and shapes of the nanocrystals are almost preserved. In homogeneous ferrite nanocrystals a rather homogeneous Co doping is achieved, whereas in nanocrystal heterostructures specific compounds of the nanocrystals can be selectively manipulated. In gold/iron-oxide core/shell nanocrystals exclusively the shell is modified by the cation exchange, whereas in FeO/CoFe2O4 nanocrystals the core undergoes severe changes. Thus, the cation exchange process enables to obtain novel nanoheterostructures with tunable magnetic properties, which are not achievable by direct synthesis. While nanocrystal materials with exchange bias effects at room temperature are of primary interest for applications in spin-valves and magneto-ectronic nanodevices based on them, the Fe2+ to Co2+ cation exchange process demonstrated here for metal-oxides can be easily expanded to further transition metal ions, for designing optimized materials for a large range of applications.
Acknowledgments
We thank Oleksandr Mykytiuk for his art work in Figure 1 and the abstract graphics. Financial support from the Austrian Science Fund FFG via the “Austrian Nano Inititative” (Project NSI-NanoShape II) and from the FWF (project SFB IRON) is acknowledged. The work was also supported by two electron microscopy centers, at ETH Zurich (EMEZ) and at EMPA. M.I.B. acknowledges financial support from Swiss SNF (Marie-Heim Vögtlin grant). We thank the staff at beamline P02 at Petra III/Hasylab Hamburg (P. Walter and A.-C. Dippel) for assistance with the synchrotron experiments. D.K. acknowledges financial support by the Austrian Academy of Science.
Supporting Information Available
Detailed experimental methods and Figures S1–S11 (STEM and TEM images, hysteresis loops, magnetization relaxation, solvent dependent magnetization, and Rutherford backscattering data). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun Y.; Xia Y. Science 2002, 298, 2176–2179. [DOI] [PubMed] [Google Scholar]
- González E.; Arbiol J.; Puntes V. F. Science 2011, 334, 1377–1380. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Mayers B. T.; Xia Y. Nano Lett. 2002, 2, 481–485. [Google Scholar]
- Sun Y.; Xia Y. Nano Lett. 2003, 11, 1569–1572. [Google Scholar]
- Lu X.; Tuan H. Y.; Chen J.; Li Z. Y.; Korgel B. A.; Xia Y. Nano Lett. 2005, 5, 2058–2062.16218737 [Google Scholar]
- Lu X.; Tuan H. Y.; Chen J.; Li Z. Y.; Korgel B. Y.; Xia Y. J. Am. Chem. Soc. 2007, 129, 1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au L.; Lu X.; Xia Y. Adv. Mater. 2008, 20, 2517–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Science 2004, 306, 1009–1012. [DOI] [PubMed] [Google Scholar]
- Robinson R. D.; Sadtler B.; Demchenko D. O.; Erdonmez C. K.; Wang L. W.; Alivisatos A. P. Science 2007, 317, 355–358. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Zheng H.; Sadtler B.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 16851–16857. [DOI] [PubMed] [Google Scholar]
- Sadtler B.; Demchenko D. O.; Zheng H.; Hughes S. M.; Merkle M. G.; Dahmen U.; Wang L. W.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 5285–5293. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. J. Am. Chem. Soc. 2010, 132, 9997–9999. [DOI] [PubMed] [Google Scholar]
- Rivest J. B.; Swisher S. L.; Fong L. K.; Zheng H.; Alivisatos A. P. ACS Nano 2011, 5, 3811–3816. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Beberwyck B, J.; Fong L. K.; Polking M. J.; Alivisatos A. P. Angew. Chem., Int. Ed. 2012, 51, 1–5. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Tang Y.; Lee K.; Ouyang M. Science 2010, 327, 1634–1638. [DOI] [PubMed] [Google Scholar]
- Miszta K.; Dorfs D.; Genovese A.; Kim M. R.; Manna L. ACS Nano 2011, 5, 7176–7183. [DOI] [PubMed] [Google Scholar]
- Rivesta J. B.; Jain P. K. Chem. Soc. Rev. 2013, 42, 89–96. [DOI] [PubMed] [Google Scholar]
- Pietryga J. M.; Werder D. J.; Williams D. J.; Casson J. L.; Schaller R. D.; Klimov V. I.; Hollingsworth J. A. J. Am. Chem. Soc. 2008, 130, 4879–4885. [DOI] [PubMed] [Google Scholar]
- Lambert K.; De Geyter B.; Moreels I.; Hens Z. Chem. Mater. 2009, 21, 778–790. [Google Scholar]
- Casavola M.; Van Huis M. A.; Bals S.; Lambert K.; Hens Z.; Vanmaeckelbergh D. Chem. Mater. 2011, 24, 294–302. [Google Scholar]
- Gupta A. K.; Gupta M. Biomaterials 2005, 26, 3995–4021. [DOI] [PubMed] [Google Scholar]
- Bulte J. W. M.; Douglas T.; Witwer B.; Zhang S. C.; Strable E.; Lewis B. K.; Zywicke H.; Miller B.; van Gelderen P.; Moskowitz B. M.; Duncan I. D.; Frank J. A. Nat. Biotechnol. 2001, 19, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Wang D.; He J.; Rosenzweig N.; Rosenzweig Z. Nano Lett. 2004, 4, 409–413. [Google Scholar]
- Scherer F.; Anton M.; Schillinger U.; Henke J.; Bergemann C.; Krüger A.; Gänsbacher B.; Plank C. Gene Ther. 2002, 9, 102–109. [DOI] [PubMed] [Google Scholar]
- Gleich B.; Weizenecker J. Nature 2005, 435, 1214–1217. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Huh Y. M.; Jun Y, W.; Seo J. W.; Jang J. T.; Song H. T.; Kim S.; Cho E. J.; Yoon H. G.; Suh J. S.; Cheon J. Nat. Med. 2007, 13, 95–99. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Jang J. T.; Choi J. S.; Moon S. H.; Noh S. H.; Kim J. W.; Kim J. G.; Kim I. S.; Park K. I.; Cheon J. Nat. Nanotechnol. 2011, 6, 418–422. [DOI] [PubMed] [Google Scholar]
- Medarova Z.; Pham W.; Farrar C.; Petkova V.; Moore A. Nat. Med. 2007, 13, 372–377. [DOI] [PubMed] [Google Scholar]
- Liong M.; Lu J.; Kovochich M.; Xia T.; Ruehm S. G.; Nel A. E.; Tamanoi F.; Zink J. I. ACS Nano 2008, 2, 889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh I.; Josephson L. Sensors 2009, 9, 8130–8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent S.; Forge D.; Port M.; Roch A.; Robic C.; Vander Elst L.; Muller R. N. Chem. Rev. 2008, 108, 2064–2110. [DOI] [PubMed] [Google Scholar]
- Jana N. R.; Chen Y.; Peng X. Chem. Mater. 2004, 16, 3931–3935. [Google Scholar]
- Kovalenko M. V.; Bodnarchuk M. I.; Lechner R. T.; Hesser G.; Schäffler F.; Heiss W. J. Am. Chem. Soc. 2007, 129, 6352–6353. [DOI] [PubMed] [Google Scholar]
- Jun Y. W.; Choi J. S.; Cheon J. Angew. Chem., Int. Ed. 2006, 45, 3414–3439. [DOI] [PubMed] [Google Scholar]
- Park J.; An K.; Hwang Y.; Park J. G.; Noh H. J.; Kim J. Y.; Park J. H.; Hwang N. M.; Hyeon T. Nat. Mater. 2004, 3, 891–895. [DOI] [PubMed] [Google Scholar]
- Ronny C.; Saunders A. E.; Banin U. Angew. Chem., Int. Ed. 2010, 49, 4878–4897. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Hou Y.; Sun S. J. Am. Chem. Soc. 2007, 129, 8696–8699. [DOI] [PubMed] [Google Scholar]
- Levin C. S.; Hofmann C.; Ali T. A.; Kelly A. T.; Morosan E.; Nordlander P.; Whitmire K. H.; Halas N. J. ACS Nano 2009, 3, 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko E. V.; Bodnarchuk M. I.; Kovalenko M. V.; Talapin D. V.; Smith R. K.; Aloni S.; Heiss W.; Alivisatos A. P. Adv. Mater. 2008, 20, 4323–4329. [Google Scholar]
- Kavich D. W.; Dickerson J. H.; Mahajan S. V.; Hasan S. A.; Park J. H. Phys. Rev. B 2008, 78, 174414. [Google Scholar]
- Skumryev V.; Stoyanov S.; Zhang Y.; Hadjipanayis G.; Givord D.; Nogués J. Nature 2003, 423, 850–853. [DOI] [PubMed] [Google Scholar]
- Pichon B. P.; Gerber O.; Lefevre C.; Florea I.; Fleutot S.; Baaziz W.; Pauly M.; Ohlmann M.; Ulhaq C.; Ersen O.; Pierron-Bohnes V.; Panissod P.; Drillon M.; Begin-Colin S. Chem. Mater. 2011, 23, 2886–2900. [Google Scholar]
- Bodnarchuk M. I.; Kovalenko M. V.; Groiss H.; Resel R.; Reissner M.; Hesser G.; Lechner R. T.; Steiner W.; Schäffler F.; Heiss W. Small 2009, 5, 2247–2252. [DOI] [PubMed] [Google Scholar]
- Wolf S. A.; Awschalom D. D.; Buhrman R. A.; Daughton J. M.; von Molnár S.; Roukes M. L.; Chtchelkanova A. Y.; Treger D. M. Science 2001, 16, 1488–1495. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zeng H.; Robinson D. B.; Raoux S.; Rice P. M.; Wang S. X.; Li G. J. Am. Chem. Soc. 2004, 126, 273–279. [DOI] [PubMed] [Google Scholar]
- Song Q.; Zhang Z. J. J. Phys. Chem B 2006, 110, 11205–11209. [DOI] [PubMed] [Google Scholar]
- Neusser S.; Grundler D. Adv. Mater. 2009, 21, 2927–2932. [Google Scholar]
- Ahrland S.; Chatt J.; Davies N. R., Q. Rev. Chem. Soc. 1958, 12, 265–276. [Google Scholar]
- Wark S. E.; Hsia C. H.; Son D. H. J. Am. Chem. Soc. 2008, 130, 9550–9555. [DOI] [PubMed] [Google Scholar]
- Pearson R. G. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- Ludwig F.; Heim E.; Schilling M. J. Appl. Phys. 2007, 101, 113909.(10 pages). [Google Scholar]
- Dieckmann R. J. Phys. Chem. Solids 1998, 59, 507–525. [Google Scholar]
- Nogués J.; Schuller I. K. J. Magn. Magn. Mater. 1999, 192, 203–232. [Google Scholar]
- Dobrynin A. N.; Ievlev D. N.; Temst K.; Lievens P.; Margueritat J.; Gonzalo J.; Afonso C. N.; Zhou S. Q.; Vantomme A.; Piscopiello E.; Van Tendeloo G. Appl. Phys. Lett. 2005, 87, 012501. [Google Scholar]
- de la Venta J.; Erekhinsky M.; Wang S.; West K. G.; Morales R.; Schuller I. K. Phys. Rev. B. 2012, 84, 134447. [Google Scholar]
- Ambrose T.; Chien C. L. Phys. Rev. Lett. 1996, 76, 1743–1746. [DOI] [PubMed] [Google Scholar]
- Kannan R.; Seehra M. S. Phys. Rev. B 1987, 35, 6847–6853. [DOI] [PubMed] [Google Scholar]
- Jette E. R.; Foote F. J. Chem. Phys. 1933, 1, 29–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.