

SUMMARY
The pancreatic serine protease chymotrypsin C (CTRC) cleaves the Leu81-Glu82 peptide bond in the calcium binding loop of human cationic trypsinogen and thereby promotes its degradation. This serves as a protective mechanism against ectopic trypsinogen activation in the pancreas. In the present study we demonstrate that cleavage of the Leu81-Glu82 peptide bond by CTRC is highly specific and other human pancreatic chymotrypsins (CTRB1, CTRB2, CTRL1) and elastases (ELA2A, ELA3A, ELA3B) do not catalyze this reaction. To elucidate the mechanistic basis for CTRC specificity, we surveyed the primary (P1) cleavage preference of these pancreatic proteases on peptide substrates. We found that CTRC cleaved after a P1 Leu with at least 10-fold higher catalytic efficiency than other enzymes tested. To assess extended subsite interactions, we introduced Ala-mutations into human cationic trypsinogen at the P3, P1' P3' and P4' amino-acid positions, where P1-P1' corresponds to Leu81-Glu82. Interestingly, CTRC mediated cleavage was stimulated 3-fold by mutation E82A and unaffected by mutations E79A and N84A, but all three mutations compromised specificity and resulted in increased cleavage by ELA2A. Mutation E85A decreased CTRC cleavage by 2-fold. Remarkably, other chymotrypsins and elastases did not cleave human cationic trypsinogen even with the L81F or L81A mutations, which introduced favorable P1 residues for these enzymes. We conclude that specific cleavage of the Leu81-Glu82 peptide bond in human cationic trypsinogen by CTRC is primarily determined by its distinctively high activity on leucyl peptide bonds, whereas the P1' Glu82, P3' Asn84 and P4' Glu85 residues serve as additional specificity determinants.
Keywords: chymotrypsin, elastase, trypsinogen, pancreas, serine protease
The human exocrine pancreas secretes several digestive serine protease precursors, namely trypsinogens, chymotrypsinogens and proelastases, which are converted to active enzymes by limited proteolysis in the duodenum [1, 2]. Trypsinogens are the most abundant proenzymes, synthesized as three isoforms in humans: cationic trypsinogen (serine protease 1, PRSS1), anionic trypsinogen (PRSS2) and mesotrypsinogen (PRSS3). The cationic isoform contributes about two-thirds and the anionic isoform about one-third of the secreted trypsinogen content in the pancreatic juice [3–5]. Mesotrypsinogen is a minor isoform [6]. Trypsinogens are activated to trypsins by the intestinal brush border enzyme enteropeptidase [2, 7]. Additionally, human cationic and anionic trypsinogens can undergo rapid autoactivation, which may facilitate their activation in the gut [8 and references therein]. Human chymotrypsinogens are expressed as four isoenzymes: chymotrypsinogen B1 (CTRB1), chymotrypsinogen B2 (CTRB2), chymotrypsinogen C (CTRC) and chymotrypsin-like enzyme 1 precursor (CTRL1), whereas human proelastases comprise proelastase 2A (ELA2A), proelastase 3A (ELA3A) and proelastase 3B (ELA3B). The archetypal proelastase 1 (ELA1) is not expressed in the human pancreas due to evolutionary mutations in its promoter region [9]. Similarly, ELA2B, a gene duplication product of ELA2A, seems to have accumulated inactivating mutations within its coding region [10]. All chymotrypsinogen and proelastase isoforms are activated to chymotrypsins and elastases by trypsin, which thus functions as the master regulator of digestive enzyme activation [2]. Under physiological conditions, digestive protease activation takes place in the intestine only, whereas pathological, premature activation in the pancreas may lead to the inflammatory disorder pancreatitis [11]. Intrapancreatic protease activation may be initiated by trypsinogen autoactivation, as enteropeptidase is not expressed in the pancreas.
Recent studies demonstrated that autoactivation of trypsinogens in humans is proteolytically regulated by CTRC by two independent and seemingly conflicting mechanisms [12]. On the one hand, CTRC stimulates autoactivation of cationic trypsinogen by processing the trypsinogen activation peptide at the Phe18-Asp19 peptide bond to a shorter form [13]. On the other hand, CTRC cleaves the Leu81-Glu82 peptide bond in the calcium binding loop of trypsinogen, which, in concert with the trypsin-mediated hydrolysis of the Arg122-Val123 peptide bond, results in the degradation of trypsinogen (Fig 1) [14, 15]. CTRC also degrades trypsin by the same mechanism albeit at a much slower rate [14, 15]. Cleavage of the Leu81-Glu82 peptide bond is partially inhibited by submillimolar concentrations of calcium prevalent in the secretory pathway and the pancreatic ducts and even more so by high millimolar concentrations present in the intestine. Impairment of CTRC-dependent regulation of trypsinogen autoactivation increases the risk for intrapancreatic trypsinogen activation and consequent pancreatitis in humans. Thus, mutations in cationic trypsinogen that inhibit trypsinogen degradation and/or increase the processing of the activation peptide by CTRC are associated with hereditary and sporadic forms of chronic pancreatitis [15 and references therein]. Similarly, loss-of-function mutations which decrease activity or secretion of CTRC are risk factors for chronic pancreatitis [12, 16].
Figure 1.

The calcium binding loop in human cationic trypsinogen and trypsin. Ribbon diagram of human cationic trypsin (Protein Data Bank code 2RA3) and schematic diagram of the calcium binding loop are shown. The amino-acid positions around the Leu81-Glu82 peptide bond that may participate in binding to CTRC are indicated, using the Schechter-Berger nomenclature where P1-P1' is the scissile peptide bond. The P1 Leu81 is highlighted in orange; other positions are shown in blue. Interactions between the Ca2+ and the protein main chain (solid lines) and side chains (dashed lines) are also indicated. The image was rendered using PyMol 1.3 (Schrödinger, LLC).
In the present study we demonstrate that cleavage of Leu81-Glu82 peptide-bond in human cationic trypsinogen by CTRC is highly specific and other pancreatic proteases do not catalyze this reaction. Furthermore, to investigate the mechanistic basis for this CTRC specificity, we ascertain the P1 preference of pancreatic chymotrypsins and elastases on peptide substrates and use Ala-mutations to examine the role of the P3, P1, P1' P3' and P4' amino-acid positions in the calcium binding loop of human cationic trypsinogen, where P1–P1' corresponds to Leu81-Glu82 (Fig 1). The results indicate that specific cleavage of the Leu81-Glu82 peptide bond by CTRC is largely determined by its unusual preference towards the P1 Leu81, while the P1' Glu82, P3' Asn84 and P4' Glu85 residues serve as additional determinants for specificity.
RESULTS
Cleavage of the Leu81-Glu82 peptide bond in the calcium binding loop of human cationic trypsinogen by human chymotrypsins and elastases
Incubation of cationic trypsinogen with catalytic concentrations (25 nM) of human CTRC in the absence of calcium resulted in rapid cleavage of the Leu81-Glu82 peptide bond in trypsinogen. The relatively stable digestion products were readily visualized by SDS-PAGE and Coomassie blue staining (Fig 2A), and the reaction could be quantitated by densitometry (Fig 2B). Remarkably, other digestive chymotrypsins (CTRB1, CTRB2, CTRL1) or elastases (ELA2, ELA3A, ELA3B) did not catalyze cleavage of this peptide bond to a detectable extent (Fig 2).
Figure 2.
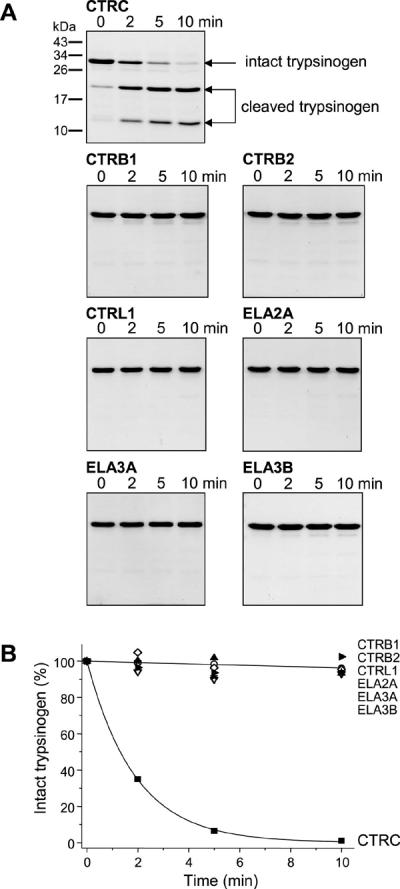
Cleavage of the Leu81-Glu82 peptide bond in human cationic trypsinogen by human pancreatic chymotrypsins and elastases. Trypsinogen (2 μM) was incubated with 25 nM of the indicated proteases in 0.1 M Tris-HCl (pH 8.0) in the presence of 20 nM SPINK1 at 37 °C (final concentrations). (A) At the indicated times, aliquots were withdrawn, precipitated with trichloroacetic acid and analyzed by reducing SDS-PAGE and Coomassie blue staining, as described in Experimental Procedures. Representative gels of two or three experiments are shown. (B) The decrease in the intensity of the trypsinogen band was quantitated by densitometry. Error bars were omitted for clarity, the error was within 10% of the mean.
Primary substrate specificity of human chymotrypsins and elastases
To determine the mechanistic basis for the high specificity of CTRC towards the Leu81-Glu82 peptide bond in human cationic trypsinogen, we characterized the P1 preference of human chymotrypsins and elastases using a library of chromogenic peptide substrates (Table 1, Fig 3). As expected, chymotrypsins cleaved after Leu, Met, Phe, Trp and Tyr with high efficiency with notable isoform-specific differences. CTRC displayed the broadest P1 specificity with comparable activity on Leu, Met, Phe and Tyr substrates. Importantly, CTRC cleaved leucyl peptide bonds with at least 10-fold higher catalytic efficiency than other chymotrypsins. CTRB1 showed a clear preference for P1 Trp and Tyr, whereas CTRB2 and CTRL1 preferred P1 Phe and Tyr. Surprisingly, human elastases all exhibited markedly lower catalytic efficiencies than chymotrypsins, suggesting that elastase activity is more dependent on prime side substrate contacts which are largely absent in the peptide substrates. ELA2A showed CTRC-like broad P1 preference and cleaved after Met, Leu, Phe and Tyr. Still, ELA2A cleaved the Leu substrate with ~70-fold lower catalytic efficiency than CTRC. In contrast to ELA2A, a P1 Ala was preferred by both ELA3A and ELA3B, although the catalytic efficiency of ELA3A was lower by an order of magnitude. ELA3B also cleaved after Val with 15-fold lower activity relative to the P1 Ala. No cleavage was detectable after P1 Gly by any of the proteases tested and only CTRC cleaved peptides with a P1 Asn, Gln, and Ile, with very low efficiency. Taken together, the results indicate that the P1 preference of human chymotrypsins and elastases is restricted and the uniquely high activity of CTRC on leucyl peptide bonds may explain the high cleavage specificity of CTRC on the Leu81-Glu82 peptide bond in trypsinogen.
Table 1.
Enzyme kinetic parameters for human pancreatic chymotrypsins and elastases on Suc-Ala-Ala-Pro-Xaa-p-nitroanilide substrates, where Xaa stands for the amino acids indicated in the table heading. Measurements were performed as described in Experimental Procedures. Blank spaces indicate no measurable activity. Although not shown, none of the proteases tested cleaved Suc-Ala-Ala-Pro-Gly-p-nitroanilide to a detectable extent.
| Ala | Asn | Gln | Ile | Leu | Met | Phe | Trp | Tyr | Val | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| CTRC | kcat | 0.3 | 3.6 | 1.1 | 0.03 | 9.2 | 14.9 | 13.2 | 0.1 | 16.6 | 0.05 |
| KM | 391.7 | 3451.6 | 738.3 | 60.5 | 11.8 | 30.4 | 22.0 | 27.7 | 19.6 | 150.4 | |
| kcat/KM | 7.7 × 102 | 1.0 × 103 | 1.5 × 103 | 5.0 × 102 | 7.8 × 105 | 4.9 × 105 | 6.0×105 | 3.6 × 103 | 8.5 × 105 | 3.3 × 102 | |
|
| |||||||||||
| CTRB1 | kcat | - | - | - | - | 3.0 | 8.0 | 23.6 | 11.0 | 127.9 | - |
| KM | - | - | - | - | 363.2 | 249.4 | 108.5 | 12.9 | 84.4 | - | |
| kcat/KM | - | - | - | - | 8.3 × 103 | 3.2 × 104 | 2.2 × 105 | 8.5 × 105 | 1.5 × 106 | - | |
|
| |||||||||||
| CTRB2 | kcat | - | - | - | - | 7.2 | 19.7 | 22.8 | 3.5 | 66.2 | - |
| KM | - | - | - | 93.2 | 140.8 | 23.2 | 35.6 | 25.3 | - | ||
| kcat/KM | - | - | - | - | 7.7 × 104 | 1.4 × 105 | 9.8 × 105 | 9.8 × 104 | 2.6 × 106 | - | |
|
| |||||||||||
| CTRL1 | kcat | - | - | - | - | 6.7 | 13.3 | 89.4 | 2.4 | 77.3 | - |
| KM | - | - | - | - | 197.5 | 331.3 | 130.6 | 97.1 | 57.2 | - | |
| kcat/KM | - | - | - | - | 3.4 × 104 | 4.0 × 104 | 6.8 × 105 | 2.5 × 104 | 1.4 × 106 | - | |
|
| |||||||||||
| ELA2A | kcat | 1.0 | - | - | - | 11.1 | 10.6 | 9.8 | - | 5.4 | - |
| KM | 3642.1 | - | - | - | 999.5 | 815.2 | 1962.5 | - | 757.1 | - | |
| kcat/KM | 2.7 × 102 | - | - | - | 1.1 × 104 | 1.3 × 104 | 5.0 × 103 | - | 7.1 × 103 | - | |
|
| |||||||||||
| ELA3A | kcat | 5.5 | - | - | - | - | - | - | - | - | - |
| KM | 2435.2 | - | - | - | - | - | - | - | - | - | |
| kcat/KM | 2.3 × 103 | - | - | - | - | - | - | - | - | - | |
|
| |||||||||||
| ELA3B | kcat | 18.9 | - | - | - | - | - | - | - | - | 4.9 |
| KM | 785.1 | - | - | - | - | - | - | - | - | 2978.6 | |
| kcat/KM | 2.4 × 104 | - | - | - | - | - | - | - | - | 1.6 × 103 | |
kcat (s−1), KM (μM), kcat/KM (M−1 s−1)
Figure 3.
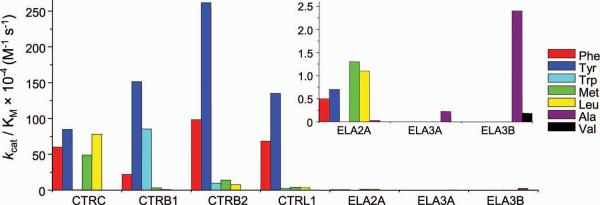
Primary cleavage specificity of human pancreatic chymotrypsins and elastases. Michaelis-Menten kinetic parameters were determined using p-nitroanilide peptide substrates with different P1 amino-acid residues, as described in Experimental Procedures and listed in Table 1. To illustrate differences in P1 preference, the specificity constants (kcat/KM values) were graphed for substrates with a P1 Ala, Leu, Met, Phe, Trp, Tyr and Val.
To validate the peptide substrate results in the context of human cationic trypsinogen, we mutated Leu81 to Ala, Gln, Met, Phe, Trp or Tyr. In agreement with the P1 specificity observed on peptide substrates, CTRC readily cleaved trypsinogen mutants L81F, L81M and L81Y at comparable rates (Fig 4). No significant cleavage by CTRC was observed with mutants L81A, L81Q and L81W (Fig 4). Unexpectedly, none of the six mutants tested was cleaved by other human chymotrypsins and elastases to an appreciable extent (not shown), indicating that besides the P1 position, other subsite interactions also contribute to CTRC specificity toward the Leu81-Glu82 peptide bond in human cationic trypsinogen.
Figure 4.

Effect of mutations of Leu81 on the cleavage of the trypsinogen calcium binding loop by CTRC. Cleavage reactions, SDS-PAGE analysis (A) and densitometric evaluation (B) were carried out as described in Experimental Procedures and in Figure 2. Representative gels of two or three experiments are shown. Error bars were omitted for clarity; the error was within 10% of the mean. Although not shown, none of the six mutants tested was cleaved by CTRB1, CTRB2, CTRL1, ELA2A, ELA3A or ELA3B.
Role of the P2, P1' and P4' Glu residues in CTRC specificity
To define the significance of secondary subsite interactions in CTRC specificity, we first mutated the bulky, negatively charged Glu79, Glu82, and Glu85 amino-acid residues to Ala (Fig 1). CTRC cleaved the three mutants at different rates. Compared to wild-type trypsinogen, cleavage of mutant E79A was unchanged; whereas mutant E82A was cleaved about 3-fold faster and mutant E85A was cleaved about 2-fold slower (Fig 5). These results indicate that the P1' Glu82 inhibits, whereas the P4' Glu85 facilitates CTRC cleavage. With the exception of ELA2A, none of the other chymotrypsins and elastases cleaved the three mutants tested. ELA2A, on the other hand, exhibited slow but measurable cleavage of mutant E79A and cleaved mutant E82A at a significant rate (Fig 5), indicating that the P1' Glu82, and to a lesser extent the P2 Glu79, inhibit ELA2A-mediated cleavage of the Leu81-Glu82 peptide bond.
Figure 5.
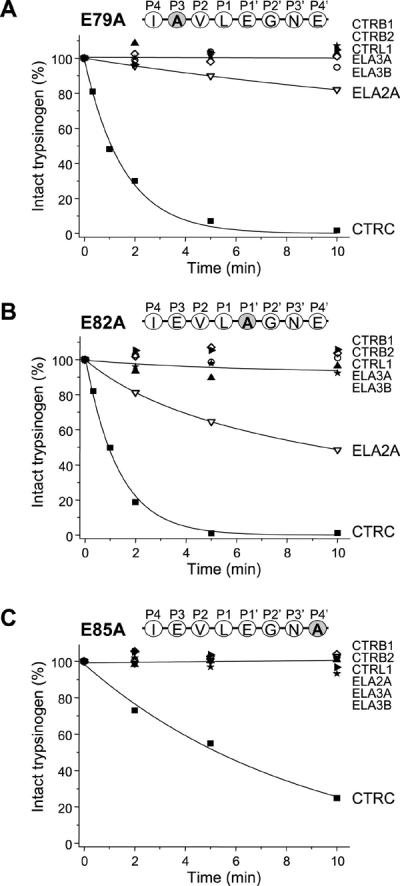
Effect of mutations of Glu79, Glu82 and Glu85 on the cleavage of the trypsinogen calcium binding loop by human pancreatic chymotrypsins and elastases. (A) Mutation of the P3 Glu79 to Ala (E79A). (B) Mutation of the P1' Glu82 to Ala (E82A). (C) Mutation of the P4' Glu85 to Ala (E85A). Cleavage reactions, SDS-PAGE analysis and densitometric evaluation were carried out as described in Experimental Procedures and in Figure 2. The average of two or three experiments is shown. Error bars were omitted for clarity; the error was within 10% of the mean.
In an attempt to compare the effect of the Glu residues on other chymotrypsins, we introduced the E79A, E82A and E85A mutations in the background of the L81F mutant where the P1 Leu was changed to Phe, preferred by all human chymotrypsins. As noted above, the L81F mutation alone did not result in cleavage of cationic trypsinogen by CTRB1, CTRB2 or CTRL1. In contrast, CTRB2 readily cleaved the double mutant L81F,E82A and slowly cleaved mutants E79A,L81F and L81F,E85A (Fig 6). No cleavage was observed by CTRB1, CTRL1 or any of the human elastases. CTRC cleaved all three L81F double mutants at rates that were comparable to those observed with the single E79A, E82A and E85A mutants containing wild-type Leu81 (cf. Figs 5 and 6).
Figure 6.
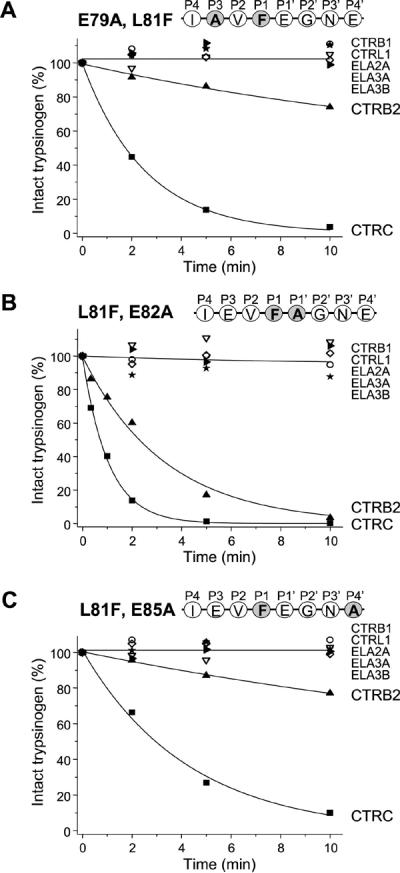
Effect of combined mutations of Leu81 and Glu79, Glu82 or Glu85 on the cleavage of the trypsinogen calcium binding loop by human pancreatic chymotrypsins and elastases. (A) Mutations of the P3 Glu79 to Ala (E79A) and the P1 Leu81 to Phe (L81F). (B) Mutations of the P1 Leu81 to Phe (L81F) and the P1' Glu82 to Ala (E82A). (C) Mutations of the P1 Leu81 to Phe (L81F) and the P4' Glu85 to Ala (E85A). Cleavage reactions, SDS-PAGE analysis and densitometric evaluation were carried out as described in Experimental Procedures and in Figure 2. The average of two or three experiments is shown. Error bars were omitted for clarity; the error was within 10% of the mean.
Role of the P3' Asn residue in CTRC specificity
Mutation of Asn84 to Ala (N84A) had no effect on the cleavage by CTRC but increased cleavage by ELA2A (Fig 7A). None of the other chymotrypsins and elastases cleaved mutant N84A; not even when the P1 Leu was changed to Phe (L81F,N84A) (Fig 7B). On the other hand, slow but measurable cleavage was observed by ELA3B when the L81A mutation was combined with N84A (Fig 7C).
Figure 7.
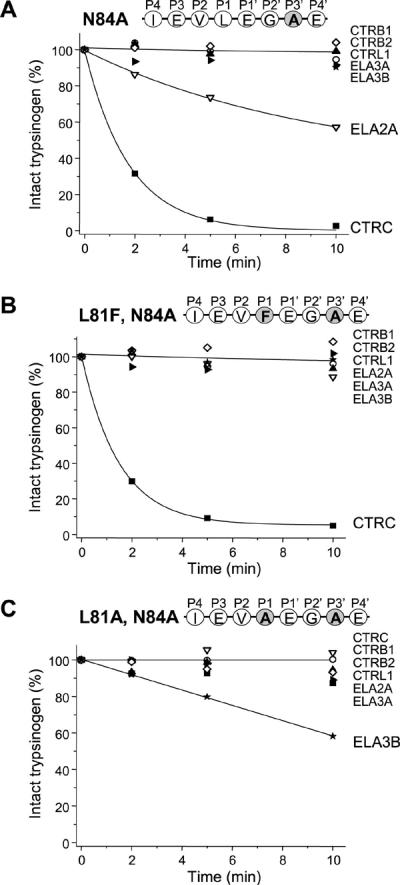
Effect of mutation of Asn84 on the cleavage of the trypsinogen calcium binding loop by human pancreatic chymotrypsins and elastases. (A) Mutation of the P3' Asn84 to Ala (N84A). (B) Mutations of the P1 Leu to Phe (L81F) and the P3' Asn84 to Ala (N84A). (C) Mutations of the P1 Leu to Ala (L81A) and the P3' Asn84 to Ala (N84A). Cleavage reactions, SDS-PAGE analysis and densitometric evaluation were carried out as described in Experimental Procedures and in Figure 2. The average of two or three experiments is shown. Error bars were omitted for clarity; the error was within 10% of the mean.
DISCUSSION
In the present study we demonstrate that the CTRC-mediated regulatory cleavage of the Leu81-Glu82 peptide bond in the calcium-binding loop of human cationic trypsinogen is highly specific and no other pancreatic chymotrypsin or elastase catalyzes this reaction. Previous studies already suggested that this cleavage was CTRC specific [14], however, these preliminary data had a number of limitations: (i) cleavage of the Leu81-Glu82 peptide bond was inferred from the activity loss of trypsin and not observed directly on gels; (ii) concentrations of enzyme preparations used were estimated from ultraviolet absorbance; and (iii) CTRL1 was not tested. Here, we used the full complement of active-site titrated human pancreatic chymotrypsins and elastases and followed cleavage of Leu81-Glu82 directly on SDS-PAGE. To clarify the mechanistic basis for this CTRC specificity, we determined the P1 preference for all human pancreatic chymotrypsins and elastases using chromogenic peptide substrates. Furthermore, we used Ala-mutations in the calcium binding loop of cationic trypsinogen to examine the role of the P3, P1, P1' P3' and P4' amino-acid positions (where P1-P1' corresponds to Leu81-Glu82) in the cleavage reaction. Taken together, the results from these two approaches indicated that (i) human chymotrypsins and elastases exhibited restricted P1 specificity and only CTRC cleaved after a P1 Leu with high activity; (ii) mutations of the P3 Glu79 (L79A), P1' Glu82 (E82A) and P3' Asn84 (N84A) weakened specificity and resulted in cleavage by ELA2A; (iii) mutation of the P4' Glu85 (E85A) decreased cleavage by CTRC; (iv) no cleavage by other chymotrypsins and elastases was observed even when Leu81 was mutated to favorable residues (L81F, L81A); (v) double mutants L81F,E82A and L81A,N84A were cleaved by CTRB2 and ELA3B, respectively. Thus, the high cleavage specificity of CTRC on the Leu81-Glu82 peptide bond is explained by its P1 leucyl preference and extended subsite interactions unfavorable to other proteases, related mostly to the P1' and P3' positions.
In a recent study we used phage display technology to characterize the amino-acid determinants underlying CTRC specificity [17]. We selected high-affinity substrate-like small protein inhibitors against CTRC from a phage library displaying variants of a natural chymotrypsin inhibitor from Schistocerca gregaria (SGPI-2). The results identified the amino acid preference of CTRC at four positions of the reactive loop, i.e. at P1, P1', P2' and P4'. With respect to the P1 residue, we found that predominantly Leu and Met was selected by phage display and inhibitors with Leu at P1 bound 10-fold stronger to CTRC than those with P1 Met. In contrast, here we demonstrated using peptide substrates (see Fig 3 and Table 1) and trypsinogen mutants (see Fig 4) that CTRC cleaved after Leu, Met, Phe and Tyr P1 residues with comparable efficiency. While an explanation for this apparent contradiction is not readily apparent, the more rigid reactive loop of inhibitors may restrict P1 preference to a larger degree than observed with the relatively flexible substrates. At position P1' mostly Met was selected in the inhibitors by phage display, although Leu and Ala also appeared in some clones, suggesting hydrophobic preference. Consistent with this notion, mutation of the P1' Glu82 to Ala (E82A) in the calcium binding loop of cationic trypsinogen increased cleavage by CTRC about 3-fold (see Fig 5). At position P2' the phage display yielded mostly Leu in the inhibitors but other amino-acids including Asp and Glu were also selected with low frequency, suggesting that CTRC is tolerant to changes at this position. Finally, selection for the P4' residue in the inhibitors was almost exclusively Glu or Asp and these acidic residues at P4' increased affinity of inhibitors to CTRC by 10-fold (versus Ala). Similarly, here we found that the presence of a Glu (versus Ala) at the P4' position in the calcium binding loop of cationic trypsinogen increased the rate of cleavage by CTRC about 2-fold (see Figs 5 and 6).
The enrichment of acidic amino-acid residues on the prime side of the scissile peptide bonds seems to be a general feature of regulatory sites cleaved by CTRC. As mentioned in the introduction, CTRC processes the trypsinogen activation peptide at the Phe18-Asp19 peptide bond, which is flanked by three Asp residues on the prime side (Asp20-Asp21-Asp22). CTRC also promotes activation of human pro-carboxypeptidases A1 and A2 by cleaving the activation peptides at the conserved Leu96-Leu97 peptide bond flanked by the acidic prime side sequences of Asp98-Glu99-Glu100 and Asp98-Lys99-Glu100, respectively [18]. On the basis of data presented here, it appears that the P1' acidic residues increase selectivity against other chymotrypsins and/or elastases, although at the expense of catalytic efficiency. In the context of phage-display selected CTRC inhibitors, the P2' acidic residues were previously shown to play a similar role [17]. The significance of P3' acidic residues has not been studied yet. Finally, the P4' acidic residues were demonstrated to increase affinity of inhibitors to CTRC and to increase cleavage efficiency of substrates by CTRC. This latter subsite interaction involves a cluster of basic amino-acid residues on the surface of human CTRC formed by Lys51, Arg56 and Arg80, which interacts with the negatively charged P4' position of inhibitors and substrates [17].
In summary, CTRC cleaves the Leu81-Glu82 peptide bond in the calcium-binding loop of human cationic trypsinogen with high specificity over other pancreatic chymotrypsins and elastases, which is determined by the unique P1 leucine preference of CTRC and extended subsite interactions on the prime side of the scissile peptide bond.
EXPERIMENTAL PROCEDURES
Nomenclature
Amino acid residues in human pancreatic proteases were numbered starting with the initiator methionine of the primary translation product; according to the recommendations of the Human Genome Variation Society. Note that the ELA (elastase) gene symbols for the five human pancreatic elastase genes have been recently changed to CELA (chymotrypsin-like elastase). In this paper we used the old ELA symbols, which may be more familiar to readers.
Plasmid construction and mutagenesis
The pTrapT7 and pcDNA3.1(−) expression plasmids harboring the coding DNA for human pancreatic digestive proteases were described previously [10, 13, 14, 19, 20]. Missense mutations in human cationic trypsinogen (PRSS1) were generated by overlap extension PCR mutagenesis and cloned in the pTrapT7 expression plasmid.
Expression, refolding and purification of trypsinogen and ELA2A proelastase
Human cationic trypsinogen and proelastase ELA2A were expressed in E. coli BL21(DE3) as cytoplasmic inclusion bodies. In vitro refolding and purification by ecotin affinity chromatography were carried out as described previously [10, 15, 19, 20]. Trypsinogen concentrations were estimated from ultraviolet absorbance at 280 nm using the extinction coefficient 37,525 M−1 cm−1. ELA2A was activated with 10 nM human anionic trypsin in 0.1 M Tris (pH 8.0), 0.05% Tween 20 (final concentrations), and active elastase concentration was determined by active site titration with ecotin, as reported recently [17].
Expression and purification of human chymotrypsinogens and proelastases ELA3A and ELA3B
Chymotrypsinogens CTRB1, CTRB2, CTRC, CTRL1 and proelastases ELA3A, ELA3B were expressed in HEK 293T cells and purified from 150–200 mL conditioned medium by ecotin affinity chromatography, using published protocols [14, 21]. Alternatively, C-terminally His-tagged forms were expressed and purified by nickel-affinity chromatography, as described elsewhere [15]. The His-tagged and non-tagged enzymes were indistinguishable functionally. CTRB1, CTRB2, CTRC, CTRL1 and ELA3B were activated with immobilized bovine trypsin in 0.1 M Tris-HCl (pH 8.0), 0.05% Tween 20, and the trypsin-beads (Pierce, Thermo Fisher Scientific, Inc., Rockford, IL) were removed by centrifugation. ELA3A was activated with 10 nM human cationic trypsin. Active protease concentrations were determined by active site titration against ecotin [17].
Enzyme kinetic measurements
Michaelis-Menten kinetic parameters of human chymotrypsins and elastases were determined on Suc-Ala-Ala-Pro-Xaa-pNA chromogenic substrates, where Xaa was Ala, Asn, Gln, Gly, Leu, Ile, Met, Phe, Trp, Tyr or Val. The peptides were purchased from Bachem Americas, Inc. (Torrance, Inc) and Peptides International, Inc. (Louisville, KY), or custom-synthesized (Asn, Gln, Tyr) by ChemPep, Inc. (Wellington, FL). Substrate concentrations were varied between 0.02 mM and 27 mM, in a final volume of 200 μL 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, 0.05% Tween 20 (final concentrations). Measurements were initiated by adding 20 μL enzyme solution to 0.02–1.5 μM final concentration. The release of the yellow p-nitroaniline was followed for 1 min at 405 nm in a SpectraMax Plus384 microplate reader (Molecular Devices, Sunnyvale, CA) and the rate of substrate cleavage was determined from the linear portions of the curves. KM and vmax values were calculated from hyperbolic fits to plots of velocity versus substrate concentration.
Analysis of trypsinogen cleavage by SDS-PAGE
Wild-type or mutant human cationic trypsinogen was incubated at 2 μM with 25 nM chymotrypsin or elastase in 0.1 M Tris-HCl (pH 8.0) containing 20 nM human SPINK1 trypsin inhibitor (final concentrations). The trypsin inhibitor was included to prevent trypsinogen autoactivation. At this concentration SPINK1 had no inhibitory effect on human chymotrypsins and elastases. Since altered calcium binding in some trypsinogen mutants may confound interpretation of the protease cleavage results, all experiments were performed in the absence of calcium. The digestion mixture was incubated at 37 °C and at given time points 75 μL aliquots were withdrawn and the reaction was stopped by adding 10% trichloroacetic acid (final concentration). The precipitate was collected by centrifugation, dissolved in 15 μL Laemmli sample buffer containing 100 mM dithiothreitol, heat denatured at 90 °C for 5 min and electrophoresed on 15% SDS-polyacrylamide minigels. Gels were stained with Coomassie blue R-250. Loss of the intact trypsinogen band was quantitated by densitometry using Quantity One 4.6.9. software (Bio-Rad, Hercules CA).
ACKNOWLEDGMENTS
This work was supported by NIH grants R01DK082412, R01DK082412-S2, and R01DK058088 (to MS-T).
REFERENCES
- 1.Chen JM, Férec C. Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas. 2000;21:57–62. doi: 10.1097/00006676-200007000-00052. [DOI] [PubMed] [Google Scholar]
- 2.Rinderknecht H. Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci. 1986;31:314–321. doi: 10.1007/BF01318124. [DOI] [PubMed] [Google Scholar]
- 3.Guy O, Lombardo D, Bartelt DC, Amic J, Figarella C. Two human trypsinogens. Purification, molecular properties, and N-terminal sequences. Biochemistry. 1978;17:1669–1675. doi: 10.1021/bi00602a014. [DOI] [PubMed] [Google Scholar]
- 4.Rinderknecht H, Renner IG, Carmack C. Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics and patients with pancreatitis and cancer of the pancreas. Gut. 1979;20:886–891. doi: 10.1136/gut.20.10.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinderknecht H, Stace NH, Renner IG. Effects of chronic alcohol abuse on exocrine pancreatic secretion in man. Dig Dis Sci. 1985;30:65–71. doi: 10.1007/BF01318373. [DOI] [PubMed] [Google Scholar]
- 6.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 7.Yamashina I. The action of enterokinase on trypsinogen. Acta Chem Scand. 1956;10:739–743. doi: 10.1016/0006-3002(56)90329-8. [DOI] [PubMed] [Google Scholar]
- 8.Kukor Z, Tóth M, Sahin-Tóth M. Human anionic trypsinogen: properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur J Biochem. 2003;270:2047–2058. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 9.Rose SD, MacDonald RJ. Evolutionary silencing of the human elastase I gene (ELA1) Hum Mol Genet. 1997;6:897–903. doi: 10.1093/hmg/6.6.897. [DOI] [PubMed] [Google Scholar]
- 10.Szepessy E, Sahin-Tóth M. Inactivity of recombinant ELA2B provides a new example of evolutionary elastase silencing in humans. Pancreatology. 2006;6:117–122. doi: 10.1159/000090031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557–1573. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J, Sahin-Tóth M. Chymotrypsin C (CTRC) mutations in chronic pancreatitis. J Gastroenterol Hepatol. 2011;26:1238–1246. doi: 10.1111/j.1440-1746.2011.06791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nemoda Z, Sahin-Tóth M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J Biol Chem. 2006;281:11879–11886. doi: 10.1074/jbc.M600124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szmola R, Sahin-Tóth M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc Natl Acad Sci USA. 2007;104:11227–11232. doi: 10.1073/pnas.0703714104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabó A, Sahin-Tóth M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J Biol Chem. 2012;287:20701–20710. doi: 10.1074/jbc.M112.360065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosendahl J, Witt H, Szmola R, Bhatia E, Ózsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, Kovács P, Blüher M, Stumvoll M, Choudhuri G, Hegyi P, te Morsche RH, Drenth JP, Truninger K, Macek M, Jr, Puhl G, Witt U, Schmidt H, Büning C, Ockenga J, Kage A, Groneberg DA, Nickel R, Berg T, Wiedenmann B, Bödeker H, Keim V, Mössner J, Teich N, Sahin-Tóth M. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabó A, Héja D, Szakács D, Zboray K, Kékesi KA, Radisky ES, Sahin-Tóth M, Pál G. High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4' acidic residues. J Biol Chem. 2011;286:22535–22545. doi: 10.1074/jbc.M111.235754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szmola R, Bence M, Carpentieri A, Szabó A, Costello CE, Samuelson J, Sahin-Tóth M. Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2. J Biol Chem. 2011;286:1819–1827. doi: 10.1074/jbc.M110.187369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahin-Tóth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275:22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 20.Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun. 2000;278:286–289. doi: 10.1006/bbrc.2000.3797. [DOI] [PubMed] [Google Scholar]
- 21.Bence M, Sahin-Tóth M. Asparagine-linked glycosylation of human chymotrypsin C is required for folding and secretion but not for enzyme activity. FEBS J. 2011;278:4338–4350. doi: 10.1111/j.1742-4658.2011.08351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]