

Abstract
Breast cancer is the most frequent cancer in women. Evidence suggests that the polyunsaturated fatty acids (PUFAs), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) affect breast cancer proliferation, differentiation and prognosis. However, the mechanism still remains unclear. In this study, the expression of transient receptor potential canonical (TRPC)3 was detected throughout the cell cytoplasm and at the cell surface of MCF-7 cells. Ca2+ entry was induced in these cells via activated TRPC3 by either the diacylglycerol analogue (OAG) or by intracellular Ca2+ store depletion. TRPC-mediated Ca2+ entry was inhibited by PUFAs including arachidonic acid (AA) and linolenic acid (LA) but not saturated fatty acids. Overexpression of the PUFA degradation enzyme, cyclooxygenase 2 (COX2), enhanced capacitative Ca2+ entry. In addition, inhibition of COX2 reduced [Ca2+]i. Nevertheless, inhibition of TRPC reduced the cell cycle S phase and cell migration, implicating a functional role for TRP-mediated Ca2+ entry in cell proliferation and invasion. Exogenous PUFA as well as a TRPC3 antagonist consistently attenuated breast cancer cell proliferation and migration, suggesting a mechanism in which PUFA restrains the breast cancer partly via its inhibition of TRPC channels. Additionally, our results also suggest that TRPC3 appears as a new mediator of breast cancer cell migration/invasion and represents a potential target for a new class of anticancer agent.
Keywords: MCF-7, TRP channel, arachidonic acid, metabolites, proliferation, migration
Introduction
Breast cancer is the most frequent cancer in women (1). Evidence suggests that polyunsaturated fatty acids (PUFAs), including arachidonic acid (AA) and linolenic acid (LA) but also eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), influence breast cancer proliferation (2–4), differentiation (3), and prognosis (5). Clinical and research data in the past 20 years reveal that cyclooxygenase (COX)2 is overexpressed in a variety of malignant tumors (6–8) and are linked to apoptosis resistance (9), invasive tumor behavior (10) and the poor prognosis of breast cancer (8,11,12). However, the mechanism of unsaturated fatty acids upon breast cancer still remains unclear.
Ca2+ is one of most important signal transduction elements in cells ranging from bacteria to neurons. The molecular identity of the membrane protein that serves to enable capacitative Ca2+ entry (CCE) following the functional depletion of intracellular Ca2+ store or activation of G-protein (13) has yet to be determined with any certainty, but the canonical, vertebrate TRPs (TRPC) are widely believed to fulfil this vital role in many cell types (14,15). Cell-specific, differential expression of TRPC has been described in many excitable and non-excitable cell types, including prostate cancer cells (16) and breast cancer cells (17,18) where they contribute and mediate the Ca2+ entry in response to multiple physico-chemical stimuli (19,20).
The PUFA, AA has been proposed to activate TRPC, in many mammalian cell types, including endothelial (21,22), epithelial and smooth muscle (23) cells, but the direct evidence for this in these cells is lacking and the proposals are based primarily upon findings in Drosophila TRP and TRP-like (TRPL) (24) and mammalian TRPV channels (25) where AA and LA induce activation, leading to Ca2+ entry.
COX acts to degrade AA. In addition, high cellular levels of COX are commonly used as a marker for malignant breast cancer (6,10,12). This suggests that AA and/or its degenerate products may play a role in this pathological process.
In this study, we found the functional expression of TRPC3 in human MCF-7 breast cancer cell-mediated Ca2+ entry. Native TRPC channels in MCF-7 cells were inhibited by PUFA. Ca2+ entry via activated TRPC was enhanced when PUFA were absent, suggesting a double-gating mechanism for TRPC that may be involved in MCF breast cancer cell proliferation and invasion.
Materials and methods
Cell culture
MCF-7 cells were grown in DMEM medium containing 10% fetal calf serum and 1% penicillin/streptomycin serum as described (9). Cells were plated onto ø13-mm coverslips and used when 60–70% confluent.
Calcium imaging
The growth medium was removed and cells were rinsed once in Earle’s balanced salts solution (EBSS; Invitrogen). Calcium-green of 50 μg AM (C3012; Invitrogen) or Fura-2 AM (F1221; Invitrogen) were dissolved in 20 μl 20% pluronic acid in DMSO (0.01 g in 50 μl DMSO stock). Before the experiment, mixtures of 1 μl dye preparation in 200 μl EBSS was applied and cells were incubated for 60 min. Prior to placing the coverslip into the recording chamber, coverslips were rinsed in EBSS to remove residual dye. Data acquisition and analysis were performed via OpenLab v.3.1.7 (Improvision Ltd., Coventry, UK). A CCD camera (ORCA-AG; Hamamatsu Ltd., Japan) was used to capture the fluorescent image by using Fura-2-AM and calcium green. In the experiments performed using Fura-2, fluorescent intensities were measured with dual-sequential-wavelength excitation at 340 and 380 nm, and emission at 510 nm. Changes in Ca2+ concentration were expressed as ratios of 340/380. Fluorescent intensity of calcium green-1 was measured with a single wavelength excitation at 488 nm and emission at 528 nm. Changes in the Ca2+ concentration were expressed as ΔF/F, where F was the fluorescence intensity when cells were at rest, and ΔF was the change in fluorescence during stimulation.
iRNA and plasmid of hCOX2
Stealth siRNA (Invitrogen) was obtained from Invitrogen. MCF-7 cells were passaged onto coverslips in 500 μl Opti-MEM (Invitrogen) one day before transfection and reached about 40–50% confluence at the time of transfection. siRNA of 20 pmol (against TRPC3) or the siRNA negative control complex, with a 1:125 final dilution of Lipofectamine 2000 (Invitrogen) was used according to the manufacturer’s instructions. The knockdown effects were examined at 48 h and the results were compared with control and control without knockdown. Results were collected from 3 different batches of MCF-7 cells. Human hCOX2 plasmids were obtained from Professor R. Kulmacz (University of Texas Health Science Center at Houston). Cells were transfected with hCOX2 by Lipofectamine 2000. The effects of transfection were examined by western blot analysis at 24 and 48 h.
RT-PCR and immunostaining
RT-PCR experiments followed standard protocols. Primers were designed with primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) for TRPC1 (NM_003304/92 bp), TRPC3 (NM_003305/157 bp), TRPC4 (NM_016179/191 bp), TRPC5 (NM_012471/108 bp), TRPC7 (NM_020389/135 bp) and the α1C subunit (NM_000719/194 bp), α1G subunit (AH_007322/135 bp) and α1H subunit of VGCCs (NM_021098/123 bp). Antibodies against TRPC1, 3, 4 and 5 were the kind gift from Professor W.P. Schilling (Case Western University, Cleveland, OH, USA). The peptide sequence (26) used to generate the antibody against TRPC3 was RRRRLQKDIEMGMGN.
Cell cycle analysis
After removal of methanol, cells were treated with a Coulter DNA-Prep reagent kit (Beckman-Coulter, France). Cells were resuspended in 40 μl of a lysing and permeabilizing reagent and 400 μl of a propidium iodide solution containing RNAse. Flow cytometry analyses were performed using a Coulter Epics Elite ESP flow cytometer (Beckman-Coulter) equipped with a 488 nm argon laser running at 15 mW. The red DNA fluorescence signal was analyzed as total (area) vs. peak signal, in order to eliminate doublets and aggregates. Data were recorded for at least 10,000 events. Cell cycle distribution was analyzed with the Multicycle-AV software (Phoenix Flow Systems, San Diego, CA, USA).
Cell survival and proliferation
Cell proliferation was determined using the tetrazolium salt reduction method (MTT). Cells were seeded at 4×104 cells/well on a 24-well plate (for a given condition on three separate experiments) and allowed to start growth for 48 h. Drugs and AA were added for 24 and 48 h at the concentrations indicated in the figure legends. Cells were incubated at 37°C with the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] and metabolically active cells reduced the dye to purple formazan. After 45 min of incubation at 37°C, the medium was discarded and MTT formazan crystals were solubilized with DMSO. Absorbance was measured in a multiwell plate spectrophotometer at 570 nm (Molecular Devices model Thermomax microplate reader, Les Ulis, France). For easier comparison between conditions, results obtained for proliferation were normalized. The means were then calculated on the daily calculated ratios. The mean of each triplicate was used to create a data point for comparing cell growth in different conditions.
Cell migration/invasion in vitro
Invasion and migration were analyzed in BD Falcon 24-well plates with 8-μm pore size polyethylene terephtalate membrane BD BioCoat cell culture inserts. For the invasion assay, the membrane was covered with a film of Matrigel (Becton-Dickinson). The upper compartment was seeded with 4×104 viable cells in DMEM with 5% FBS ± drugs/AA and the lower compartment was filled with DMEM supplemented with 10% FBS as a chemo-attractant ± drugs/AA. After 48 h at 37°C, cells remaining on the upper side on the membrane were removed with a cotton swab and the cells that had migrated and were attached to the lower side were stained with hematoxylin for 2 min and counted on the total surface of the insert using light microscopy at x200 magnification.
Solution and chemicals
EBSS for calcium imaging recording contained: NaCl 116.3 mM, NaH2PO4 1 mM, KCl 5.3 mM, MgCl2 1 mM, CaCl2 1.8 mM, NaHCO3 26 mM, D-glucose 5.5 mM, HEPES 10 mM. The EGTA solution for calcium imaging recording contained: NaCl 116.3 mM, NaH2PO4 1 mM, KCl 5.3 and 1.8 mM, NaHCO3 26 mM, D-glucose 5.5 mM, HEPES 10 mM, EGTA 0.2 mM. All solutions were prepared fresh on the day of experimentation.
Chemicals dissolved in ethanol or DMSO were made up as 1,000 times stock solutions. All 1X chemical solutions were made with an appropriate bath solution on the day of experimentation. Solutions such as TG, AA, ETYA, were kept in the dark and on ice and added into a syringe which led to the recording chamber when necessary. The solvent, ethanol at the same dilution, was tested alone in controls and had no effects on calcium entry evoked either by store depletion or OAG stimulation.
Results
TRPC3 and the α1H subunit of the voltage gated calcium channel (VGCC) expressed in MCF-7 breast cancer cells
Expression of the TRPC3, α1H (T type) but not the α1C or α1G subunits of the voltage-gated calcium channel (VGCC) was observed in MCF-7 cells (Fig. 1).
Figure 1.
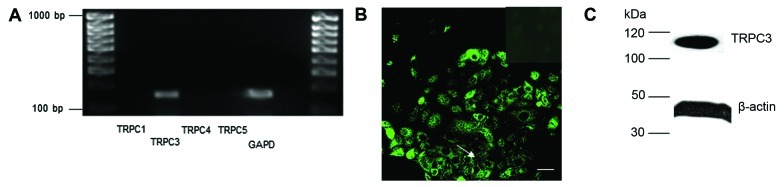
Expression of TRPC3 in MCF-7 cells. (A) The expression of TRPC channels in MCF-7 cells was examined by RT-PCR. The PCR reaction yielded RNA transcript, of expected size, for TRPC3 but not TRPC1, 4 or 5. GAPD positive controls were found in each sample. (B) Immunostaining of TRPC3 in MCF-7 breast cancer cells. TRPC3 expression was seen throughout the cell cytoplasm and cell membrane. Arrows indicate location of antibody in cell membrane. Inset at the top right corner shows peptide negative control (26). Scale bar, 40 μm. (C) Western blot analyses showing the presence of TRPC3 protein in MCF-7 breast cancer cells.
The spatial distribution of TRPC3 protein in MCF-7 cells was determined by immunohistochemistry, as previously reported in other cell types (27). Abundant, non-discrete TRPC3 expression throughout the cell cytoplasm and at the cell surface was observed (Fig. 1B). Western blot analysis consistently demonstrated abundant expression of TRPC3 in MCF-7 cells (Fig. 1C).
Native TRPC3 in MCF-7 breast cancer cells activated by DAG and store depletion
Recombinant human TRPC3 can be directly activated by diacylglycerol (DAG) (28,29). In MCF-7 cells, OAG (100 μM), a synthetic DAG, induced a significant intracellular Ca2+ elevation in an extracellular calcium-dependent manner (Fig. 2). Recent reports have confirmed that TRPC3 could also mediate store operated calcium entry (SOCs) (30). Activation of native TRPC3 via store depletion by SERCA pump inhibitors, such as TG, has been reported in HSG (31) and epithelial cells (32). In MCF-7 cells, store depletion by TG (1 μM) and EGTA bath led to significant Ca2+ elevations (Fig. 2A).
Figure 2.

Polyunsaturated fatty acids inhibited Ca2+ entry through TRPC3 channels. (A) Ca2+ entry was evoked by store depletion with thapsigargin (TG, 1 μM) and EGTA (blue line). 2-APB (pink line, 100 μM) induced a transient Ca2+ elevation following a rapid decrease. [Ca2+]i elevation was inhibited by arachidonic acid (AA, yellow line, 10 μM), linonic acid (LA, 10 μM) and ETYA (brown line, 20 μM). (B) The dashed horizontal line indicates the basal Ca2+ level. TG (1 μM) elevated cellular Ca2+ levels that were rapidly reduced to below basal levels by wash out with EGTA (200 μM). Application of arachidonic acid (AA, 10 μM) prevented the overshooting response seen in (A) and only returned Ca2+ to the basal level, indicating a block of the TRPC-mediated Ca2+ entry pathway by AA. Wash off of AA with EBSS caused an overshooting response to occur that could be blocked, as previously, by application of AA (10 μM). (C) Log dose response curve of varying concentrations of AA upon the OAG-induced Ca2+ elevation. Responses were quantified as the area under the Ca2+ response curve and normalized by determining the area in the presence of AA, expressed as a fraction of the control response. Curve fit was with a single sigmoidal function. The inlet shows an example of different concentration-dependent effects of AA upon Ca2+ entry elicited by OAG (50 μM). (D) Western blot analysis demonstrates that protein levels of TRPC3 channels gradually diminished after 48 h. (E) Ca2+ entry evoked by store depletion was significantly reduced 48 h after incubation of MCF-7 cells with RNAi against TRPC3, . Results were averaged from 3 coverslips except where indicated.
AA and LA directly inhibited calcium entry via native TRPC3
The Ca2+ elevation induced by store depletion was almost quenched by bath application of 10 μM AA (Fig. 2A, yellow line) or 10 μM LA (Fig. 2A, blue line). 2APB, as a common antagonist of TRPC3, at the concentration of 100 μM, caused a transient calcium peak followed by decay (Fig. 2A, pink line) to a level similar to that in the presence of AA or LA. In addition, the CCE following Ca2+ replacement after EGTA, could be prevented by 10 μM AA and CCE was observed after removal, by washout, of the AA inhibition (Fig. 2B).
Effect of PUFA on TRPC3 not via the degenerated products of PUFA
Exogenous AA can freely penetrate membranes, but will be challenged and degraded by endogenous intracellular cyclooxygenases (COX) or lipooxygenases (LOX). To exclude the possibility that the inhibition effect is due to degenerated metabolites of AA rather than AA itself, 14-eicosatetraynoic acid (ETYA, 20 μM), a competitive analogue of AA resistant to LOX and other degradative enzymes, was used to mimic the effect of AA. ETYA, like AA, inhibited CCE induced by store depletion (Fig. 2A, brown line), suggesting that PUFA interacts directly with TRPC channels.
The effect of fatty acids on ion channels is sometimes attributed to non-specific influences upon cell membranes (33,34). Polyunsaturated fatty acids may, for example, increase membrane fluidity. AA at a concentration between 25–100 μM directly affected channel proteins via a change of the lipid environment (34). We observed that application of 500 μM AA induced a massive, irreversible Ca2+ elevation, presumably following the collapse of cell integrity. We therefore maintained PUFA concentrations below the threshold (15 μM) for such effects. Additionally, CCE was also inhibited by the double-bonds unsaturated fatty acid, e.g. LA (5 and 10 μM) and the mono-bond unsaturated fatty acid, oleic acid (10 μM). However, the saturated fatty acid, stearic acid (20 μM), had no effect upon induced CCE.
To confirm that CCE in MCF-7 cells was mediated via TRPC3 channels, we examined Ca2+ entry in MCF-7 cells by knocking down TRPC3 (shealth RNAi; Invitrogen) (Fig. 2D and E). A negative control iRNA was utilized as a control to discount the non-specific effect of TRPC3 iRNA. Transfected cells gradually lost the TRPC3 channels and after 48 h, detectable TRPC3 protein was almost eliminated (Fig. 2D). In parallel to the decrease of TRPC3 protein, calcium elevation evoked by store depletion was also significantly reduced; suggesting that calcium elevation induced by store depletion was mediated via the TRPC3 channels.
Dose response of AA inhibition upon calcium entry via TRPC3
Ca2+ entry in MCF-7 cells could be evoked by application of OAG (Fig. 2C). AA in a range from 0.1 to 20 μM was employed to study the inhibitory effect of AA on OAG induced Ca2+ elevation. A single, sigmoidal dose-response curve was fitted according to data (Fig. 2C) and gave an IC50 of AA upon OAG induced Ca2+ elevation of 1.78±0.17 μM.
Regulation of endogenous AA in MCF-7 and its effect on Ca2+ entry via TRPC3
There are generally two principal sources in AA generation, i) cytosolic phospholipase A2 (PLA2) releasing AA from appropriate phospholipids (35–37) and ii) fatty acid amide hydrolase (FAAH) degenerating endogenous anandamide and relatives into AA. The AA metabolism diverges down two main pathways, the COX and LOX pathways (38).
cPLA2, FAAH and COX2 were demonstrated to be present in MCF-7 breast cancer cells by western blot analyses (Fig. 3A). Accordingly, reduction of endogenous PUFA could be achieved by either inhibition of cPLA2, FAAH or overexpression of COX2. Inhibition COX2 or LOX could induce the elevation of endogenous PUFA. Consistent with our results above, reduction of endogenous PUFA by the cPLA2 antagonist (AACOCF3), FAAH antagonist (PMSF), and overexpression of COX2 significantly enhanced Ca2+ entry via TRPC3. Niflumic acid, the antagonist of COX2, reduces degeneration of PUFA, resulting in elevation of endogenous PUFA. Ca2+ entry via TRPC3 was reduced by pre-incubation with niflumic acid (Fig. 3D).
Figure 3.

Modulation of endogenous PUFA and its effect on TRPC3 mediated Ca2+ entry. (A) Western blot analyses demonstrate the presence of fatty acid hydoxylase (FAAH), cytoplasmic phospholipase A (cPLA) and COX2 in MCF-7 breast cancer cells. (B) COX2 protein levels were dramatically increased (almost 3-fold) 24 h after cells were transfected with COX2 plasmid. (C) Ca2+ entry evoked by store depletion was enhanced from control (pink line) by reduction of AA generation e.g. AACOCF3 (yellow line, 10 μM), COX2 expression (blue line). Nifedipine (purple line, 20 μM) did not affect Ca2+ entry but niflumic acid (blue line, 100 μM) as a COX2 inhibitor, slightly reduced the Ca2+ entry induced by store depletion. (D) All results were normalized to the controls. AACOCF3, PMSF (50 μM), and overexpression of COX2 significantly enhanced Ca2+ entry via activated TRPC induced by store depletion. Meanwhile niflumic acid reduced Ca2+ entry via TRPC induced by store depletion. Indomethacin and nifedipine did not affect Ca2+ entry via TRPC3.
Effect of TRPC antagonism on MCF-7 cell proliferation and invasion
The common antagonist of TRP channels, 2-APB, was used to investigate the effects of these channels on the proliferation of MCF-7 breast cancer cells. 2-APB (50 μM) significantly reduced the S phase of the cell cycle (Fig. 4A-i), and cell proliferation (MTT test; 100 μM), whilst D609, an inhibitor of phospholipase C, had no effect (Fig. 4A-ii). 2-APB also reduced MCF-7 cell migration and invasion in a dose-dependent manner (Fig. 4A-iii and iv). The effects of increasing concentration of AA on cell migration are shown in Fig. 4B. Taken together, these results suggest that cellular AA inhibits Ca2+ entry via TRPC3, thus interfering with the Ca2+ requirement for cell proliferation and invasion.
Figure 4.

(A) Antagonists of TRPC and AA interfer with the cell cycle and the migration of MCF-7 breast cancer cells. (i) 2-APB caused a dose-dependent and selective decrease in the S phase of the MCF-7 cell cycle compared to the vehicle control. (ii) D609 had no effect on the cell cycle parameters measured. (iii) 2-APB caused a significant decrease in the migration of MCF-7 cells. (iv) 2-APB caused a dose-dependent and significant decrease in the invasion of MCF-7 cells. (B) The normalized bar figure shows the effects of different concentrations of AA on cell migration.
Discussion
In this study, we found that TRPC3 channels were highly expressed in MCF-7 breast cancer cells. Not only DAG but also store depletion activated TRPC3 to mediate Ca2+ entry. [Ca2+]i was thereafter crucial to determine breast cancer cell proliferation and migration. PUFAs, such as AA and LA, but not saturated fatty acids, inhibited Ca2+ entry mediated by TRPC3. Channel activation and removal of PUFAs were required to allow Ca2+ entry via TRPC. Endogenous local PUFAs regulated by cellular generation and degeneration pathways therefore played an important role in mediating [Ca2+]i via TRPC. PUFAs as well as the TRPC3 antagonist attenuated breast cancer cell proliferation and migration, suggesting a mechanism in which PUFAs restrain breast cancer via inhibition of TRPC channels.
TRP channels play a key role in the regulation of intracellular Ca2+ in multiple cell types e.g. sperm (39), smooth muscle (40) and prostate cancer cells (41) in response to multiple stimuli. At least four of the TRPC family members (TRPC1, 2, 4 and 5) are known to be activated by store depletion, while TRPC3 and 6 are generally regarded to be activated by DAG (28,29). However, native TRPC3 in different cells e.g. avian pre-B cells (42), ROS 17/2.8 (43) and PS1 prostate cancer cells (44) are also reported to be activated by store depletion. The proportion of activation in these cells, induced by DAG or by store depletion, may vary with different levels of TRPC3 expression. It has been reported that expression levels of the TRPC3 channel might determine the actual activation mechanism (45). Inconsistent to previous studies, we found that TRPC3 in MCF-7 breast cancer cells are activated by DAG as well as by store depletion. The expression of TRP in MCF-7 cells may vary (18) due to the culture methods. In hBDC, TRPC1, TRPC6, TRPM7, TRPM8 and TRPV6 channels were overexpressed in hBDA compared to the adjacent non-tumoral tissue. TRPC1, TRPM7 and TRPM8 expression strongly correlated with proliferative parameters, and TRPV6 was mainly overexpressed in invasive breast cancer cells (18). However, in the MCF-7 cell line in our laboratory, TRPC1 was not expressed.
PUFAs have been found to directly activate Drosophila TRP and TRPL (24) as well as TRPV in vascular endothelial cells (25). However, there is little evidence to show that PUFA directly activates native TRPC in mammalian tissues rather than in expression systems. Interestingly, Ca2+ entry mediated by AA has been reported (46–48), but it is ascribed to a novel receptor-activated calcium entry pathway, ARC (49,50), as there appeared to be mutual antagonism of CCE and ARC (51–53). In our experiment, the application of PUFA alone did not cause calcium elevation, and selective TRPC3 iRNA inhibited the calcium elevation in response to store depletion. We therefore ruled out the possibility of calcium entry via ARC. Other studies on the effect of AA on voltage-gated Kv channels suggest that AA closed Kv channels by introducing conformational alternations in selective filter regions of the channel (54). How PUFAs interact with the TRPC3 protein within internal membranes, at present, is still unclear.
The lack of specific antagonists for TRP channels creates difficulties in determining the molecular basis of TRP channels. 2-APB has a broad effect on TRPC, IP3 and TRPV. The silent response of MCF-7 cells to CAP, the most potent TRPV agonist, ruled out the possible involvement of TRPV in the mediation of calcium entry in MCF-7 cells. Consistently, western blot analyses against TRPV1 in MCF-7 cells failed to detect the expected protein, compared to positive controls from rat brain. In addition, the potent inhibition of RNAi against TRPC3 on the Ca2+ entry induced by store depletion suggests that the inhibition of AA on Ca2+ entry occurred via its effect on TRPC3.
The concentration of free AA in resting cells is universally described as low but varies from cell to cell, AA concentrations in the resting leukocyte have been measures at 0.5–1 μM as opposed to at 15 μM in the islets of Langerhans (55). This resting concentration can be varied dynamically by either increases, for example via activation of G-protein coupled receptors and phospholipase A2 (56), or decreases via AA degenerative pathways mediated principally by COX and LOX. As there are multiple buffer systems for AA, the free intracellular AA concentrations correlating with the bath AA concentration are not easily determined.
As expected, reduction of endogenous AA by inhibition of cPLA2, FAAH and overexpression of COX2 increase Ca2+ entry evoked by store depletion. However, the effect of increasing endogenous AA by inhibition of COX2 and/or LOX was not of significance, suggesting some unknown mechanism. In addition, inhibition of FAAH was complicated, because AEA alone causes calcium elevations (data not shown).
In addition, COX2 has been shown to be overexpressed in a variety of malignant tumors (6,7) and linked to apoptosis resistance (9), invasive tumor behavior (10) and the poor prognosis of breast cancer (12). Our findings suggest a mechanism whereby AA, through direct inhibition of Ca2+ entry via TRPC3 channels may play a role in cancer cell proliferation and invasion.
In conclusion, we have demonstrated that polyunsaturated fatty acids directly inhibit TRPC3 in MCF-7 cells and is a potent mechanism for regulating Ca2+ entry and [Ca2+]i. Our data suggests a role for such regulation in breast cancer metastasis. Thus, TRPC3 appears as a new mediator of breast cancer cell migration/invasion and represents a potential target for a new class of anticancer agents.
Acknowledgments
This study was supported by grants from the Scientific Research Projects of Scientific and Technology Bureau of Shanghai (09411952800) to H.Z. This study was partly supported in part by the Dean’s Initiative Fund, University of Birmingham and BBSRC to Y.G.
Abbreviations:
- TRP
transient receptor potential;
- PUFA
polyunsaturated fatty acids;
- CCE
capacitative Ca2+ entry;
- VGCC
voltage-gated calcium channel;
- SER
smooth endoplasmic reticulum;
- TG
tharpsigargin;
- AA
arachidonic acid;
- LA
linoleic acid;
- COX
cyclooxygenases;
- LOX
lipooxygenases;
- ETYA
14-eicosatetraynoic acid;
- EPA
eicosapentaenoic acid;
- DHA
docosahexaenoic acid.
References
- 1.Weigelt B, van’t Veer LJ. Hard-wired genotype in metastatic breast cancer. Cell Cycle. 2004;3:756–757. doi: 10.4161/cc.3.6.926. [DOI] [PubMed] [Google Scholar]
- 2.Noguchi M, Rose DP, Earashi M, Miyazaki I. The role of fatty acids and eicosanoid synthesis inhibitors in breast carcinoma. Oncology. 1995;52:265–271. doi: 10.1159/000227471. [DOI] [PubMed] [Google Scholar]
- 3.Chamras H, Ardashian A, Heber D, Glaspy JA. Fatty acid modulation of MCF-7 human breast cancer cell proliferation, apoptosis and differentiation. J Nutr Biochem. 2002;13:711–716. doi: 10.1016/s0955-2863(02)00230-9. [DOI] [PubMed] [Google Scholar]
- 4.De Petrocellis L, Melck D, Palmisano A, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yee LD, Young DC, Rosol TJ, Vanbuskirk AM, Clinton SK. Dietary (n-3) polyunsaturated fatty acids inhibit HER-2/neu-induced breast cancer in mice independently of the PPARγ ligand rosiglitazone. J Nutr. 2005;135:983–988. doi: 10.1093/jn/135.5.983. [DOI] [PubMed] [Google Scholar]
- 6.Singh B, Lucci A. Role of cyclooxygenase-2 in breast cancer. J Surg Res. 2002;108:173–179. doi: 10.1006/jsre.2002.6532. [DOI] [PubMed] [Google Scholar]
- 7.Zha S, Gage WR, Sauvageot J, et al. Cyclooxygenase-2 is up-regulated in proliferative inflammatory atrophy of the prostate, but not in prostate carcinoma. Cancer Res. 2001;61:8617–8623. [PubMed] [Google Scholar]
- 8.Horia E, Watkins BA. Complementary actions of docosahexaenoic acid and genistein on COX-2, PGE2 and invasiveness in MDA-MB-231 breast cancer cells. Carcinogenesis. 2007;28:809–815. doi: 10.1093/carcin/bgl183. [DOI] [PubMed] [Google Scholar]
- 9.Liou JY, Aleksic N, Chen SF, Han TJ, Shyue SK, Wu KK. Mitochondrial localization of cyclooxygenase-2 and calcium-independent phospholipase A(2) in human cancer cells: Implication in apoptosis resistance. Exp Cell Res. 2005;306:75–84. doi: 10.1016/j.yexcr.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Arun B, Goss P. The role of COX-2 inhibition in breast cancer treatment and prevention. Semin Oncol. 2004;31:22–29. doi: 10.1053/j.seminoncol.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 11.Murff HJ, Shu XO, Li H, et al. Dietary polyunsaturated fatty acids and breast cancer risk in Chinese women: a prospective cohort study. Int J Cancer. 2011;128:1434–1441. doi: 10.1002/ijc.25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denkert C, Winzer KJ, Hauptmann S. Prognostic impact of cyclooxygenase-2 in breast cancer. Clin Breast Cancer. 2004;4:428–433. doi: 10.3816/cbc.2004.n.006. [DOI] [PubMed] [Google Scholar]
- 13.Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol. 2004;559:685–706. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilius B. Store-operated Ca2+ entry channels: still elusive! Sci STKE 2004. 2004:e36. doi: 10.1126/stke.2432004pe36. [DOI] [PubMed] [Google Scholar]
- 15.Montell C. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. 2001 doi: 10.1126/stke.2001.90.re1. Sci STKE 2001: re1. [DOI] [PubMed] [Google Scholar]
- 16.Sydorenko V, Shuba Y, Thebault S, et al. Receptor-coupled, DAG-gated Ca2+-permeable cationic channels in LNCaP human prostate cancer epithelial cells. J Physiol. 2003;548:823–836. doi: 10.1113/jphysiol.2002.036772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Hiani Y, Ahidouch A, Roudbaraki M, Guenin S, Brule G, Ouadid-Ahidouch H. Calcium-sensing receptor stimulation induces nonselective cation channel activation in breast cancer cells. J Membr Biol. 2006;211:127–137. doi: 10.1007/s00232-006-0017-2. [DOI] [PubMed] [Google Scholar]
- 18.Dhennin-Duthille I, Gautier M, Faouzi M, et al. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell Physiol Biochem. 2011;28:813–822. doi: 10.1159/000335795. [DOI] [PubMed] [Google Scholar]
- 19.Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- 20.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 21.Mottola A, Antoniotti S, Lovisolo D, Munaron L. Regulation of noncapacitative calcium entry by arachidonic acid and nitric oxide in endothelial cells. FASEB J. 2005;19:2075–2077. doi: 10.1096/fj.05-4110fje. [DOI] [PubMed] [Google Scholar]
- 22.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 23.Nuttle LC, Ligon AL, Farrell KR, Hester RL. Inhibition of phospholipase A2 attenuates functional hyperemia in the hamster cremaster muscle. Am J Physiol. 1999;276:H1289–H1294. doi: 10.1152/ajpheart.1999.276.4.H1289. [DOI] [PubMed] [Google Scholar]
- 24.Chyb S, Raghu P, Hardie RC. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 26.Goel M, Sinkins WG, Schilling WP. Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem. 2002;277:48303–48310. doi: 10.1074/jbc.M207882200. [DOI] [PubMed] [Google Scholar]
- 27.Buniel MC, Schilling WP, Kunze DL. Distribution of transient receptor potential channels in the rat carotid chemosensory pathway. J Comp Neurol. 2003;464:404–413. doi: 10.1002/cne.10798. [DOI] [PubMed] [Google Scholar]
- 28.Zitt C, Obukhov AG, Strubing C, et al. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J Cell Biol. 1997;138:1333–1341. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 30.Kaznacheyeva E, Glushankova L, Bugaj V, et al. Suppression of TRPC3 leads to disappearance of store-operated channels and formation of a new type of store-independent channels in A431 cells. J Biol Chem. 2007;282:23655–23662. doi: 10.1074/jbc.M608378200. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem. 2005;280:21600–21606. doi: 10.1074/jbc.C400492200. [DOI] [PubMed] [Google Scholar]
- 32.Bandyopadhyay BC, Swaim WD, Liu X, Redman RS, Patterson RL, Ambudkar IS. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J Biol Chem. 2005;280:12908–12916. doi: 10.1074/jbc.M410013200. [DOI] [PubMed] [Google Scholar]
- 33.Meves H. Modulation of ion channels by arachidonic acid. Prog Neurobiol. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt H, Meves H. Modulation of neuronal calcium channels by arachidonic acid and related substances. J Membr Biol. 1995;145:233–244. doi: 10.1007/BF00232715. [DOI] [PubMed] [Google Scholar]
- 35.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 36.Ackermann EJ, Kempner ES, Dennis EA. Ca(2+)-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J Biol Chem. 1994;269:9227–9233. [PubMed] [Google Scholar]
- 37.Dennis EA. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem Sci. 1997;22:1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- 38.Steele VE, Hawk ET, Viner JL, Lubet RA. Mechanisms and applications of non-steroidal anti-inflammatory drugs in the chemoprevention of cancer. Mutat Res. 2003;523–524:137–144. doi: 10.1016/s0027-5107(02)00329-9. [DOI] [PubMed] [Google Scholar]
- 39.Jungnickel MK, Marrero H, Birnbaumer L, Lemos JR, Florman HM. Trp2 regulates entry of Ca2+ into mouse sperm triggered by egg ZP3. Nat Cell Biol. 2001;3:499–502. doi: 10.1038/35074570. [DOI] [PubMed] [Google Scholar]
- 40.Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circ Res. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- 41.Vanden Abeele F, Lemonnier L, Thebault S, et al. Two types of store-operated Ca2+ channels with different activation modes and molecular origin in LNCaP human prostate cancer epithelial cells. J Biol Chem. 2004;279:30326–30337. doi: 10.1074/jbc.M400106200. [DOI] [PubMed] [Google Scholar]
- 42.Putney JW, Jr, Trebak M, Vazquez G, Wedel B, Bird GS. Signalling mechanisms for TRPC3 channels. Novartis Found Symp. 258:123–133. discussion 133–139, 155–159, 263–266, 2004. [PubMed] [Google Scholar]
- 43.Baldi C, Vazquez G, Calvo JC, Boland R. TRPC3-like protein is involved in the capacitative cation entry induced by 1alpha,25-dihydroxy-vitamin D3 in ROS 17/2.8 osteoblastic cells. J Cell Biochem. 2003;90:197–205. doi: 10.1002/jcb.10612. [DOI] [PubMed] [Google Scholar]
- 44.Thebault S, Zholos A, Enfissi A, et al. Receptor-operated Ca2+ entry mediated by TRPC3/TRPC6 proteins in rat prostate smooth muscle (PS1) cell line. J Cell Physiol. 2005;204:320–328. doi: 10.1002/jcp.20301. [DOI] [PubMed] [Google Scholar]
- 45.Vazquez G, Wedel BJ, Trebak M, St John Bird G, Putney JW., Jr Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem. 2003;278:21649–21654. doi: 10.1074/jbc.M302162200. [DOI] [PubMed] [Google Scholar]
- 46.Shuttleworth TJ. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J Biol Chem. 1996;271:21720–21725. doi: 10.1074/jbc.271.36.21720. [DOI] [PubMed] [Google Scholar]
- 47.Shuttleworth TJ, Thompson JL. Muscarinic receptor activation of arachidonate-mediated Ca2+ entry in HEK293 cells is independent of phospholipase C. J Biol Chem. 1998;273:32636–32643. doi: 10.1074/jbc.273.49.32636. [DOI] [PubMed] [Google Scholar]
- 48.Mignen O, Shuttleworth TJ. I(ARC), a novel arachidonate-regulated, noncapacitative Ca(2+) entry channel. J Biol Chem. 2000;275:9114–9119. doi: 10.1074/jbc.275.13.9114. [DOI] [PubMed] [Google Scholar]
- 49.Mignen O, Thompson JL, Shuttleworth TJ. Ca2+ selectivity and fatty acid specificity of the noncapacitative, arachidonate-regulated Ca2+ (ARC) channels. J Biol Chem. 2003;278:10174–10181. doi: 10.1074/jbc.M212536200. [DOI] [PubMed] [Google Scholar]
- 50.Shuttleworth TJ, Thompson JL, Mignen O. ARC channels: a novel pathway for receptor-activated calcium entry (Review) Physiology (Bethesda) 2004;19:355–361. doi: 10.1152/physiol.00018.2004. [DOI] [PubMed] [Google Scholar]
- 51.Luo D, Broad LM, Bird GS, Putney JW., Jr Mutual antagonism of calcium entry by capacitative and arachidonic acid-mediated calcium entry pathways. J Biol Chem. 2001;276:20186–20189. doi: 10.1074/jbc.M100327200. [DOI] [PubMed] [Google Scholar]
- 52.Mignen O, Thompson JL, Shuttleworth TJ. Reciprocal regulation of capacitative and arachidonate-regulated noncapacitative Ca2+ entry pathways. J Biol Chem. 2001;276:35676–35683. doi: 10.1074/jbc.M105626200. [DOI] [PubMed] [Google Scholar]
- 53.Huang GN, Zeng W, Kim JY, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 54.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 55.Brash AR. Arachidonic acid as a bioactive molecule. J Clin Invest. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun GY, Xu J, Jensen MD, et al. Phospholipase A2 in astrocytes: responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Mol Neurobiol. 2005;31:27–42. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]