

Abstract
Recurrent alterations in promoter methylation of tumor suppressor genes (TSGs) and LINE1 (L1RE1) repeat elements were previously reported in pheochromocytoma and abdominal paraganglioma. This study was undertaken to explore CpG methylation abnormalities in an extended tumor panel and assess possible relationships between metastatic disease and mutation status. CpG methylation was quantified by bisulfite pyrosequencing for selected TSG promoters and LINE1 repeats. Methylation indices above normal reference were observed for DCR2 (TNFRSF10D), CDH1, P16 (CDKN2A), RARB, and RASSF1A. Z-scores for overall TSG, and individual TSG methylation levels, but not LINE1, were significantly correlated with metastatic disease, paraganglioma, disease predisposition, or outcome. Most strikingly, P16 hypermethylation was strongly associated with SDHB mutation as opposed to RET/MEN2, VHL/VHL, or NF1-related disease. Parallel analyses of constitutional, tumor, and metastasis DNA implicate an order of events where constitutional SDHB mutations are followed by TSG hypermethylation and 1p loss in primary tumors, later transferred to metastatic tissue. In the combined material, P16 hypermethylation was prevalent in SDHB-mutated samples and was associated with short disease-related survival. The findings verify the previously reported importance of P16 and other TSG hypermethylation in an independent tumor series. Furthermore, a constitutional SDHB mutation is proposed to predispose for an epigenetic tumor phenotype occurring before the emanation of clinically recognized malignancy.
Keywords: Molecular genetics, Gene regulation, Metastasis, Adrenal medulla, Endocrine therapy
Introduction
Pheochromocytomas are catecholamine-secreting tumors of the chromaffin cells of the adrenal medulla. Extra-adrenal abdominal paragangliomas (here referred to as abdominal paraganglioma or paraganglioma) are related to neuroendocrine tumors in chromaffin cells distributed along the sympathetic ganglia throughout the abdomen. There is an increasing appreciation for the hereditary background of these tumors; indeed, known predisposing gene variations are present in more than 25% of pheochromocytoma and paraganglioma patients (Neumann et al. 2002). Four syndromes are closely associated with pheochromocytomas and paragangliomas: MEN2, VHL, neurofibromatosis type 1, and familial paraganglioma syndrome types 1, 3, and 4, which are caused by constitutional mutations in the RET, VHL, NF1, and SDHD, SDHC, and SDHB genes respectively (Elder et al. 2005). Recently, constitutional mutations of SDHAF2 (SDH5) and SDHA were also identified in paraganglioma and of TMEM127 and MAX in pheochromocytoma (Hao et al. 2009, Burnichon et al. 2010, Qin et al. 2010, Yao et al. 2010, Comino-Méndez et al. 2011).
Others and we have previously shown that promoter hypermethylation in tumor suppressor genes (TSGs) is a prominent feature of pheochromocytoma and paraganglioma (Cascon et al. 2004, Margetts et al. 2005, Geli et al. 2007, Geli et al. 2008, Kiss et al. 2008, Muscarella et al. 2008, Sandgren et al. 2010a). P16 (CDKN2A) and its novel transcript variant P16G are important tumor suppressors that act through the retinoblastoma pathway to regulate the cell cycle (Lin et al. 2007). Knocking out the mouse equivalent of the INK4A locus, Ink4a/Arf, causes a substantial increase in the severity of the disease phenotype in pheochromocytoma-prone mice (You et al. 2002). While this region is rarely lost in human pheochromocytomas and paragangliomas, we found that hypermethylation of the residing P16 gene is strongly associated with malignancy – 4/5 cases with P16 hypermethylation were classified as malignant, while only 1/44 tumors without evidence of malignancy harbored P16 hypermethylation (Kiss et al. 2008). We further found concerted hypermethylation in the promoters of the CDH1, DCR2 (TNFRSF10D), p16INK4A (CDKN2A), RASSF1A, and RARB genes in the same panel of 55 pheochromocytomas and paragangliomas (Geli et al. 2008). Coordinated hypermethylation of multiple TSGs was observed in agreement with a CpG island methylator phenotype (CIMP) originally described for colon cancer (Toyota et al. 1999). Interestingly, CIMP was associated with malignancy (4/5 cases with CIMP had developed metastases) and with SDHB mutation (SDHB mutation was detected in 4/5 cases with CIMP, Geli et al. (2008) and Kiss et al. (2008)).
In this study, we aimed to evaluate the possibility that predisposing mutation and TSG hypermethylation are associated events and to substantiate the evidence that TSG hypermethylation is indicative of malignant behavior. We have therefore characterized TSG promoter hypermethylation in relation to global CpG methylation, analogized by assessing methylation in the LINE1 (L1RE1) retrotransposon element, and in relation to disease predisposition in an extended tumor panel of pheochromocytomas and paragangliomas. Notably and as our previous findings indicated a presence of CIMP in strong association with malignant paragangliomas with SDHB mutation (Geli et al. 2008, Kiss et al. 2008), the present panel included additional cases with known mutation in predisposing genes and/or associated syndromes, paragangliomas, and metastatic disease. Furthermore, comparisons were made with non-tumorous material with regard to genetic and epigenetic events in a subset of cases. Our aims were to i) verify the occurrence of TSG hypermethylation in an independent tumor series; ii) assess the temporal relation of methylation to tumor development; iii) determine the chronological relation to genetic alterations; iv) evaluate the relation to mutations in predisposing genes; and v) assess associations to metastatic disease.
Materials and methods
Patients and clinical samples
All samples were collected with informed consent and ethical approval from patients surgically treated for pheochromocytoma or abdominal paraganglioma at Sahlgrenska University Hospital, Göteborg or Karolinska University Hospital, Stockholm, Sweden (Table 1). Fresh frozen tumor samples and matching non-tumor tissues were obtained from the respective endocrine Biobanks and matching leukocytes from peripheral venous samples. In this study, tumors were classified according to the World Health Organization criteria (DeLellis 2004, Tischler 2008) whereby only cases with metastases were regarded as malignant.
Table 1.
Clinical and genetic details for pheochromocytoma/paraganglioma cases
| Parameter | This study | ||
|---|---|---|---|
| Phenotype | Series A | Series Ba | Series A+B |
| No of cases | |||
| Patients | 39 | 54 | 93 |
| Tumors | 40 | 56 | 96 |
| Samples studied | |||
| Blood/normal tissues | – | 5 | |
| Primary tumors | 38 | 54 | 92 |
| Metastases | 2 | 2 | 4 |
| Gender | |||
| Female | 20 | 31 | 51 |
| Male | 18 | 23 | 41 |
| Age at diagnosis | |||
| Range (years) | 17–81 | 13–77 | 13–81 |
| Syndrome/mutation | |||
| SDHB | 6 | 4 | 10 |
| MEN2/RET | 9 | 3 | 12 |
| NF1 | 5 | 2 | 7 |
| VHL/VHL | 2 | 1 | 3 |
| Diagnosis | |||
| Pheochromocytoma | 27 | 43 | 70 |
| Paraganglioma | 12 | 11 | 23 |
| Metastasized | 7 | 7 | 14 |
| Without metastasis | 32 | 47 | 79 |
| Bilateral/multiple | 7 | 6 | 13 |
| Survival | |||
| DOD | 4 | 2 | 6 |
| DOC | 7 | 6 | 13 |
| A | 27 | 45 | 72 |
DOD, dead of disease; DOC, dead of other cause; A, alive, no reported disease.
Data for tumors from Geli et al. (2008) and Kiss et al. (2008).
Tumor series A (Tables 1 and 2) constituted of 38 primary tumors and two metastases (Khorram-Manesh et al. 2005, Wängberg et al. 2006, Muth et al. 2012) was analyzed for promoter methylation and SDHB/D mutation in this study (Table 2). For Series B (Table 1), clinical/genetic details and methylation status for tumor samples have been previously published (Edström Elder et al. 2003, Geli et al. 2008, Kiss et al. 2008, Sandgren et al. 2010b). In this study, mutation screenings and methylation quantifications in Series B were carried out in matching normal samples from a subset of cases (Supplementary Table S1, see section on supplementary data given at the end of this article). Histopathologically evaluated normal adrenal medullary DNA samples purchased from Clinomics (Watervliet, NY, USA) were used as non-tumor references in methylation analyses.
Table 2.
Results from methylation quantifications of promoter regions in tumor Series A
| DCR2 (% meth) | CDH1 (%) | NORE1A (%) | P16 (%) | RARB (%) | RASSF1A (%) | LINE-1 (%) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case no. | Tumor type | Metastatic disease | Mutation/syndrome | CIMP type | Z-scorea | MetI | Range | MetI | Range | MetI | Range | MetI | Range | MetI | Range | MetI | Range | MetI | Range |
| AG-1 | Pheo | No | MEN2 | – | −0.2 | 4 | 1–8 | 2 | 1–4 | 2 | 1–6 | 1 | 1–2 | 3 | 2–4 | 17 | 6–36 | 46 | 37–54 |
| AG-2 | Paraga | Met | SDHB | – | 0.5 | 15 | 11–26 | 11 | 4–18 | 3 | 1–7 | 10 | 8–13 | 4 | 3–7 | 7 | 7–8 | 75 | 74–77 |
| AG-3 | Pheo | No | RET | – | −0.4 | 3 | 1–7 | 3 | 1–15 | 2 | 1–6 | 1 | 1–1 | 3 | 2–4 | 2 | 1–3 | 68 | 65–73 |
| AG-4 | Pheo | No | NF1 | – | 0.1 | 21 | 6–33 | 4 | 1–6 | 2 | 2–5 | 2 | 0–9 | 3 | 0–5 | 10 | 7–15 | 75 | 71–77 |
| AG-5 | Pheo | No | NF1 | – | −0.4 | 4 | 2–9 | 3 | 1–6 | 2 | 1–6 | 1 | 1–2 | – | – | 3 | 2–3 | 71 | 66–75 |
| AG-6 | Paraga | No | SDHB | – | 0.2 | 12 | 6–20 | 8 | 2–20 | 3 | 1–6 | 9 | 3–17 | 2 | 1–3 | 11 | 5–20 | 77 | 75–78 |
| AG-7 | Pheo | No | NF1 | – | −0.2 | 13 | 4–22 | 4 | 1–7 | 2 | 1–5 | 1 | 1–2 | 2 | 1–3 | 7 | 3–11 | 74 | 70–76 |
| AG-8 | Pheo | No | RET | – | −0.5 | 2 | 1–4 | 3 | 1–5 | 2 | 1–5 | 1 | 1–2 | 2 | 1–3 | 2 | 1–3 | 76 | 71–81 |
| AG-9 | Paraga | Met | SDHB | CIMP | 2.8 | 49 | 22–64 | 4 | 1–7 | 6 | 3–11 | 50 | 40–57 | 21 | 4–37 | 42 | 39–47 | 68 | 64–74 |
| AG-10 | Pheo | No | – | – | 0.1 | 6 | 1–15 | 4 | 1–20 | 2 | 1–5 | 1 | 0–2 | 2 | 0–3 | 35 | 30–42 | 72 | 70–76 |
| AG-11 | Pheo | No | VHL | – | −0.3 | 4 | 1–5 | 4 | 1–7 | 2 | 1–4 | 1 | 1–2 | 3 | 2–3 | 2 | 1–3 | 80 | 76–83 |
| AG-12 | Pheo | No | VHL | – | −0.3 | 6 | 1–12 | 5 | 1–10 | 2 | 1–6 | 1 | 1–1 | 2 | 1–3 | 1 | 1–1 | 77 | 75–78 |
| AG-13 | Paraga | No | – | – | 0.5 | 25 | 6–43 | 5 | 2–8 | 2 | 1–5 | 29 | 15–49 | 2 | 0–4 | 12 | 8–16 | 78 | 75–81 |
| AG-14 | Paraga | Met | SDHB | CIMP | 2.8 | 50 | 34–58 | 24 | 5–53 | 8 | 3–27 | 40 | 32–47 | 2 | 2–4 | 28 | 25–33 | 79 | 78–79 |
| AG-15 | Pheo | No | NF1 | – | −0.3 | 3 | 1–5 | 4 | 2–5 | 2 | 1–4 | 1 | 1–2 | – | – | 13 | 4–33 | 73 | 68–76 |
| AG-16 | Paraga | No | – | – | −0.4 | 3 | 1–4 | 3 | 2–5 | 2 | 1–6 | 1 | 1–1 | – | – | 8 | 4–15 | 76 | 72–78 |
| AG-17 | Pheo | No | RET | – | 0.0 | 7 | 2–13 | 5 | 2–8 | 4 | 1–11 | 1 | 1–2 | 4 | 3–5 | 3 | 2–4 | 77 | 75–79 |
| AG-18a | Paraga | Met | – | – | 0.6 | 13 | 7–18 | 5 | 2–8 | 2 | 1–6 | 21 | 18–25 | – | – | 30 | 27–33 | 72 | 70–73 |
| AG-18b | Paraga | – | – | – | 16 | 3–34 | 4 | 2–8 | 4 | 2–11 | 1 | 1–2 | 3 | 2–6 | 3 | 2–4 | 76 | 73–79 | |
| AG-19 | Pheo | No | RET | – | −0.3 | 5 | 2–11 | 2 | 1–5 | 3 | 1–11 | 4 | 1–14 | 2 | 0–3 | 2 | 1–3 | 70 | 66–74 |
| AG-20 | Paraga | No | SDHB | CIMP | 1.3 | 45 | 17–69 | 4 | 1–6 | 2 | 1–7 | 40 | 14–59 | 3 | 2–5 | 32 | 15–51 | 64 | 61–70 |
| AS-21 | Paraga | Met | SDHB | – | 0.6 | 27 | 6–48 | 6 | 2–8 | 2 | 1–6 | 8 | 4–12 | 8 | 2–25 | 17 | 8–28 | 76 | 70–74 |
| AS-22 | Pheo | No | RET | – | −0.4 | 4 | 0–11 | 3 | 0–7 | 2 | 0–8 | 2 | 0–6 | 0 | 0–0 | 9 | 1–30 | 62 | 57–66 |
| AS-23 | Pheo | No | – | – | −0.3 | 2 | 0–14 | 8 | 0–29 | 2 | 0–6 | 0 | 0–1 | 1 | 0–4 | 9 | 0–21 | 64 | 57–70 |
| AS-24 | Pheo | No | NF1 | – | −0.6 | 5 | 2–11 | 5 | 3–8 | 0 | 0–4 | 2 | 0–4 | 1 | 0–4 | 2 | 0–4 | 61 | 58–63 |
| AS-25 | Pheo | No | MEN2 | – | −0.7 | 2 | 0–6 | 1 | 0–5 | 1 | 0–3 | 0 | 0–1 | 0 | 0–3 | 8 | 6–11 | 51 | 46–57 |
| AS-26 | Pheo | No | RET | – | −0.4 | 11 | 4–17 | 2 | 0–7 | 1 | 0–4 | 2 | 1–3 | 2 | 0–5 | 7 | 1–20 | 59 | 57–61 |
| AS-27 | Pheo | No | RET | – | −0.5 | 1 | 0–3 | 6 | 0–45 | 1 | 0–6 | 0 | 0–1 | 0 | 0–4 | 0 | 0–1 | 63 | 58–68 |
| AS-28 | Pheo | No | – | – | −0.3 | 5 | 2–11 | 6 | 3–9 | 1 | 0–4 | 2 | 0–4 | 3 | 0–9 | 2 | 0–4 | 63 | 59–65 |
| AS-29 | Pheo | No | – | – | −0.3 | 10 | 0–47 | 2 | 0–4 | 2 | 0–5 | 2 | 1–4 | 2 | 0–8 | 11 | 4–34 | 59 | 53–63 |
| AS-30 | Pheo | No | – | – | −0.5 | 1 | 0–4 | – | – | 1 | 0–5 | 0 | 0–1 | 0 | 0–4 | 12 | 4–24 | 61 | 58–65 |
| AS-31 | Pheo | No | – | – | 0.2 | 16 | 3–58 | 6 | 0–23 | 1 | 0–5 | 1 | 0–1 | 0 | 0–4 | 38 | 22–50 | 56 | 53–58 |
| AS-32 | Pheo | No | – | – | −0.2 | 2 | 0–5 | 14 | 4–34 | 1 | 0–4 | 1 | 0–3 | 0 | 0–0 | 3 | 3–5 | 63 | 58–67 |
| AS-33 | Pheo | No | – | – | −0.5 | 11 | 0–24 | 3 | 0–7 | 1 | 0–4 | 1 | 0–1 | 0 | 0–4 | 1 | 1–2 | 66 | 64–67 |
| AS-34 | Pheo | No | – | – | −0.4 | 19 | 5–35 | 3 | 0–5 | 1 | 0–5 | 1 | 1–2 | 1 | 0–3 | 8 | 5–11 | 64 | 60–67 |
| AS-35 | Pheo | No | – | – | −0.5 | 1 | 0–4 | 4 | 0–15 | 1 | 0–5 | 2 | 0–8 | 1 | 0–6 | 1 | 1–1 | 60 | 56–63 |
| AS-36 | Pheo | No | – | – | −0.3 | 9 | 0–32 | 2 | 0–3 | 1 | 0–5 | 0 | 0–1 | 0 | 0–0 | 24 | 4–39 | 54 | 47–57 |
| AS-37 | Paraga | No | – | – | −0.4 | 2 | 0–5 | 3 | 0–7 | 2 | 0–7 | 1 | 0–5 | 1 | 0–5 | 12 | 0–38 | 68 | 65–74 |
| AS-38 | Paraga | Met | – | – | −0.2 | 3 | 1–6 | – | – | 3 | 0–7 | 1 | 0–1 | 0 | 0–2 | 14 | 6–17 | 53 | 49–55 |
| AS-39 | Paraga | No | – | – | −0.3 | 3 | 0–5 | 7 | 3–10 | 1 | 0–4 | 2 | 0–3 | 4 | 2–6 | 0 | 0–1 | 66 | 64–67 |
| Reference values | |||||||||||||||||||
| Cutoff (%) | >30 | >30 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >30 | >30 | |||||||
meth, methylation; range, minimum–maximum value detected at an individual. CpG G-18a and G-18b are primary tumor and metastasis from the same patient.
DNA extractions
Genomic DNA was extracted from tissue and blood samples applying ChargeSwitch gDNA Mini Tissue Kit (Invitrogen/Life Technologies Corporation). A few of the non-tumor reference cases were extracted using a standard method with proteinase K digestion, phenol–chloroform extraction, and ethanol precipitation. DNA quality and concentrations were assessed using a NanoDrop Spectrophotometer (ND-1000).
Sequencing of the SDHB and SDHD genes
The coding exons and exon–intron junctions of the SDHB and SDHD genes were sequenced in tumor and normal DNA samples. SDHB sequences were amplified by PCR as eight different fragments and SDHD in four different reactions under previously described experimental conditions (Baysal et al. 2000, Benn et al. 2003, Castellano et al. 2006). Primer sequences and amplification details are given in Supplementary Table S2, see section on supplementary data given at the end of this article. PCR amplicons were purified by Exozap-IT (USB, GE Healthcare, Pittsburgh, PA, USA) and sequenced at the KIGene facility at KI, Stockholm, Sweden, using the ABI 3730 DNA Analyzer system (Applied Biosystems). Sequences were subsequently analyzed with the CodonCode and SeqScape Software (Applied Biosystems). Detected sequence alterations were verified and re-sequenced by forward and reverse sequencing of the involved exons.
SDHB was sequenced in 22 tumors from Series A (AG-1, -2, -6, -9, -10, -13, -14, -16, -18a, -18b, and -20; AS-21, -23, -28, -32, -33, -34, -35, -37, -38, -39, and -40). These included all paragangliomas and all pheochromocytomas previously described as malignant from Series A. Further, SDHB was sequenced in eight tumors from Series B (BS-7, -26, -30, -33, -36, -41, -44, and -45), including paragangliomas and malignant pheochromocytomas. In addition, non-tumorous DNA was sequenced in five cases in Series B with previously reported SDHB tumor mutations (Kiss et al. 2008) to assess whether mutations were constitutional or not. SDHD was sequenced in ten tumors (AS-23, -28, -32, -33, -34, -35, -37, -38, -39, and -40) including samples previously described as malignant in Series A and one tumor in Series B (BS-2) that featured CIMP but where no SDHB mutation could be detected (Geli et al. 2008, Kiss et al. 2008).
Mutation status of predisposing genes
Cases with a hereditary form of the disease were identified by mutation screening of constitutional DNA or in some cases based on the clinical presentation as reported in Edström Elder et al. (2003) and Muth et al. (2012). In addition, cases with SDHB mutations were identified by sequencing of tumor DNA and by verification of constitutional mutations in samples from blood or normal tissues. Results from tumor analyses have been published for Series B in Geli et al. (2008) and Kiss et al. (2008).
Pyrosequencing of promoter regions
Promoter methylation density was quantified in tumor and normal DNA samples for the TSGs DCR2, CDH1, NORE1A, P16, RARB, and RASSF1A and for LINE1 repeat elements. All assays were performed according to the PyroMark Assay Database, with the exception of P16 and LINE1 that were available as analysis kits from Qiagen. Genomic DNA was sodium bisulfite modified with the EZ DNA Methylation Kit (Zymo Research Corporation, Orange, CA, USA). Target regions were amplified by PCR from 25 to 50 ng DNA using 0.2 mM of the forward and reverse primers detailed in Supplementary Table S2, 0.2 mM dNTPs, 1.6 units of HotStarTaq, 10× PCR buffer (Qiagen), and for DCR2 3.0 mM MgCl2 (Qiagen). Subsequent pyrosequencing was carried out using sequencing primers and annealing temperatures listed in Supplementary Table S2 and Biotage/Qiagen PyroMark equipment (Qiagen).
Methylation indices (MetI) were calculated as the mean of all CpGs assayed for a TSG, and in addition, methylation densities at individual CpGs were considered. Cutoffs for hypermethylation were based on previous analyses of normal adrenal medullary samples (Geli et al. 2008, Kiss et al. 2008, Kiss et al. 2012). Hence, cutoffs for hypermethylation were set at >10% for CDH1, NORE1A, P16, and RARB and at >30% for DCR2 and RASSF1A, the elevated cutoffs being based on observations of higher levels of intrinsic methylation in normal samples for the latter two.
Statistical analyses
To allow comparison between samples in relation to various parameters, Z-scores were calculated for each sample with reference to each TSG, mean of all TSGs, and LINE1 in the following way: (mean CpG methylation density for each sample – mean methylation density for that promoter in the tumor panel)/s.d. of that methylation density. This negated large differences in mean MetIs between different genes and allowed direct comparison of gene methylation levels, which are expressed as s.d.s from the mean for the particular gene. STATISTICA 10.0 Software (Statsoft, Inc., Tulsa, OK, USA) was used for all statistical calculations (Supplementary Table S3, see section on supplementary data given at the end of this article). Z-scores were compared for cases with different clinical and genetic features – summarized in Table 3 – including age, tumor type, predisposing mutation/syndrome, metastasis, outcome, and relation to CIMP. Mann–Whitney U test and Kruskal–Wallis one-way ANOVA test were used to compare groups of continuous data; Fisher's exact test was used for comparisons of categorical data, and Spearman's rank order correlation was used to assess correlations between continuous data sets. Survival curves for hypermethylated and unmethylated cases in Series A+B were compared by log-rank test for P16 and RASSF1A, and results were illustrated graphically by Kaplan–Meier plots. P values <0.05 were considered as statistically significant. One of the metastases from Series A (AG-18b) was also represented by its primary tumor and was therefore excluded from the statistical calculations.
Table 3.
Correlations between Z-scores and clinical/genetic characteristics
| Promoter | Gender | Age at surgery | Mutation | Tumor type | Metastatic disease | Outcome | CIMP |
|---|---|---|---|---|---|---|---|
| Series A | |||||||
| Mean Z-score | NS | NS | <0.007 | <0.0002 | <0.0004 | 0.01 | <0.005 |
| DCR2 | NS | NS | <0.01 | 0.01 | <0.02 | NS | <0.005 |
| CDH1 | NS | NS | NS | NS | <0.04 | NS | NS |
| NORE1A | NS | NS | NS | 0.002 | <0.006 | 0.01 | 0.01 |
| P16 | 0.04 | NS | 0.005 | 0.001 | <0.05 | NS | <0.005 |
| RARB | NS | NS | 0.02 | <0.005 | NS | NS | <0.05 |
| RASSF1A | NS | NS | 0.005 | <0.008 | 0.002 | 0.009 | 0.01 |
| LINE1 | 0.003 | <0.05 | NS | NS | NS | NS | NS |
| Series A+B | |||||||
| Mean Z-score | NS | NS | 0.0002 | <0.00001 | 0.0006 | <0.0009 | <0.000001 |
| DCR2 | <0.002 | NS | 0.0003 | <0.0002 | 0.01 | <0.02 | <0.000001 |
| CDH1 | NS | NS | <0.01 | 0.03 | 0.04 | 0.03 | NS |
| NORE1A | NS | NS | NS | 0.01 | 0.005 | <0.04 | 0.02 |
| P16 | NS | NS | 0.0002 | 0.00002 | 0.002 | <0.02 | <0.000001 |
| RARB | NS | NS | 0.003 | <0.008 | <0.05 | NS | <0.0003 |
| RASSF1A | NS | NS | 0.0002 | 0.0004 | <0.0004 | <0.0008 | <0.000001 |
| LINE1 | <0.002 | NS | NS | NS | NS | NS | NS |
NS, not significant.
Results
Promoter hypermethylation in DCR2, CDH1, NORE1A, P16, RARB, and RASSF1A
Thirty-eight primary tumors and two metastases in Series A were assayed for promoter hypermethylation in multiple CpGs of DCR2, CDH1, NORE1A, P16, RARB, and RASSF1A by pyrosequencing. Three primary tumors fulfilled the criteria of CIMP phenotype, i.e. exhibited increased MetI for three or more genes (AG-9, -14, and -20; Table 2). Hypermethylation with MetI above cutoff was observed for all genes except NORE1A, and in addition, some tumors exhibited increased methylation at ≥1 CpG without raising the MetI above cutoff (Table 2). Increased MetI was identified in three primary tumors for DCR2, three tumors for CDH1, five tumors for P16, one tumor for RARB, and five tumors for RASSF1A. In addition, hypermethylation at single CpG(s) was observed for DCR2 (in four additional primary tumors), CDH1 (seven tumors), NORE1A (five tumors), P16 (four tumors), and RASSF1A (six tumors). In the two metastases, hypermethylation was not observed in regard to MetI, although hypermethylation above cutoff was observed at single CpGs in several of the genes (AG-18b and AS-21; Table 2).
For all promoters, the distribution of methylation densities was relatively even without obvious involvement of single CpGs (Fig. 1). However, the methylation levels varied largely between genes; DCR2, P16, and RASSF1A showed the highest levels, while methylation was less pronounced for CDH1, NORE1A, and RARB (Fig. 1). For DCR2 and RASSF1A, the higher densities also coincided with somewhat higher levels of methylation in normal references, while this was not the case for P16.
Figure 1.

Methylation densities of promoter regions for individual tumor suppressor genes measured by pyrosequencing for pheochromocytomas and paragangliomas in Series A. Scatterplots illustrate methylation levels (%) at individual CpG sites assayed in each tumor sample. Cutoff levels for hypermethylation determined from normal adrenal medulla are indicated by arrows for DCR2 (>30%), CDH1 (>10%), NORE1A (>10%), P16 (>10%), RARB (>10%), and RASSF1A (>30%).
Association of TSG hypermethylation with clinical and genetic features
Methylation densities in cases with different clinical and genetic features from Series A and B were compared using Z-scores calculated for the mean of all TSGs as well as for each individual TSG. Importantly, higher mean Z-score was significantly associated with mutation (SDHB), tumor type (paraganglioma), metastatic disease, death of disease, and CIMP (Fig. 2; Table 3). High Z-scores for individual TSG promoters were also significantly associated with different clinical and genetic features including SDHB mutation (all save NORE1A), paraganglioma tumor type (all), metastasis (all), and death of disease (all except RARB, with RASSF1A being highly significant) (Table 3). Supplementary Table S3 details the outcome of the statistical tests performed.
Figure 2.
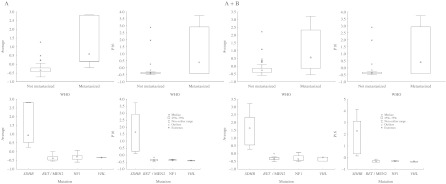
Boxplots illustrate the significant associations in Series A and A+B between increased mean Z-scores and metastatic disease and SDHB mutation, as well as between high P16 Z-score and SDHB mutation.
TSG hypermethylation in relation to SDHB mutation
The heat map in Fig. 3 illustrates the patterns of TSG hypermethylation in cases with different forms of heritable disease. Altogether, 32 primary tumors and two metastases from the combined Series A+B were from patients with a hereditary form of the disease based on (familial) syndromic presentation and/or identified germ-line mutations. The 11 SDHB-positive tumors from ten cases exhibited promoter hypermethylation for two or more of the investigated genes concerning MetI or the maximum for at least one individual CpG. Specifically, increased methylation was observed in 11/11 tumors for P16, 9/11 for DCR2, 7/11 for RASSF1A, 6/11 for CDH1, 5/11 for RARB, and 4/11 for NORE1A (Fig. 3). For P16, DCR2, and RASSF1A, the hypermethylation was, with few exceptions, pronounced, with MetI above cutoff. By contrast, tumors from patients associated with RET/MEN2 (n=12), NF1 (n=7), or VHL/VHL (n=3) exhibited increased methylation for individual CpGs in nine instances only, and MetI did not exceed background level. Taken together, the combined observations in Series A+B strongly support an association between SDHB mutation and TSG hypermethylation, with P16 being the most frequently involved.
Figure 3.

Association between SDHB mutation, tumor suppressor gene hypermethylation, and metastasis. Methylation densities are illustrated for individual tumors in Series A+B from patients with predisposing mutations and/or syndromic disease related to SDHB, RET/MEN2, NF1, or VHL/VHL. Tumors with MetI levels above cutoff are indicated by dark blue boxes, above cutoff for one or more CpG sites by light blue boxes, and below cutoff levels by green boxes. Cutoff levels for individual genes are >30% for DCR2 and RASSF1A and >10% for CDH1, NORE1A, P16, and RARB. Cases with metastasis are indicated to the right. *Metastasis from the tumor in the row above.
Order of genetic events and TSG hypermethylation in tumor development
To evaluate the relationship between occurrence of genetic and epigenetic events in the tumor development, matched samples of normal and tumor tissues were compared in five cases from Series B (Fig. 4; Supplementary Table S1). All five primary tumors and two metastases from the five patients have previously been reported to carry increased promoter MetI in two to five of the investigated genes DCR2, CDH1, NORE1A, P16, RARB, or RASSF1A (Geli et al. 2007, 2008, Kiss et al. 2008). In four of the five cases, three or more TSG promoters showed increased MetI in agreement with a CIMP phenotype. By contrast, pyrosequencing of the same promoters in constitutional DNA revealed only very low levels of CpG methylation in all five cases (Supplementary Table S1).
Figure 4.

Schematic illustrations of genetic and epigenetic events detected in normal tissue, primary tumors, and metastasis from tumor Series B. Genetic events concerning constitutional SDHB mutations and somatic loss of 1p in primary tumors are illustrated above the flow charts. Detection of promoter hypermethylation – reflected in increased MetI – and methylation above the cutoff for one or more CpG only (in parenthesis) are indicated below for the TSG promoter concerned (see Supplementary Table S1 for exact values). Samples assayed are shown in bold and by solid lines, while metastases not assayed are indicated by dotted lines.
Four of the five cases carried a constitutional inactivating mutation of the SDHB gene that was present in normal, tumor, and metastasis analyzed. In mutated cases, double peaks of mutated and wild-type sequences were observed in constitutional DNA, in agreement with heterozygous state of the mutations (Supplementary Figure S1, see section on supplementary data given at the end of this article). The occurrence of genetic and epigenetic events from constitutional tissue to primary tumor and metastasis is outlined in Fig. 4 for the five cases. Hence, SDHB mutations were present constitutionally, while TSG hypermethylation and CIMP were acquired events first observed in primary tumors together with loss of chromosomal region 1p encompassing the SDHB locus, and subsequently retained to metastatic tissue.
Global methylation of LINE1 repeat elements
MetI levels for LINE1 repeat elements varied from 46 to 80% in the 39 primary tumors in Series A, and the values at the individual CpGs ranged from 37 to 83% (Table 2). The two metastases exhibited MetI of 71 and 76% (range 70–79%). These results overlap with those in normal adrenal references, where MetI at 68–71% and individual CpGs methylation within the range 65–76% are observed. LINE1 Z-scores differed significantly between the genders (higher mean value in males).
Discussion
We here report significantly higher mean TSG methylation levels in paragangliomas, metastasizing tumors, SDHB-mutated tumors, and cases with poor survival (Fig. 2; Table 3). Taken together, these findings in the novel tumor Series A powerfully substantiate similar observations from Series B (Geli et al. 2008).
We have previously defined CIMP as mean CpG hypermethylation above the normal range cutoff in three or more TSG promoters. To herein provide a more detailed assessment of the epigenetic events in these tumors, we have also considered methylation at individual CpGs (Fig. 1; Table 2). Z-scores denoting individual TSG methylation levels, as well as mean Z-scores for all TSGs in each case, were compared with clinical and genetic features (Table 3).
Indeed, individual TSG hypermethylation was salient in paragangliomas, being especially prominent in tumors classified as malignant. In the combined Series A+B, 7/11 metastasized paragangliomas exhibited MetI above cutoff in two or more of the assessed TSG promoters (Fig. 5). Further, 4/12 paragangliomas without metastases carried SDHB mutations – strongly linked to malignant behavior and metastatic potential (Boedeker 2011) – three of which also exhibited MetI above cutoff for two or more TSGs (Fig. 5). In contrast, TSG hypermethylation proved infrequent in pheochromocytomas; 5/54 cases featured MetI above cutoff in a single TSG each (Fig. 5). The most frequently hypermethylated genes were DCR2 (eight tumors), P16 (11 tumors (AG-6 exhibited elevated MetI, but was nonetheless. Five percentage points too low to reach cutoff)), and RASSF1A (nine tumors), indicating that epigenetic modifications of these genes is an important facet of malignancy in paragangliomas.
Figure 5.

Comparison of promoter hypermethylation at individual tumor suppressor gene promoters in paraganglioma and pheochromocytoma with and without metastasis from tumor combined Series A+B. Methylation levels are indicated as dark blue boxes for MetI above cutoff, light blue boxes refer to one or more CpG sites above cutoff, and green boxes denote promoters without detected hypermethylation. Cutoff levels for individual genes were applied at >30% (DCR2 and RASSF1A) or >10% (CDH1, NORE1A, P16, and RARB). *AG-6 displays elevated P16 MetI just under cutoff.
We found a very strong association between SDHB mutation and hypermethylation of several TSGs in the novel Series A (Fig. 2; Table 3), in agreement with our previous reports of an association to P16 hypermethylation (Kiss et al. 2008). The Z-score box plots in Fig. 2, grouped by syndrome/mutation, further highlight the over-representation of TSG CpG methylation coinciding with SDHB mutation compared with other genetic variants. P16 hypermethylation was most frequently seen with and was unequivocally associated with SDHB mutation (Fig. 2; Table 2). Furthermore, for the paraganglioma AG-6 with SDHB mutation, the MetI for P16 was elevated and close to the nominal 10% cutoff value, and hypermethylation above cutoff was observed for individual CpGs. Similarly, the SDHB-mutated paraganglioma lymph node metastasis AS-21 exhibited a MetI of 8% (range 4–12%) for P16. This observation is perhaps indicative of a developing phenotype or a result of tumor heterogeneity. By contrast, MetI levels for P16 were very low (1 or 2%) in tumors without detectable SDHB mutation. Figure 3 further illustrates that in the combined Series A+B, concerted epigenetic events occur in relation to SDHB mutation but are not observed in association with other predisposing syndromes. Survival analyses of combined Series A+B demonstrated significantly shorter survival in patients with primary tumors that displayed P16 MetI >10% compared with those with MetI below cutoff (Fig. 6).
Figure 6.

Kaplan–Meyer plots illustrating the significant association between short disease-related (A) and overall (B) survival and hypermethylation of P16 based on data from combined Series A+B.
In Series A as well as in combined Series A+B, hypermethylation of TSGs was found in association with paraganglioma tumor type, development of metastases, and mutation of the predisposing gene SDHB. While metastasis and SDHB mutations are known to be correlated, the question arises whether hypermethylation also occurs in SDHB wild-type metastatic tumors. SDHB mutations were not identified in 6 of the 14 metastatic tumors in this study. Among these, only two paragangliomas had hypermethylation. Significant methylation in RASSF1A alone was detected when analyzing TSG methylation in all metastatic tumors without apparent SDHB involvement (P≥0.02). Hence, increased TSG methylation does not appear to be frequently involved in sporadic, malignant cases or in conjunction with other predisposing mutations. However, it is presently unknown whether metastatic tumors with mutations other than SDHB harbor TSG methylation. We see no such involvement in benign tumors with known mutations other than SDHB (Fig. 3).
SDHB mutations are known to be frequently associated with malignant forms of paraganglioma (Ricketts et al. 2010), raising questions about causes and consequences in relation to these abnormalities and their clinical effects. In four cases with constitutional SDHB mutations in Series B, TSG hypermethylation was absent in constitutional DNA – indicating that the TSG hypermethylation is tumor-specific – and first observed in primary tumors in conjunction with loss of 1p encompassing the SDHB gene locus (Fig. 4). Furthermore, acquired TSG hypermethylation was observed in four malignant primary paragangliomas before the development of metastasis and one case where metastasis had not developed before surgery (BS-10; Fig. 4). Importantly, these findings implicate that TSG hypermethylation is not a secondary consequence of a malignant tumor state. Furthermore, they would imply that heterozygous SDHB inactivating mutations do not confer detectable TSG hypermethylation at the constitutional level, at least not for the tissues/genes assessed here. However, it is a theoretical possibility that heterozygous SDHB inactivation could lead to TSG hypermethylation in cancer progenitor cells of the target tissue and subsequently be selected for at tumor transformation.
The data presented here strongly indicate that SDHB inactivation and TSG CpG hypermethylation are associated; an attractive possibility being that SDHB inactivation in fact causes TSG hypermethylation. To our knowledge, this would represent the first instance where a known hereditary cancer syndrome (familial paraganglioma syndrome type 4) is linked to an epigenetic phenotype. The link between SDH loss and epigenetic remodeling represented by histone modifications was recently shown (Smith et al. 2007, Cervera et al. 2009). Importantly, the breakdown of the mitochondrial respiratory chain by loss of SDH function leads to accumulation of succinate, which causes inhibition of α-ketoglutarate-dependent enzymes (that normally produce succinate as a byproduct; Lee et al. 2005, Selak et al. 2005). Among them are the jumonji-domain histone demethylases (Smith et al. 2007) that regulate histone H3 and H4 lysine and arginine methylation (Agger et al. 2008, Cervera et al. 2009). This would overture a general inhibition of histone demethylase activity (Cervera et al. 2009), likely affecting chromatin on a global level. It is plausible that these histone modifications and the epigenetic perturbances observed in the current project have a joint causality. However, we observe a very specific pattern of promoter CpG methylation in our tumor panel, while global methylation patterns analogized by LINE1 remain heterogeneous regardless of SDHB status. This specific methylative inactivation of apoptotic and antiproliferative genes might instead mirror a physiological attempt to counter the state of pseudo-hypoxia, induced by SDH dysfunction and described in Pollard et al. (2005) and Cervera et al. (2008). Indeed, recent immunohistochemical studies have shown that tumors from patients with SDHB, SDHC, and SDHD mutations lack SDHB immunoreactivity and that SDH activity is abolished in SDHB- and SDHD-mutated cases but not in connection with SDHC (van Nederveen et al. 2009). These observations contrast those in tumors from patients with predisposing mutations in other genes. Based on the results, the authors recommended that screening for SDH gene mutations should be carried out in cases with negative SDHB immunohistochemistry (van Nederveen et al. 2009). Another implication from these interesting findings is that the increased risk of malignancy associated with SDHB would not be a direct result of SDHB protein loss but relate to other molecular abnormalities perhaps occurring at the transcriptional level.
Recognition of constitutional mutation carriers among pheochromocytoma and paraganglioma patients is of clinical importance but in principle requires extensive mutation screenings in a large group of patients – as several genes are involved, and hereditary predisposition is often present in spite of a negative family history. The findings presented here suggest that methylation quantification for P16 promoter CpGs could be a valuable clinical tool in the assessment of paragangliomas and that cases with P16 hypermethylation should be genetically screened for SDHB mutations.
In summary, our results associated mutation of the SDHB gene to alterations in TSG methylation in paragangliomas. We here propose that epigenetic inactivation of TSGs is an important component in familial paraganglioma syndrome and suggest inquiries into the use of demethylating agents as a means to combat malignant paragangliomas. Future analyses should also encompass the assessment of gene-specific methylation in SDHB-deficient paragangliomas using methylation arrays. We further propose the use of P16 methylation assessment as an additive tool in identification of patients for SDHB mutation screenings.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/ERC-12-0267.
Acknowledgements
Dedicated to the memory of Håkan Ahlman, 1947–2012. The authors wish to thank Stefano Caramuta for expert assistance with survival analyses.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The study was financially supported by grants from Swedish Cancer Society, Swedish Research Council, Göran Gustafsson Foundation for Research in Natural Sciences and Medicine, Gustav V Jubilee Foundation, Karolinska Institutet, and Stockholm County Council.
References
- Agger K, Christensen J, Cloos PAC, Helin K. The emerging functions of histone demethylases. Current Opinion in Genetics & Development. 2008;18:159–168. doi: 10.1016/j.gde.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, Mey A, Taschner PEM, Rubinstein WS, Myers EN. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Benn DE, Croxson MS, Tucker K, Bambach CP, Richardson AL, Delbridge L, Pullan PT, Hammond J, Marsh DJ, Robinson BG. Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene. 2003;22:1358–1364. doi: 10.1038/sj.onc.1206300. [DOI] [PubMed] [Google Scholar]
- Boedeker C. Paragangliomas and paraganglioma syndromes. Laryngo-Rhino-Otologie. 2011;90:S56–S82. doi: 10.1055/s-0030-1270447. [DOI] [PubMed] [Google Scholar]
- Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A. SDHA is a tumor suppressor gene causing paraganglioma. Human Molecular Genetics. 2010;19:3011. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascon A, Ruiz-Llorente S, Fraga M, Leton R, Telleria D, Sastre J, Diez JJ, Diaz-Guerra GM, Perez JAD, Benitez J. Genetic and epigenetic profile of sporadic pheochromocytomas. Journal of Medical Genetics. 2004;41:e30–e30. doi: 10.1136/jmg.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano M, Mori L, Giacche M, Agliozzo E, Tosini R, Panarotto A, Cappelli C, Mulatero P, Cumetti D, Veglio F. Genetic mutation screening in an Italian cohort of nonsyndromic pheochromocytoma/paraganglioma patients. Annals of the New York Academy of Sciences. 2006;1073:156–165. doi: 10.1196/annals.1353.016. [DOI] [PubMed] [Google Scholar]
- Cervera AM, Apostolova N, Crespo FL, Mata M, McCreath KJ. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Research. 2008;68:4058. doi: 10.1158/0008-5472.CAN-07-5580. [DOI] [PubMed] [Google Scholar]
- Cervera AM, Bayley JP, Devilee P, McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modifications in mammalian cells. Molecular Cancer. 2009;8:89. doi: 10.1186/1476-4598-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genetics. 2011;43:663–667. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- DeLellis RA 2004 Tumors of the adrenal gland. In World Health Organization Classification of Tumors. Pathology & Genetics, pp136–166. Eds RA DeLellis, RV Lloyd, PU Heitz & C Eng. Lyon, France: IARC Press.
- Edström Elder E, Hjelm Skog AL, Höög A, Hamberger B. The management of benign and malignant pheochromocytoma and abdominal paraganglioma. European Journal of Surgical Oncology. 2003;29:278–283. doi: 10.1053/ejso.2002.1413. [DOI] [PubMed] [Google Scholar]
- Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. Journal of Surgical Oncology. 2005;89:193–201. doi: 10.1002/jso.20177. [DOI] [PubMed] [Google Scholar]
- Geli J, Kiss N, Lanner F, Foukakis T, Natalishvili N, Larsson O, Kogner P, Höög A, Clark GJ, Ekström TJ. The Ras effectors NORE1A and RASSF1A are frequently inactivated in pheochromocytoma and abdominal paraganglioma. Endocrine-Related Cancer. 2007;14:125–134. doi: 10.1677/ERC-06-0031. [DOI] [PubMed] [Google Scholar]
- Geli J, Kiss N, Karimi M, Lee JJ, Bäckdahl M, Ekström TJ, Larsson C. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: association to malignant behavior. Clinical Cancer Research. 2008;14:2551–2559. doi: 10.1158/1078-0432.CCR-07-1867. [DOI] [PubMed] [Google Scholar]
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorram-Manesh A, Ahlman H, Nilsson O, Friberg P, Oden A, Stenström G, Hansson G, Stenquist O, Wängberg B, Tisell LE. Long-term outcome of a large series of patients surgically treated for pheochromocytoma. Journal of Internal Medicine. 2005;258:55–66. doi: 10.1111/j.1365-2796.2005.01504.x. [DOI] [PubMed] [Google Scholar]
- Kiss N, Geli J, Lundberg F, Avci C, Velazquez-Fernandez D, Hashemi J, Weber G, Höög A, Ekström T, Bäckdahl M. Methylation of the p16INK4A promoter is associated with malignant behavior in abdominal extra-adrenal paragangliomas but not pheochromocytomas. Endocrine-Related Cancer. 2008;15:609–621. doi: 10.1677/ERC-07-0285. [DOI] [PubMed] [Google Scholar]
- Kiss NB, Kogner P, Johnsen JI, Martinsson T, Larsson C, Geli J. Quantitative global and gene-specific promoter methylation in relation to biological properties of neuroblastomas. BMC Medical Genetics. 2012;13:83. doi: 10.1186/1471-2350-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG., Jr Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Lin Y, Diccianni M, Kim Y, Lin H, Lee C, Lin R, Joo S, Li J, Chuang T, Yang A. Human p16γ, a novel transcriptional variant of p16INK4A, coexpresses with p16INK4A in cancer cells and inhibits cell-cycle progression. Oncogene. 2007;26:7017–7027. doi: 10.1038/sj.onc.1210507. [DOI] [PubMed] [Google Scholar]
- Margetts C, Astuti D, Gentle D, Cooper W, Cascon A, Catchpoole D, Robledo M, Neumann H, Latif F, Maher E. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel–Lindau and sporadic phaeochromocytomas. Endocrine-Related Cancer. 2005;12:161–172. doi: 10.1677/erc.1.00865. [DOI] [PubMed] [Google Scholar]
- Muscarella P, Bloomston M, Brewer AR, Mahajan A, Frankel WL, Ellison EC, Farrar WB, Weghorst CM, Li J. Expression of the p16INK4A/Cdkn2a gene is prevalently downregulated in human pheochromocytoma tumor specimens. Gene Expression. 2008;14:207–216. doi: 10.3727/105221608786883825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth A, Abel F, Jansson S, Nilsson O, Ahlman H, Wängberg B. Prevalence of germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: a population-based study in western Sweden. World Journal of Surgery. 2012;36:1389–1394. doi: 10.1007/s00268-012-1430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EMCA, Sleddens HF, Derkx P, Rivière J, Dannenberg H. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncology. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann HPH, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K. Germ-line mutations in nonsyndromic pheochromocytoma. New England Journal of Medicine. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- Pollard P, Briere J, Alam N, Barwell J, Barclay E, Wortham N, Hunt T, Mitchell M, Olpin S, Moat SJ. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Human Molecular Genetics. 2005;14:2231. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nature Genetics. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, Cole TR, Armstrong R, Kumar V, Morrison PJ. Tumor risks and genotype–phenotype–proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Human Mutation. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- Sandgren J, Andersson R, Rada-Iglesias A, Enroth S, Åkerström G, Dumanski JP, Komorowski J, Westin G, Wadelius C. Integrative epigenomic and genomic analysis of malignant pheochromocytoma. Experimental & Molecular Medicine. 2010a;42:484. doi: 10.3858/emm.2010.42.7.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren J, de Ståhl TD, Andersson R, Menzel U, Piotrowski A, Nord H, Bäckdahl M, Kiss NB, Brauckhoff M, Komorowski J. Recurrent genomic alterations in benign and malignant pheochromocytomas and paragangliomas revealed by whole-genome array comparative genomic hybridization analysis. Endocrine-Related Cancer. 2010b;17:561–579. doi: 10.1677/ERC-09-0310. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-[α] prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Smith EH, Janknecht R, Maher LJ. Succinate inhibition of α-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Human Molecular Genetics. 2007;16:3136. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Archives of Pathology & Laboratory Medicine. 2008;132:1272–1284. doi: 10.1043/1543-2165(2008)132[1272:PAEPU]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JPJ. CpG island methylator phenotype in colorectal cancer. PNAS. 1999;96:8681. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wängberg B, Muth A, Khorram-Manesh A, Jansson S, Nilsson O, Forssell-Aronsson E, Tisell L, Ahlman H. Malignant pheochromocytoma in a population-based study. Annals of the New York Academy of Sciences. 2006;1073:512–516. doi: 10.1196/annals.1353.054. [DOI] [PubMed] [Google Scholar]
- Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Pérez L, King EE, Toledo RA, Ercolino T, Rapizzi E, Ricketts CJ. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. Journal of the American Medical Association. 2010;304:2611–2619. doi: 10.1001/jama.2010.1830. [DOI] [PubMed] [Google Scholar]
- You MJ, Castrillon DH, Bastian BC, O'Hagan RC, Bosenberg MW, Parsons R, Chin L, DePinho RA. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. PNAS. 2002;99:1455. doi: 10.1073/pnas.022632099. [DOI] [PMC free article] [PubMed] [Google Scholar]