

Abstract
Smooth muscle cells (SMCs) contract to perform many physiological functions, including regulation of blood flow and pressure in arteries, contraction of the pupils, peristalsis of the gut and voiding of the bladder. SMC lineage in these organs is characterized by cellular expression of the SMC isoform of α-actin, encoded by the ACTA2 gene. We report here on a unique and de novo mutation in ACTA2, R179H, that causes a syndrome characterized by dysfunction of SMCs throughout the body, leading to aortic and cerebrovascular disease, fixed dilated pupils, hypotonic bladder, malrotation and hypoperistalsis of the gut and pulmonary hypertension.
Keywords: ACTA2, alpha-actin, smooth muscle cell, thoracic aortic aneurysm, moyamoya disease, congenital mydriasis
INTRODUCTION
The walls of arteries and hollow visceral organs, including the stomach, colon, bladder, airways and uterus, are lined with smooth muscle cells (SMCs) that contract to perform these organs’ various functions. These contractile cells are also found in the irises of the eye, the hair follicles and lacrimal ducts. Although the SMCs found in these various organs differ in terms of their embryonic origin and physiologic properties, the differentiation into functional SMCs is defined by expression of contractile proteins, including the SMC-specific isoform of the contractile protein α-actin (ACTA2) [Martin et al., 2007]. While this isoform is the primary actin in vascular SMCs, the SMC-specific muscular δ-actin (ACTG2) is the predominant isoform expressed in intestinal, urogenital, and respiratory SMCs [Gabbiani et al., 1981].
Heterozygous missense mutations in ACTA2 cause a predisposition to a variety of vascular diseases, including thoracic aortic aneurysms and dissections, early onset coronary artery disease (CAD) and strokes [Guo et al., 2007; Guo et al., 2009; Morisaki et al., 2009]. In families with inherited ACTA2 mutations, carriers can present with aortic disease, CAD or strokes, but none of the mutations identified are fully penetrant for any of these vascular diseases. Interestingly, correlations have emerged between specific ACTA2 mutations and particular vascular diseases. For example, R118Q and R149C predispose primarily to aortic disease and early onset CAD. In contrast, ACTA2 mutations altering R258 are associated with aortic disease, patent ductus arteriosus (PDA) and very early onset cerebrovascular disease, including Moyamoya disease, which is an early onset stroke syndrome characterized by bilateral occlusion of the distal internal carotid arteries and collateral vessel formation [Scott and Smith, 2009].
In mice, deficiency of ACTA2 leads to normal development but decreased contractility of arterial segments and hypotension [Schildmeyer et al., 2000]. It has been proposed that ACTA2 missense mutations in humans disrupt α-actin polymerization and lead to decreased contractility of aortic SMCs, and this dysfunction leads to thoracic aortic disease [Milewicz et al., 2008]. Accumulating evidence suggests a different etiology for the occlusive disease of smaller vessels associated with ACTA2 mutations. Examination of coronary and intracardiac arteries, and arteries in the vasa vasorum of the aorta from ACTA2 mutation patients shows increased number of SMCs located in the intimal or medial layers leading to a reduction in lumen diameter [Guo et al., 2009]. In addition, SMCs harboring ACTA2 mutations proliferate more rapidly than control SMCs in vitro. Taken together, these data suggest that mutations in SMC α-actin have a gain of function effect leading to increased vascular SMC proliferation and, consequently, the stenosis or occlusion of arteries.
We report here patients with a de novo mutation in ACTA2 not previously identified in our cohort of families with aortic disease. These patients have a unique clinical syndrome characterized by SMC dysfunction in organs throughout the body, including decreased contractile function in the iris, bladder and gastrointestinal tract, and SMC proliferative lesions in the vasculature. In addition, this ACTA2 mutation appears to represent the most severe end of the spectrum of ACTA2 mutations based on the very early onset and highly penetrant vascular diseases in affected patients.
MATERIALS AND METHODS
The study protocol was approved by the Institutional Review Board of the University of Texas Health Science Center at Houston. Three families (proband and parents) had genetic testing at a clinical laboratory. Two families had testing in our research laboratory. DNA sequencing was performed according to protocol described previously [Guo et al., 2007]. Pathology slides of the aorta were obtained for two patients (C and D, Table I) and of the lungs (patient D) and stained with Movat and Trichome, respectively. Medical records including imaging studies, surgical reports, pathology reports and physicians’ notes were obtained and reviewed.
Table I.
Clinical characteristics of patients with the ACTA2 R179H mutation.
| Patient | A | B | C | D | E | |
|---|---|---|---|---|---|---|
| Age | d. 11 y.o.a | 14 y.o. | 17 y.o. | 11 y.o. | 26 y.o. | |
|
| ||||||
| Gender | female | male | male | female | female | |
|
| ||||||
| CV | PDA, age at repair | +, 5 months | +, 20 days | +, 18 months | +, 4 months | +, 4 months |
| TAA, age at repair | + | + | +, 12 yrs | +, 10 yrs | +, 11 yrs | |
| Dilated pulmonary arteries |
+ | + | + | |||
| Other CV findings | Dilatation of aortic arch, brachiocephalic, common carotid and left subclavian arteries, ASD |
Left brachial artery AVM |
Aortic coarctation | Dilatation of aortic arch, suprarenal abdominal aorta, brachiocephalic and common carotid arteries |
||
| Bilateral stenoses of the terminal ICA |
+ | + | + | + | + | |
| Bilateral dolichoectatic ICA |
+ | + | + | + | + | |
| Bilateral periventricular WMH |
+ | + | + | + | + | |
| Other CNS findings | Microaneurysms in the retina |
Colpocephaly, basilar artery stenosis |
Posterior communicating arteries and right ophthalmic artery occlusion |
ACA and MCA distribution infarctions |
||
|
| ||||||
| Ocular | Congenital mydriasis |
+ | + | + | + | + |
|
| ||||||
| GI | Malrotation | − | − | + | + | − |
| Hypoperistalsis | − | − | + | + | − | |
|
| ||||||
| GU | Hypotonic bladder | + | + | + | + | + |
| Other | Undescended testes |
|||||
|
| ||||||
| Pulm | Tachypnea at birth | + | + | + | + | |
| Pulmonary hypertension |
+b | + | + | |||
| Lung disease | Hyperinflation of the lung |
Chronic interstitial pneumonitis |
Dysgenic cystic lung disease |
Undefined | ||
Symbols are as follows: +, present; −, absent; blank, not known. Abbreviations are as follows: d, died; ASD, atrial septal defect; ICA, internal carotid arteries; WMH, white matter hyperintensities; AVM, arterial venous malformation; ACA, anterior cerebral artery; MCA, middle cerebral artery.
cause of death not determined at autopsy
bilateral lung transplant at 18 months of age for primary pulmonary hypertension.
RESULTS
We describe here five patients of Northern European descent with a novel heterozygous ACTA2 missense mutation, R179H. Sequencing of the parents’ DNA confirmed that these mutations were independent de novo events in all five patients. These patients, ranging from 11 to 26 years of age, had similar clinical findings including vascular diseases consistent with the defined spectrum of arterial complications of previously reported ACTA2 mutations, but R179H appears to be a more severe mutation based on the very early onset and high penetrance of the vascular diseases (Table I). In addition, these patients have unique clinical complications characterized by SMC dysfunction in organs throughout the body, including decreased contractile function in the iris, bladder and gastrointestinal tract. Clinical reports were published on two of these patients (Table I, B and D) but with partial reporting of the clinical features and no identification of the causative mutation [Khan et al., 2004; Lemire et al., 2004].
The de novo ACTA2 R179H mutation caused ascending aortic aneurysms and PDA in all five patients. Large PDAs were diagnosed in infancy and required surgical closure. Patient D also had surgical repair of an aortic coarctation at the age of 4 months. All patients subsequently developed fusiform ascending aortic aneurysms extending to the arch during childhood, with the diameter of the ascending aorta greater than the sinuses of Valsalva, and three patients underwent aortic surgical repair between 10 and 12 years of age. Aneurysms of the arteries originating from the arch and the suprarenal abdominal aortic aneurysm were also identified. Pathologic analysis of the ascending aorta was similar in two of the patients (Figure 1). Fibrocellular intimal SMC proliferation was evident, along with significant SMC proliferation in the subintimal medial layer associated with fragmentation of the elastic fibers. Changes that are more typical of medial degeneration, such as proteoglycan accumulation and loss of elastic fibers, were evident in the middle of the medial layer. Stenosis of the vasa vasorum due to SMC proliferation was also noted (Fig 1, Panel E).
Figure 1.
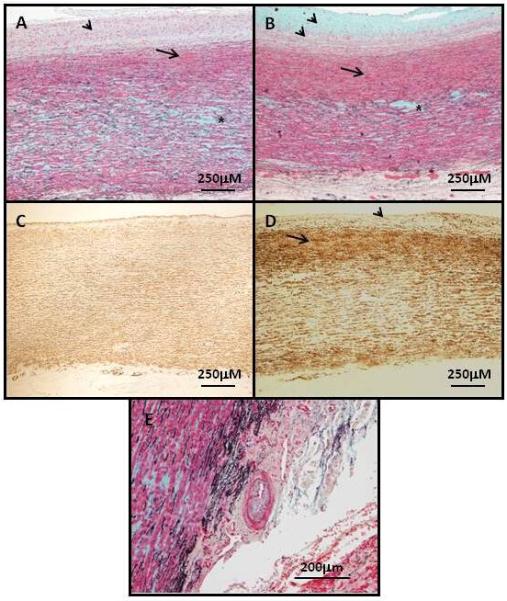
Panel A and B show the aortic pathology of the ascending aneurysms associated with ACTA2 R179H mutations (patients D and C, respectively). Movat staining of the aortas shows significant fibroproliferative lesions in the intima (arrowheads) characterized by SMCs (red) and extracellular matrix accumulation, including collagens (yellow) and proteoglycans (blue). In the medial layer of the aorta, there is subintimal proliferation of SMCs (arrows) and medial degeneration with proteoglycan accumulation and loss of elastic fibers (black), which is most apparent in the middle of medial layer (asterisks). Panel C is an age-matched control aorta and panel D is an ascending aneurysm (patient C) immunostained with α-actin to identify SMCs. These panels confirm intimal SMC hyperplasia (arrowheads) and increased SMCs in the subintimal medial layer (arrows) in the patient compared with the control. Panel E shows an artery in the aortic vasa vasorum with intimal fibrocellular proliferation (the thin black line in the artery is the internal elastic lamina, which demarcates the intimal from the medial layer). Magnification is indicated on the images.
In addition to the aortic disease, all patients had cerebrovascular abnormalities. MRA and angiograms in all of these patients showed fusiform dilatation of the internal carotid arteries (ICA) from the cavernous to the clinoidal segments (Fig 2, Panel A). In addition, the terminal region of the ICA showed mild to moderate tapering indicative of stenosis of the artery and consistent with changes observed in Moyamoya disease. These stenoses often extended into the M1 and A1 segments of the middle cerebral and the anterior cerebral arteries. Patients A and B [Khan et al., 2004] underwent neurosurgical bypass for cerebral revascularization for Moyamoya disease.
Figure 2.
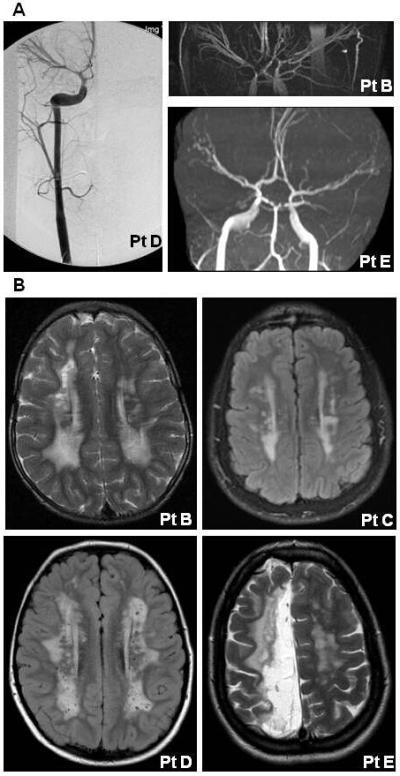
Panel A shows angiogram and MRAs images from three patients with the ACTA2 R179H mutation. These images illustrate the bilateral ectasia of internal carotid arteries from the cavernous to the clinoidal segments and stenosis of the carotid terminus in these patients. Panel B shows MRIs from four patients illustrating increased T2 signal intensity in the periventricular white matter bilaterally. In addition, MRI of patient E shows white matter changes consistent with an ischemic stroke in the distribution of the anterior cerebral artery.
All patients also had white matter abnormalities characterized by a similar pattern of increased T2 signal intensity on MRI in the periventricular white matter (Fig 2, Panel B). The Virchow-Robin spaces were mildly prominent. In addition to the periventricular white matter changes, the oldest patient (E) also had white matter changes in the distribution of the middle and anterior cerebral arteries consistent with a prior ischemic stroke. Patient C had colpocephaly with a thin corpus callosum and somewhat small cerebellar vermis, and was diagnosed with developmental delay.
All patients also had disrupted function of SMC-dependent organs that have not been previously associated with ACTA2 mutation, including the irises, bladder and gastrointestinal tract. All had congenital mydriasis, which is a rare condition characterized by aplasia of the iris sphincter muscles and hypofunction or hypoplasia of the dilator muscles, resulting in fixed dilated pupils from birth. They were all diagnosed with a hypotonic bladder, which was variably associated with dilated ureters, calyces or renal pelves, hydronephrosis, vesicoureteral reflux, and recurrent urinary tract infections. Two of the patients had congenital intestinal malrotation, one of whom required resection of a mesenteric band for an obstructed jejunum five days after birth. These same patients were diagnosed with hypoperistalsis of the gut. Patient D had gallstones that spontaneously resolved and subsequently presented with hydrops of the gallbladder without evidence of residual gallstones. Biopsies of the esophagus, stomach and small intestine revealed normal ganglionic cells and no specific neural or smooth muscle pathology.
Four patients were noted to have tachypnea at birth. Patient A required supplemental oxygen and positive pressure after birth and had an unexplained pulmonary infiltrate and severely dilated pulmonary arteries. The pulmonary complications resolved over the next two years. The tachypnea resolved in Patient B with ligation of the PDA at 20 days of age but he was noted at age 14 years to have hyperinflation of the upper lung segment, a hypoplastic lower segment and a dilated pulmonary trunk. Patient D was noted to have evidence of cystic lung disease as an infant. She had a negative sweat test for cystic fibrosis and normal alpha-1-antitrypsin, karyotype and chromosome 22q11 microdeletion analyses. Lung biopsy at 4 months of age confirmed alveolar dysgenesis, with a pattern of alveolar formation suggestive of a developmental defect (Fig 3, Panel A). The biopsy of the pulmonary vasculature showed marked changes consistent with pulmonary hypertension (grade II – III out of IV) but it was not clear whether these changes were primary or secondary to the dysgenesis (Fig 3, Panel B and C). Patient C was diagnosed with primary pulmonary hypertension and underwent bilateral lung transplant at the age of 18 months. Lung pathology at the time of transplant showed pulmonary arterial hypertensive changes (grade III-IV) with SMC hyperplasia and neointimal fibrocellular proliferative lesions.
Figure 3.
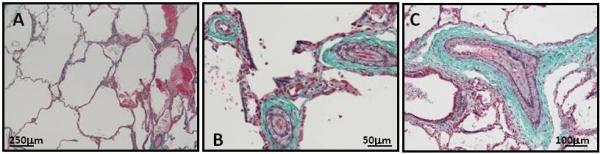
Panel A illustrates the emphysematous changes in the lungs in patient D (modified Trichrome staining elastin purple and collagen blue). Panel B and C show the increased thickness of the medial layer and subintimal proliferation of SMCs in the pulmonary vasculature of the same patient (dark purple lines surrounding the artery lumen are the internal and external elastic laminas, demarcating the intimal and medial layers and the medial and adventitial layers, respectively).
DISCUSSION
In summary, the patients described here with a unique ACTA2 mutation have a syndrome characterized by ascending aortic aneurysms, PDA, cerebrovascular disease, periventricular white matter lesions, congenital mydriasis, hypotonic bladder, malrotation and hypoperistalsis of the gastrointestinal tract and pulmonary hypertension-associated vascular changes. Other less common complications require further study to confirm an association with this mutation and include a cystic lung disease, undescended testes, gallstones, arterial venous malformation and aortic coarctation. There are previously published clinical reports of children with similar findings [Adès et al., 1999; Graf and Jungherr, 2002; Kato et al., 1999; Khan et al., 2004; Lindberg and Brunvand, 2005]. Dysmorphic facies and joint, skeletal and skin abnormalities were reported to be absent in these patients, which is surprising given that dermal myofibroblasts express ACTA2. In addition, the livedo reticularis present in a subset of ACTA2 mutation patients was not reported to be present in these patients. Although the clinical findings are pleiotropic, the complications involve organs dependent on SMCs for their function. In fact, the congenital mydriasis, urinary retention and intestinal hypoperistalsis most likely result from a loss of SMC contraction. In addition, it has been proposed that ascending aortic aneurysm formation results, at least in part, from loss of aortic SMC contractile function [Milewicz et al., 2008].
The occlusive vascular diseases found in these patients, including stenosis of the terminal internal carotids in a Moyamoya pattern and pulmonary vascular changes, are not well explained by SMC contractile dysfunction. The occlusive vascular pathology previously described in other patients with ACTA2 mutations is characterized by increased SMCs in both the intimal and medial layers of arteries, suggesting that excessive SMC proliferation leads to the greater-than-expected incidence of ischemic stroke and Moyamoya disease in these patients. A similar vascular pathology of SMC proliferation in the neointimal layer is described in the occluded carotid arteries of Moyamoya disease patients [Masuda et al., 1993]. The pulmonary vascular lesions in the patients reported here show a similar neointimal and medial SMC proliferation. It is interesting to note that the aortic pathology at the time of aneurysm repair for Patients C and D also showed significant intimal and medial SMC proliferation, raising the possibility that SMC proliferation may also contribute to aneurysm formation.
The periventricular white matter lesions identified in these patients would suggest a leukodystrophy with gliosis or demyelination originating from abnormalities in nervous tissue. A connection between neural development and SMC function is already suggested by mutations in filamin A, a cytoskeletal protein that links actin filaments to membrane receptors. Filamin A mutations cause a syndrome characterized by periventricular nodular heterotopia due to defective neuronal migration and thoracic aortic disease [Sheen and Walsh, 2005]. However, since arterial occlusive lesions occur with ACTA2 mutations, it is possible that these white matter changes result from the occlusion of small vessels. It is important to note that similar periventricular findings are observed in adult patients with occlusive small vessel disease as a result of CADASIL syndrome, diabetes or chronic hypertension [van, 1998]. Another possible explanation is that these MRI findings are watershed lesions resulting from chronic occlusion of the internal carotid artery, and similar lesions have been described in Moyamoya disease [Hasuo et al., 1998].
The pulmonary problems in these patients were already present in infancy. The etiology of the cystic lung disease and chronic interstitial pneumonitis is not known. Patient C was diagnosed with primary pulmonary hypertension, but whether the similar pulmonary vascular changes in two other patients were primary or secondary is not clear. It is intriguing to speculate that the pulmonary vascular pathology results from disruption of the maturational changes of the pulmonary vasculature that occur at birth. In utero, the pulmonary vasculature maintains high vascular resistance, in part due to proliferative and poorly differentiated SMCs in the arteries, causing thickening of the walls [Haworth, 1995]. The resistance falls rapidly after inhalation at birth and is associated with flattening and spreading of the individual SMCs and differentiation and decreased numbers of SMCs overall, resulting in decreased thickness in the arterial walls. The abnormal thickening of the medial layer of the pulmonary arteries observed in these patients suggests that postnatal regression of pulmonary vascular SMCs had not occurred and that these SMCs may have continued to proliferate in the intimal lesions.
The urinary and gastrointestinal problems found in these patients are similar to complications of megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS; OMIM 249210), an autosomal recessive condition typically lethal shortly after birth. This disorder is characterized by megacystis and hydronephrosis, microcolon/short bowel with malrotation and hypoperistalsis. The underlying genetic defect has not been identified but the overlap of MMIHS with the ACTA2 R179H phenotype suggests a defect of SMC contractile function, possibly from recessive or de novo ACTA2 mutations. Interestingly, congenital mydriasis has been reported in a patient with MMIHS [Narayanan et al., 2007].
SMCs express a number of different isoforms of actin, with SMC specific α-actin and δ-actin being the predominant forms expressed, in addition to lesser amounts of cytoplasmic β- and δ-actin [Rubenstein, 1990; Vandekerckhove and Weber, 1978]. The amount of each isoform found in SMCs varies according to the organ. In vascular SMCs, α-actin is the major isoform expressed, accounting for approximately 60% of the cellular actin [Fatigati and Murphy, 1984]. In contrast, the major actin isoform in intestinal and bladder SMCs is muscular δ-actin [Kim et al., 1994]. Therefore, the disruption of intestine and bladder function in these patients due to mutant α-actin is surprising. It is possible that the mutant α-actin disrupts the polymerization or function of the muscular δ-actin, or that α-actin plays a more important role in the contraction of these organs than previously concluded from isoform expression levels. The observation that mice lacking smooth muscle α-actin have decreased contractility of the bladder supports the second hypothesis [Zimmerman et al., 2004]. Since R179 is located close to a key protein-protein interaction site on the macromolecular surface of α-actin, the mutation may also disrupt critical interaction and disrupt downstream signaling events necessary for SMC function [Gerthoffer, 2005].
The R179H mutation is the most common recurrent mutation identified in ACTA2 in our cohort of patients with thoracic aortic disease. Analysis of the nucleotide sequence around the mutation failed to identify a mutable motif to explain the increased frequency of this mutation [Rogozin and Pavlov, 2003]. Therefore, the recurrent identification of this mutation may be due to a recruitment bias resulting from the unique phenotype in these patients, specifically the fixed dilated pupils, early onset aortic disease and Moyamoya-like cerebrovascular disease.
In summary, the recurrent de novo ACTA2 missense mutation R179H leads to more severe and penetrant vascular disease than with previously reported inherited ACTA2 mutations. In addition, this mutation is associated with novel clinical features affecting SMC-dependent organs not described for other ACTA2 mutations. Since the vascular diseases in the patients described here overlap with Marfan syndrome and vascular Ehlers Danlos syndrome, many of these patients were classified as having an underlying connective tissue disease. The identification of the causative gene as ACTA2, along with the constellation of findings involving organs dependent on SMCs for function, suggests that these patients should instead be classified as having a unique syndrome globally disrupting SMC function, what may essentially be a smooth muscular dystrophy. As such, the phenotype of these patients aids in predicting the spectrum of clinical complications resulting from gene mutations disrupting smooth muscle function.
Addendum added during revision of the manuscript.
We have identified two additional patients with the ACTA2 R179H mutation but do not have complete medical records for these individuals. The first patient is a male who was previously reported in the literature [Adès et al, 1999] and had the following features: PDA, surgically ligated at 3 months of age, bilateral iris hypoplasia diagnosed at 3 months of age, bilateral inguinal hernia repair at 6 months of age, absence of one testis, asthma during childhood, hypoplasia of the terminal phalanges of both thumbs with mild shortening of the distal phalanges of the fingers, descending aortic dissection at age 14 years, and sudden death at age 17 years. The second patient is a woman who is currently 27 years old, in whom the following clinical features are reported by the patient and physician: gastrointestinal malrotation, PDA, congenital mydriasis, asthma as a child, gallstones diagnosed at 16 years of age, ascending aortic aneurysm extending to the arch and the brachiocephalic artery (repaired at the age of 25 years), andsuprarenal aortic aneurysm. Cerebrovascular imaging at 22 years of age showed focal stenosis of the right supraclinoid carotid artery and an aplastic right A1 segment of the middle cerebral artery, stretched appearance of all the intracranial arteries, and colpocephaly.
ACKNOWLEDGMENTS
Supported by grants from the NIH, P50HL083794-01 (D.M.M.) and RO1 HL62594 (D.M.M.), the Vivian Smith Foundation (D.M.M.) and the Doris Duke Charitable Trust (D.M.M.). We would like to thank the patients and their families for participating in this study and the following physicians and genetic counselors who helped assess and obtain medical records for these patients: F. Sessions Cole, M.D., Patricia A. Wolff, M.D., Stuart C. Sweet, M.D., Linda Manwaring, M.S., Hope Chipman, M.S. and Dani Bishay, M.S.
REFERENCES
- Adès LC, Davies R, Haan EA, Holman KJ, Watson KC, Sreetharan D, Cao SN, Milewicz DM, Bateman JF, Chiodo AA, Eccles M, McNoe L, Harbord M. Aortic dissection, patent ductus arteriosus, iris hypoplasia and brachytelephalangy in a male adolescent. Clin Dysmorphol. 1999;8:269–276. [PubMed] [Google Scholar]
- Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem. 1984;259:14383–14388. [PubMed] [Google Scholar]
- Gabbiani G, Schmid E, Winter S, Chaponnier C, de CC, Vandekerckhove J, Weber K, Franke WW. Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci U S A. 1981;78:298–302. doi: 10.1073/pnas.78.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerthoffer WT. Actin cytoskeletal dynamics in smooth muscle contraction. Can J Physiol Pharmacol. 2005;83:851–856. doi: 10.1139/y05-088. [DOI] [PubMed] [Google Scholar]
- Graf MH, Jungherr A. Clinicopathologic reports, case reports, and small case series: congenital mydriasis, failure of accommodation, and patent ductus arteriosus. Arch Ophthalmol. 2002;120:509–510. [PubMed] [Google Scholar]
- Guo DC, Pannu H, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Adès L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete S, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasuo K, Mihara F, Matsushima T. MRI and MR angiography in moyamoya disease. J Magn Reson Imaging. 1998;8:762–766. doi: 10.1002/jmri.1880080403. [DOI] [PubMed] [Google Scholar]
- Haworth SG. Development of the normal and hypertensive pulmonary vasculature. Exp Physiol. 1995;80:843–853. doi: 10.1113/expphysiol.1995.sp003892. [DOI] [PubMed] [Google Scholar]
- Kato K, Tomura N, Takahashi S, Hirano H, Watarai J, Sawaishi Y, Takada G. A case of moyamoya-like vessels combined with brain anomaly. Radiat Med. 1999;17:373–377. [PubMed] [Google Scholar]
- Khan N, Schinzel A, Shuknecht B, Baumann F, Ostergaard JR, Yonekawa Y. Moyamoya angiopathy with dolichoectatic internal carotid arteries, patent ductus arteriosus and pupillary dysfunction: a new genetic syndrome? Eur Neurol. 2004;51:72–77. doi: 10.1159/000076248. [DOI] [PubMed] [Google Scholar]
- Kim YS, Wang Z, Levin RM, Chacko S. Alterations in the expression of the beta-cytoplasmic and the gamma-smooth muscle actins in hypertrophied urinary bladder smooth muscle. Mol Cell Biochem. 1994;131:115–124. doi: 10.1007/BF00925947. [DOI] [PubMed] [Google Scholar]
- Lemire BD, Buncic JR, Kennedy SJ, Dyack SJ, Teebi AS. Congenital mydriasis, patent ductus arteriosus, and congenital cystic lung disease: new syndromic spectrum? Am J Med Genet A. 2004;131:318–319. doi: 10.1002/ajmg.a.30341. [DOI] [PubMed] [Google Scholar]
- Lindberg K, Brunvand L. Congenital mydriasis combined with aneurysmal dilatation of a persistent ductus arteriosus Botalli: a rare syndrome. Acta Ophthalmol Scand. 2005;83:508–509. doi: 10.1111/j.1600-0420.2005.00496.x. [DOI] [PubMed] [Google Scholar]
- Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282:36112–36120. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- Masuda J, Ogata J, Yutani C. Smooth-Muscle Cell-Proliferation and Localization of Macrophages and T-Cells in the Occlusive Intracranial Major Arteries in Moyamoya Disease. Stroke. 1993;24:1960–1967. doi: 10.1161/01.str.24.12.1960. [DOI] [PubMed] [Google Scholar]
- Milewicz DM, Guo D, Tran-Fadulu V, Lafont A, Papke C, Inamoto S, Pannu H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, Yoshimuta T, Okajima T, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Ishibashi-Ueda H, Morisaki T. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD) Hum Mutat. 2009;30:1406–1411. doi: 10.1002/humu.21081. [DOI] [PubMed] [Google Scholar]
- Narayanan M, Murphy MS, Ainsworth JR, Arul GS. Mydriasis in association with MMIHS in a female infant: evidence for involvement of the neuronal nicotinic acetylcholine receptor. J Pediatr Surg. 2007;42:1288–1290. doi: 10.1016/j.jpedsurg.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- Rubenstein PA. The functional importance of multiple actin isoforms. Bioessays. 1990;12:309–315. doi: 10.1002/bies.950120702. [DOI] [PubMed] [Google Scholar]
- Schildmeyer LA, Braun R, Taffet G, Debiasi M, Burns AE, Bradley A, Schwartz RJ. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Walsh CA. Periventricular heterotopia: new insights into Ehlers-Danlos syndrome. Clin Med Res. 2005;3:229–233. doi: 10.3121/cmr.3.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van GJ. Leukoaraiosis and vascular dementia. Neurology. 1998;51:S3–S8. doi: 10.1212/wnl.51.3_suppl_3.s3. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J, Weber K. At least six different actins are expressed in a higher mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J Mol Biol. 1978;126:783–802. doi: 10.1016/0022-2836(78)90020-7. [DOI] [PubMed] [Google Scholar]
- Zimmerman RA, Tomasek JJ, McRae J, Haaksma CJ, Schwartz RJ, Lin HK, Cowan RL, Jones AN, Kropp BP. Decreased expression of smooth muscle alpha-actin results in decreased contractile function of the mouse bladder. J Urol. 2004;172:1667–1672. doi: 10.1097/01.ju.0000139874.48574.1b. [DOI] [PubMed] [Google Scholar]