

Abstract
Objective
Tolvaptan, an oral antagonist of the vasopressin V2 receptor, has been found to improve hyponatremia in patients with mixed etiologies. This study analyzed a subgroup of patients with the syndrome of inappropriate antidiuretic hormone secretion (SIADH) to evaluate the efficacy and safety of tolvaptan in this group.
Design and patients
Hyponatremic patients in the SALT-1 and SALT-2 studies with a diagnosis of SIADH were identified based on clinical diagnosis by individual study investigators. Subjects were randomized to receive oral placebo (n=52) or tolvaptan 15 mg daily, with further titration to 30 and 60 mg daily, if necessary, based on the response of serum [Na+] (n=58).
Results
In patients with SIADH, improvement in serum [Na+] was significantly greater (P<0.0001) with tolvaptan than placebo over the first 4 days of therapy as well as the entire 30-day study, with minimal side effects of increased thirst, dry mouth, and urination. Only 5.9% of tolvaptan-treated patients had overly rapid correction of hyponatremia as defined by current guidelines. After discontinuation of tolvaptan, serum [Na+] declined to values similar to placebo. A significant positive treatment effect favoring tolvaptan on the physical component, and a near-significant trend on the mental component, was found using the SF-12 Health Survey. Tolvaptan was associated with a significantly reduced incidence of fluid restriction.
Conclusions
Results for the SIADH subgroup were analogous to those of the combined SALT population regarding efficacy and safety but demonstrated a greater improvement in the physical component of the SF-12 Health Survey than in the full mixed etiology SALT patient group.
Introduction
Hyponatremia is the most common electrolyte abnormality encountered in clinical practice, affecting up to 15–28% of hospitalized patients across numerous studies over the last several decades throughout the world (1, 2). The high prevalence of hyponatremia in hospitalized patients reflects the increased vulnerability of this patient population to disruptions of body fluid homeostasis. In addition to its frequency, hyponatremia is also important because it has been associated with worse clinical outcomes across the entire range of inpatient care, from the general hospital population (3) to those treated in the intensive care unit (4). In virtually every disease ever studied, the presence of hyponatremia has been found to be an independent risk factor for increased mortality, from congestive heart failure to tuberculosis to liver failure (2).
Tolvaptan is a novel, orally administered, selective, non-peptide arginine vasopressin (AVP) receptor (AVPR) antagonist that blocks binding of AVP to V2 receptors in the distal nephron, thereby preventing the antidiuresis caused by circulating AVP. AVPR antagonists, also called ‘vaptans’, produce an electrolyte-free water excretion, or aquaresis, without significantly affecting renal sodium and potassium excretion (5). The net result is a reduction in body water without loss of body electrolytes, which leads to an increase in the serum [Na+]. Because >90% of patients with euvolemic and hypervolemic hyponatremia have non-suppressed levels of AVP in their blood (6, 7), AVPR antagonists directly attack the underlying pathophysiology of most cases of dilutional hyponatremia.
Two randomized, placebo-controlled, double-blind studies (SALT-1 and SALT-2) examined the effect of tolvaptan on hypervolemic and euvolemic hyponatremia of different etiologies and found that tolvaptan was an efficacious and safe therapy for the treatment of hyponatremia of diverse origin (8). Based on these phase 3 studies, tolvaptan was approved by the FDA for the treatment of euvolemic and hypervolemic hyponatremia in the United States in 2008, and by the EMEA for the treatment of hyponatremia due to syndrome of inappropriate antidiuretic hormone (SIADH) in the European Union in 2009.
The goal of this study was to examine whether the major findings of the SALT trials in a mixed population of different etiologies also applied to the subgroup of patients in SALT-1 and SALT-2 diagnosed with SIADH. In particular, we evaluated the efficacy and safety of tolvaptan therapy in this subgroup of hyponatremic patients from the SALT studies, including the patient responses to the SF-12 Health Survey administered before and after 30 days of treatment with either tolvaptan or placebo.
Methods
Study design
SALT-1 and SALT-2 were randomized, double-blind, placebo-controlled, phase 3 studies assessing inpatient/ outpatient use of tolvaptan for hyponatremia of diverse origin. Hyponatremia was defined as a non-artifactual serum [Na+] of <135 mmol/l. Patients were eligible to participate in the trials if they were 18 years of age or older and had euvolemic or hypervolemic hyponatremia, often in association with chronic heart failure, cirrhosis, or SIADH. Eligible patients were randomly assigned to once-daily oral placebo (223 patients) or oral tolvaptan (225 patients) for 30 days. Initial dosage of the study drug was 15 mg, which could be increased from 15 to 30 mg or from 30 to 60 mg during the initial 4 days of therapy according to a regimen designed for slow correction of serum [Na+] to ≥135 mmol/l. All patients were hospitalized for the first day of the study, but continued hospitalization was based on the determination of each investigator. Detailed information on the study design, inclusion and exclusion criteria, dose titration scheme, disposition, demographics, and outcomes of the SALT trials are available in a prior publication (8).
The institutional review board or ethics committee at each site approved the study protocol and ensured that written informed consent was obtained from all patients. The study adhered to the Ethical Principles for Medical Research Involving Human Subjects, World Medical Association Declaration of Helsinki.
The current analysis examined the subgroup of patients with a diagnosis of SIADH from the original SALT studies. Patients were identified based on a primary clinical diagnosis of SIADH made by individual study investigators, as recorded on their case report forms. This group is referred to as ‘investigator-diagnosed’ SIADH. Because the SALT trials included patients with congestive heart failure and cirrhosis as well as SIADH, an elevated urine sodium concentration was not used as an inclusion criterion, because this would have eliminated many of the patients with secondary hyperaldosteronism due to heart failure or hepatic cirrhosis. In order to verify that the findings in patients with a clinical diagnosis of SIADH were analogous to those who more formally met the usual criteria for this diagnosis (9), the efficacy data from a subgroup of the SIADH patients who had elevated urine sodium levels (>20 mmol/l) in a 24 h urine collection on the first day of tolvaptan or placebo therapy were also analyzed and compared with the investigator-diagnosed group. A total of 24 patients in the tolvaptan group and 25 patients in the placebo group met the criteria of elevated urine sodium levels (tolvaptan: mean urine sodium 123±65 mmol/l, range 32–290 mmol/l; placebo: mean urine sodium 109±64 mmol/l, range 23–228 mmol/l). This subgroup is referred to as ‘lab-diagnosed’ SIADH. Although this subgroup represented only approximately half of the patients who had a clinical diagnosis of SIADH, this is not surprising in view of the fact that the only urine sodium data available was from the first day of tolvaptan or placebo treatment, where a stimulated aquaresis would be expected to dilute the measured urine sodium concentration. All SALT patients, regardless of diagnosis, were confirmed to have normal TSH and morning cortisol levels, measured by a central laboratory, at baseline prior to entry into the study. Although baseline urine osmolalities were not measured, patients with psychogenic polydipsia and uncontrolled hyperglycemia were excluded from the SALT studies.
Assessments
Patients were evaluated at baseline, 8 h after the first administration of the study drug, and pre-dose on days 2, 3, 4, 11, 18, 25, 30, and 37. Study drugs were withheld after day 30, and the effect of discontinuation of the study drug was assessed on day 37.
Efficacy assessments included the change in the average daily area under the curve (AUC) for the serum [Na+] from baseline to day 4 and from baseline to day 30, the absolute serum [Na+] at each visit, and the percentage of patients with serum [Na+] that had normalized (>135 mmol/l) at each visit. Other end points included fluid intake and output on day 1, institution of fluid restriction or use of i.v. saline as rescue therapy, and the change from baseline in scores on the Physical Component Summary (PCS) and Mental Component Summary (MCS) of the Medical Outcomes Study 12-item Short-Form (SF-12) General Health Survey.
Safety was assessed at all visits by adverse events, vital signs, and directed physical examinations (i.e. based on the patient’s symptoms and diagnosis). Adverse events were defined as any new medical problem or exacerbation of an existing medical problem according to the Medical Dictionary for Regulatory Activities (MedDRA, registered trademark of the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA)). Patients were instructed to report such events to an investigator at each visit with a non-leading question. Each investigator was required to assess and report to the sponsor the seriousness and severity of each event and whether the event was potentially associated with the study drug. The sponsor then reported such events to the appropriate regulatory authorities and to the study’s independent safety oversight committee.
Statistical analysis
All reported P values are two sided. The change in the average daily AUC for the serum [Na+] from baseline to day 4 and from baseline to day 30 was calculated as the AUC for each patient, divided by the observation period (4 or 30 days), minus the baseline value. The serum [Na+] changes in the two study groups were compared with an analysis of covariance (ANCOVA) model in which the group assignment and baseline stratification factors were covariates. Serum [Na+] changes were compared between study groups with the use of the ANCOVA model and the covariates noted above. The percentage of patients in whom serum [Na+] normalized (>135 mmol/l) or in whom fluid restricted was analyzed with the Cochran–Mantel–Haenszel test.
Fluid loss, fluid intake, and fluid balance (total intake minus total output) on day 1 were evaluated with the use of an ANOVA model, with the assigned study group and baseline stratification factors as covariates. The PCS and MCS scales of the SF-12 Health Survey (ranges, 5–69 for the physical component and 8–73 for the mental component, with higher scores indicating better functioning) were derived with the use of weights provided in the SF-12 General Health Survey manual (10, 11). The SF-12 General Health Survey was chosen as a patient-reported outcome for overall health status because it has been validated in numerous clinical studies. The physical component assesses physical functioning, bodily pain, physically limited accomplishment, and general health and the mental component assesses vitality, social functioning, emotionally limited accomplishment, calmness, and sadness. An absolute shift from baseline of 3 units was considered a minimal clinically important difference (12).
Results
Patients
A total of 448 patients received study medication in the SALT trials, 110 of whom had a primary diagnosis of SIADH derived by the investigator using standard clinical criteria (Fig. 1). Of the 52 SIADH patients who were randomly assigned to oral tolvaptan, 51 received therapy and were included in the safety and efficacy analyses. All 58 SIADH subjects randomly assigned to oral placebo received therapy and were included in the safety and efficacy analyses. In each treatment group, 42 patients completed the full 30-day treatment period. Reasons for subject discontinuations are described in Fig. 1 and below (see Safety). Demographic characteristics of patients in both treatment groups were similar (Table 1). In the tolvaptan and placebo groups, 30 (57.7%) and 29 (50.0%) patients respectively had marked hyponatremia (defined as serum [Na+]<130 mmol/l). The remainder had moderate hyponatremia (≥130 and<135 mmol/l). As expected, the great majority of patients in both treatment groups were adjudged to be euvolemic by clinical examination (86.5 and 89.7% in the tolvaptan and placebo groups respectively). Because the presence of any edema was a basis for classifying patients as hypervolemic, some patients with SIADH with edema that was not caused by heart failure or cirrhosis would have been classified as hypervolemic rather than euvolemic.
Figure 1.

Flow diagram for the combined SIADH subgroups in SALT-1 and SALT-2.
Table 1.
Demographics and patient characteristics.
| Tolvaptan | Placebo | |
|---|---|---|
| Total patients, n (%) | 52 (100) | 58 (100) |
| Mean age, years (±S.D.) | 64 (15) | 65 (14) |
| Race, n (%) | ||
| White | 45 (86.5) | 52 (89.7) |
| Black | 3 (5.8) | 5 (8.6) |
| Hispanic | 2 (3.8) | 1 (1.7) |
| Asian | 1 (1.9) | 0 (0.0) |
| Other | 1 (1.9) | 0 (0.0) |
| Baseline serum [Na+], n (%) | ||
| <130 mmol/l | 30 (57.7) | 29 (50.0) |
| ≥130 mmol/l | 22 (42.3) | 29 (50.0) |
Efficacy
The primary efficacy outcome in SALT was the change in the average daily AUC for the serum [Na+] from baseline to day 4 and from baseline to day 30. For the SIADH subgroup, patients on tolvaptan exhibited a highly significant (P<0.0001) improvement in this parameter relative to placebo at day 4 (5.28±3.35 vs 0.47±2.81 mmol/l respectively) and day 30 (8.07 ±4.55 vs 1.89±4.13 mmol/l) (Fig. 2, left). The subgroup of patients with lab-diagnosed SIADH who had a mean urine sodium level >20 mmol/l on the first day of the study showed similar results at day 4 (4.61±1.97 vs 0.96±2.78 mmol/l; P<0.0001) and day 30 (6.28 ±3.17 vs 2.03±4.37 mmol/l; P<0.0001) (Fig. 2, left). Because 17 of the 49 patients in the lab-diagnosed SIADH group were on diuretics at baseline, the lab-diagnosed subgroup was re-analyzed excluding these 17 patients; the results were not significantly different from the analysis of the entire lab-diagnosed group.
Figure 2.
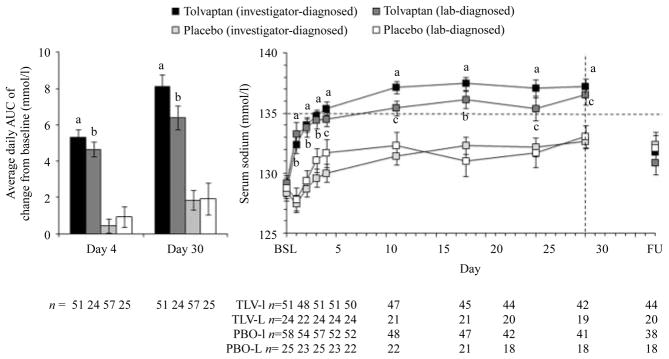
Average daily AUC of change from baseline in serum [Na+] (LOCF, left) and serum [Na+] (patients on treatment, right) in the combined SIADH subgroups of SALT-1 and SALT-2. Investigator-diagnosed patients are those who received a primary diagnosis of SIADH from the investigator. Lab-diagnosed patients are those with an investigator diagnosis of SIADH who also had a urine sodium concentration >20 mmol/l during the first day of therapy. BSL, baseline; FU, 7-day follow-up visit. aP<0.0001, tolvaptan (investigator diagnosed) versus placebo (investigator diagnosed); bP≤0.001, tolvaptan (lab diagnosed) versus placebo (lab diagnosed); cP≤0.029, tolvaptan (lab diagnosed) versus placebo (lab diagnosed). Error bars are ±S.E.M.
The tolvaptan group had significantly higher mean serum [Na+] than the placebo group at all post-baseline time points (Fig. 2, right). This was true in the population with investigator-diagnosed SIADH, as well as those with lab-diagnosed SIADH who had elevated urine sodium levels. In the tolvaptan group, mean serum [Na+] rose to >135 mmol/l in ~3–4 days and remained there throughout the treatment phase. In the placebo group, serum [Na+] remained <135 mmol/l throughout the study period. Withdrawal of tolvaptan therapy resulted in the re-establishment of baseline hyponatremic serum [Na+] within 7 days, consistent with the results of the earlier SALT studies (8), which showed that the aquaretic effect of tolvaptan (excretion of electrolyte-free water) was required to maintain normal serum [Na+] in patients with chronic hyponatremia. Finally, the increase in mean serum [Na+] in the tolvaptan group relative to the placebo group was associated with a higher frequency of patients with normalized serum [Na+] (>135 mmol/l) at all time points measured, including as early as 8 h after initiation of treatment (Fig. 3).
Figure 3.
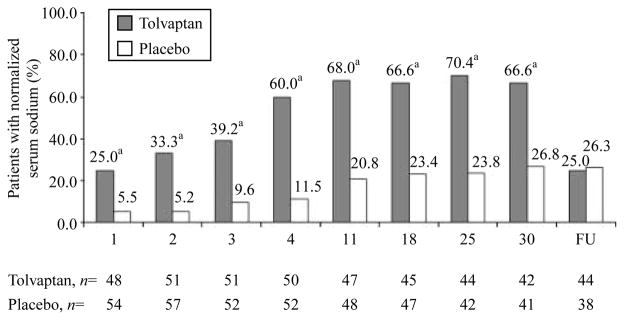
Serum [Na+] normalization rates in the combined SIADH subgroups of SALT-1 and SALT-2 (investigator diagnosed) compared with placebo at all days of measurement across the 30-day treatment period as well as 7 days after cessation of therapy (FU). aP<0.05 compared with placebo group.
Urine output and fluid intake were monitored in patients who had their urine collected for ≥22 and ≤26 h over the first day (Fig. 4). Relative to the placebo group, the tolvaptan group exhibited both larger mean fluid intake (2016±1234 vs 1563±966 ml; P=0.049) and larger mean urine output (3057±1701 vs 1758±928 ml; P<0.001). However, because the treatment effects favoring tolvaptan were greater for urine output than fluid intake, patients on tolvaptan significantly lost more net fluid over the first study day than those on placebo (1109±1259 vs 220±857 ml; P<0.001).
Figure 4.

Urine output, fluid intake, and fluid balance in patients who had their urine collected for ≥ 22 and ≤ 26 h in the combined SIADH subgroups of SALT-1 and SALT-2 (investigator diagnosed). Fluid balance was calculated as fluid intake minus urine output without correction for insensible losses, which were assumed to be equivalent in both groups. Error bars are ±S.E.M.
Of the 51 patients treated with tolvaptan, four (7.8%) required institution of a fluid restriction (defined as ≤1000 ml/day) at any time in the 30-day treatment period, as compared with eight of 58 (13.7%) patients on placebo (P=0.494, χ2 test).
Patient’s self-assessed health status was determined at baseline and day 30 using the SF-12 General Health Survey (Table 2). Over the 30-day study period, the tolvaptan group showed significantly greater improvement in mean PCS scores than the placebo group (3.64±9.55 vs −0.16±8.85; P=0.019). The tolvaptan group also showed a greater improvement in mean MCS scores (5.47±12.01 vs −0.45±9.66); the difference approached closely, but did not reach, statistical significance (P=0.051).
Table 2.
SF-12 General Health Survey summary scores in the combined SIADH subgroups in SALT-1 and SALT-2. Positive changes from baseline indicate improvement.
| Tolvaptan mean ±S.D. (n) |
Placebo mean ± S.D. (n) |
P value | |
|---|---|---|---|
| PCS score | |||
| Baseline | 34.82±10.76 (49) | 34.15±10.06 (54) | – |
| Day 30 | 39.91±10.68 (40) | 34.54±9.67 (41) | – |
| Change from baseline | 3.64±9.55 (39) | −0.16±8.85 (41) | 0.019 |
| MCS score | |||
| Baseline | 44.90±11.56 (49) | 47.12±10.91 (54) | – |
| Day 30 | 51.02±11.81 (40) | 48.47±11.86 (41) | – |
| Change from baseline | 5.47±12.01 (39) | −0.45±9.66 (41) | 0.051 |
Safety
SIADH patients in the SALT trials were exposed to 1396 patient-days (3.8 patient-years) of tolvaptan therapy and 1425 patient-days (3.9 patient years) of placebo therapy (Table 3). Profiles of potentially drug-related adverse events were relatively similar between the two treatment groups (Table 3). Thirst and dry mouth, the most common tolvaptan-related adverse events in the SALT trials (8), occurred in nine (18%) and eight (16%) patients respectively on tolvaptan and five (9%) and six (10%) patients respectively on placebo in this SIADH subgroup analysis. The potentially drug-related adverse events of dizziness, vomiting, hypotension, and nasopharyngitis occurred at slightly higher rates in the placebo group.
Table 3.
Investigator-assessed treatment-emergent adverse events.
| Tolvaptan | Placebo | |
|---|---|---|
| Treated subjects, n | 51 | 58 |
| Drug exposure | ||
| Patient-days | 1396 | 1425 |
| Most common potentially drug-related AEs, n (%)a | ||
| Thirst | 9 (17.6) | 5 (8.6) |
| Dry mouth | 8 (15.7) | 6 (10.3) |
| Headache | 7 (13.7) | 5 (8.6) |
| Peripheral edema | 6 (11.8) | 7 (12.1) |
| Urinary frequency | 5 (9.8) | 4 (6.9) |
| Weakness | 5 (9.8) | 3 (5.2) |
| Fatigue | 5 (9.8) | 2 (3.4) |
| Nausea | 4 (7.8) | 3 (5.2) |
| Constipation | 4 (7.8) | 2 (3.4) |
| Dizziness | 3 (5.9) | 7 (12.1) |
| Diarrhea | 3 (5.9) | 4 (6.9) |
| Hypokalemia | 3 (5.9) | 3 (5.2) |
| Chest pain | 3 (5.9) | 1 (1.7) |
| Insomnia | 2 (3.9) | 3 (5.2) |
| Vomiting | 1 (2.0) | 5 (8.6) |
| Hypotension | 1 (2.0) | 4 (6.9) |
| Nasopharyngitis | 1 (2.0) | 4 (6.9) |
AEs, adverse events.
Occurring in >5% of patients in either treatment group.
In the tolvaptan and placebo groups, ten (19%) and 16 (28%) patients respectively discontinued from the trial before completing the 30-day treatment period. Of these, five (10%) on tolvaptan and seven (12%) on placebo withdrew specifically for adverse experiences. No patient in the tolvaptan group experienced a serious adverse event considered by the investigator to be related to study medication. Four patients in the placebo group experienced six potentially drug-related serious adverse events (rash, vomiting, dyspnea, peripheral edema, blood creatinine increase, and hematocrit decrease). In this population of patients, four deaths were reported (one in the tolvaptan group and three in the placebo group); none of the deaths were considered to be treatment related.
Of the 51 patients treated with tolvaptan, three (5.9%) exceeded protocol recommended correction limits of an increase in serum [Na+]>12 mmol/l over the first 24 h of correction and >18 mmol/l over the first 48 h of correction: one with a correction of 13 mmol/l and two with a correction of 14 mmol/l over the first 24 h of therapy. All three of the patients with overly rapid correction had marked hyponatremia (baseline serum [Na+]<130 mmol/l). None of the overly corrected patients were reported to exhibit any neurological symptoms suggestive of osmotic demyelination.
Length of stay (LOS) was not designated as an outcome measure in the SALT studies, but post hoc analysis of this variable is possible because the SALT protocol only mandated 1 day of hospital stay in order to collect a 24 h urine following the first day of tolvaptan or placebo dosing. The need to continue hospitalization for all further days of was left to the discretion of each individual investigator. Mean hospital LOS for the combined SALT-1 and SALT-2 SIADH populations was slightly lower for patients on tolvaptan, but this difference was not significant: tolvaptan=4.98 ±6.61 versus placebo=6.19±7.89 days (P=0.326). However, analysis by severity of hyponatremia indicated that there were differences in LOS depending on the starting serum [Na+]. Patients with mild hyponatremia, defined as a serum [Na+] ≥130 mmol/l, had shorter LOS (tolvaptan=3.00±1.24 versus placebo= 4.61±6.20 days, P=0.212) compared with patients with severe hyponatremia, defined as a serum [Na+]<130 mmol/l (tolvaptan=6.40±8.39 versus placebo=8.00±9.31 days, P=0.576). Although neither of these analyses reached statistical significance, analysis of the investigator-diagnosed SIADH group combined with patients classified as ‘Other’ (i.e. all patients not meeting clinical criteria for diagnosis of CHF or cirrhosis), demonstrated a significantly shortened LOS favoring tolvaptan in the severely hyponatremic group (tolvaptan=4.70±3.89 versus placebo= 8.40±9.67 days, P=0.045).
Discussion
The results of this subgroup analysis of the SALT trials confirm that the efficacy and safety results reported previously for the mixed hyponatremia population in SALT-1 and SALT-2 (8) are applicable to patients with a diagnosis of SIADH as well. The superiority of tolvaptan over placebo was apparent for all of the end points related to improvement in serum [Na+] in the SIADH subgroup (average daily AUC of change from baseline in serum [Na+] at days 4 and 30, mean serum [Na+] at each visit, time to serum [Na+] normalization, percentage of subjects with serum [Na+] normalization at days 4 and 30, and categorical change in serum [Na+] at days 4 and 30). Only relatively acute studies reporting the effects of AVPR antagonists to correct hyponatremia in small numbers of patients with SIADH have been reported previously (13, 14). The present analysis, therefore, represents the largest number of patients with SIADH studied chronically using an orally active, AVP V2 receptor antagonist to correct hyponatremia. The mean increases in serum [Na+] in the SIADH subgroup treated with tolvaptan were quantitatively analogous to the increases observed in this subgroup over a much longer (up to 3-year) period of treatment in the SALTWATER open-label extension studies (15). Most side effects were due to the physiological effect of the drug, namely increased urination, dry mouth, and thirst.
In addition to the efficacy in increasing serum [Na+], the effect of tolvaptan on the SF-12 General Health Survey was examined. In contrast to the combined SALT analysis where no effect of the drug on the PCS was observed (8), it is notable that this subgroup analysis of patients with SIADH did show a modest but clinically and statistically significant improvement in the PCS of the tolvaptan-treated group after 30 days of therapy (Table 2). Although the current analysis does not allow assessment of the reasons for this difference, it can be speculated that patients with SIADH have fewer symptoms that are related to underlying co-morbidities than do patients with heart failure and cirrhosis and thus are more likely to perceive improvement in physical symptoms that are primarily related to the hyponatremia itself. Similar to the combined SALT analysis, in which a demonstrable effect on the MCS was observed (8), an improvement in the MCS of the SIADH subgroup also occurred. The MCS improvement was equivalent in the SIADH subgroup of patients (+5.47±12.01) compared with the mixed SALT patient group (+6.09±11.79), both of which are clearly in a clinically relevant range. However, the SIADH subgroup approached but did not meet the criteria for statistical significance compared with placebo (P=0.051), likely due to the reduced size of the SIADH subgroup available for this analysis. Thus, improvements in patient-related symptoms were of equal or greater magnitude in the SALT SIADH patients as in the mixed etiology SALT total patient group.
The SALT studies were the first studies of patients with SIADH conducted primarily in the outpatient setting without mandated restriction of fluid, or of frequently used medications such as diuretics. Thus, the results of these studies are readily applicable to common clinical practice. In this regard, it is also significant that only 7.8% of SIADH patients on tolvaptan required institution of a fluid restriction (defined as ≤1000 ml/day) at any time in the 30-day treatment period, as compared with 13.7% patients on placebo. Independent of the improvement of serum [Na+], the efficacy of tolvaptan to decrease the need for fluid restriction would represent a significant clinical benefit to patients with chronic SIADH.
A continued concern of all treatments used to correct hyponatremia is the potential to cause osmotic demyelination with overly rapid correction of the hyponatremia (16). Multiple clinical studies have provided guidance on the correction rates that increase the risk of this adverse outcome, and current clinical guidelines recommend that correction rates should not exceed 12 mmol/l over the first 24 h of correction and 18 mmol/l over the first 48 h of correction (17–19). All past and future methods used to correct hyponatremia must be assessed for their ability to stay within these limits. In this regard, it is notable that only 5.9% of patients treated with tolvaptan exceeded these limits: one with a correction of 13 mmol/l and two with a correction of 14 mmol/l over the first 24 h of therapy. As for the combined SALT studies, none of these patients exhibited any neurological symptoms suggestive of osmotic demyelination, which has been true for all studies of vasopressin receptor antagonists to date. Nonetheless, it is prudent for clinicians to be vigilant when using tolvaptan to correct patients with chronic hyponatremia. Following labeling instructions to avoid fluid restriction during the phase of active correction will help to avoid overly rapid correction by allowing the patients’ thirst to mitigate the increases in serum [Na+] as a result of pronounced aquaresis; the usefulness of this strategy to guard against overly rapid correction is evident from the balance studies during the first day of therapy, where the tolvaptan-treated SIADH patients had a significantly greater fluid intake of ~450 ml/24 h than the placebo-treated patients (Fig. 4). However, as with all therapies used to correct chronic hyponatremia, measuring serum [Na+] at frequent intervals is mandatory in order to interrupt the correction if accepted guidelines are exceeded. Generally, measurement of serum [Na+] every 6–8 h is sufficient, but frequency should be increased if additional risk factors predisposing to osmotic demyelination are present, including alcoholism, malnutrition, hypokalemia, and a very low starting serum [Na+] (18).
Recently published estimates place direct costs of treating hyponatremia between US $1.6 and $3.6 billion, annually, ~70% of which occur in hospital (20). The treatment of some of these cases of hyponatremia with an orally active, AVPR antagonist could potentially decrease hospital admissions as well as decrease length of hospital stays, with the accompanying economic implications. Although the SALT studies were neither designed nor powered to study length of hospital stay, it is of interest that there was a nominally greater shortening of hospital stay in the tolvaptan-treated patients, and this difference achieved statistical significance when the severely hyponatremic patients with investigator-diagnosed SIADH and those classified as ‘Other’ were combined. Definitive proof of this possibility will require additional clinical trials to specifically evaluate this outcome.
A number of limitations of this analysis should be recognized. First, the SALT studies were not designed to evaluate SIADH as a separate entity but rather as one of the several etiologies of dilutional hyponatremia. Hence, rigorous criteria for a diagnosis of SIADH as classically defined were not required for entry into the studies. Nonetheless, the analysis of the SIADH subgroup with elevated urine sodium measurements after tolvaptan administration (lab-diagnosed SIADH) confirms that the results in this group do not significantly differ from the larger group with a clinical diagnosis of SIADH (investigator-diagnosed SIADH). Furthermore, the investigator determination of a clinical diagnosis of SIADH is likely more relevant to actual medical practice, and therefore indicative of the patients who will be considered potential treatment candidates for tolvaptan therapy. Secondly, all post hoc analyses of subgroups from a larger study are compromised by lesser power to detect differences because of smaller numbers of patients in each subgroup. This was evidenced in this study by a lack of statistical significance in the improvement of the MCS score of the SF-12 despite an equivalent numerical improvement as was seen in the mixed SALT group. Nonetheless, the primary end points of correction in serum [Na+] remain highly significant despite the smaller numbers of patients in the SIADH subgroup, however defined. Thirdly, both SALT studies excluded patients with serum [Na+]<120 mmol/l who had neurological symptoms, because it was felt that it would be inappropriate to place such at-risk patients into a placebo-controlled study. Thus, the low incidence of overly rapid correction observed in these studies (5.9%) may not be applicable to patients with lower starting serum [Na+], who have a greater chance to exceed the recommended guidelines for maximum corrections. Finally, some of the findings, and specifically the LOS results, were never intended to be an end point in the SALT studies. Thus, although intriguing, these must be considered preliminary findings that will need to be confirmed in subsequent studies specifically designed to evaluate this end point.
In summary, in the largest trials of efficacy and safety of treatment of hyponatremia yet published, the vasopressin V2 receptor antagonist tolvaptan was superior to placebo in raising and maintaining serum [Na+] in patients with SIADH when added to standard therapy. Tolvaptan was generally well tolerated, having side effects generally consistent with its physiological activity, and could be safely titrated to achieve the desired rate and degree of serum [Na+] correction while avoiding overly rapid correction in most patients. Clinical benefits of tolvaptan therapy included a decreased need for fluid restriction and improved self-reported physical and mental symptoms. Other potential benefits of tolvaptan therapy, including decreased length of hospital stay and resource utilization, were not specifically assessed by virtue of the study design of the SALT protocols, but LOS was significantly decreased in a subgroup of euvolemic patients with more severe hyponatremia. These and other potential benefits, such as decreases in morbidity and mortality, remain to be evaluated by future clinical trials.
Acknowledgments
Funding
Otsuka Pharmaceutical Development and Commercialization, Inc., provided the financial support for this study.
We thank the patients involved in the SALT-1 and SALT-2 trials for their participation and contribution; the study investigators, subinvestigators, and nurses (including those at the NIH National Center for Research Resources General Clinical Research Centers that participated in these studies); the study monitors, and the data coordinators and their managers; C Zimmer, J Ouyang, S Shoaf, A Ozaki, and Y Yamamura for their scientific advice; D Kostic and David Norris for assistance in the preparation of the manuscript; and the members of the Safety Oversight Committee (SALT-1 and SALT-2): R Schrier, chair; D G Bichet (Hôpital du Sacre-Coeur de Montreal, Montreal); M Yarborough (University of Colorado, Denver); and J Ouyang (Otsuka Pharmaceutical Development and Commercialization, Inc., Rockville, MD, USA).
Footnotes
Declaration of interest
S Adler has no disclosures to report. FS Czerwiec and Q Zhao are employees of Otsuka Pharmaceutical Development and Commercialization, Inc. JG Verbalis reports having served as a consultant to Otsuka, Astellas, Cardiokine, Ferring Research Institute, Sanofi- Aventis, and Yamanouchi Pharma America and having received grant support from Yamanouchi Pharma America, Astellas, and Otsuka. RW Schrier reports having served as a consultant to Otsuka, Astellas, Bayer, and Amgen. T Berl reports having served as a consultant to Bayer and Astellas and having received grant support from Otsuka. No other potential conflict of interest relevant to this study was reported.
References
- 1.Hawkins RC. Age and gender as risk factors for hyponatremia and hypernatremia. Clinica Chimica Acta. 2003;337:169–172. doi: 10.1016/j.cccn.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. American Journal of Medicine. 2006;119:S30–S35. doi: 10.1016/j.amjmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Terzian C, Frye EB, Piotrowski ZH. Admission hyponatremia in the elderly: factors influencing prognosis. Journal of General Internal Medicine. 1994;9:89–91. doi: 10.1007/BF02600208. [DOI] [PubMed] [Google Scholar]
- 4.Bennani SL, Abouqal R, Zeggwagh AA, Madani N, Abidi K, Zekraoui A, Kerkeb O. Incidence, causes and prognostic factors of hyponatremia in intensive care. Revue de Médecine Interne. 2003;24:224–229. doi: 10.1016/S0248-8663(02)00811-1. [DOI] [PubMed] [Google Scholar]
- 5.Ohnishi A, Orita Y, Okahara R, Fujihara H, Inoue T, Yamamura Y, Yabuuchi Y, Tanaka T. Potent aquaretic agent. A novel nonpeptide selective vasopressin 2 antagonist (OPC-31260) in men. Journal of Clinical Investigation. 1993;92:2653–2659. doi: 10.1172/JCI116881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zerbe R, Stropes L, Robertson G. Vasopressin function in the syndrome of inappropriate antidiuresis. Annual Review of Medicine. 1980;31:315–327. doi: 10.1146/annurev.me.31.020180. 001531. [DOI] [PubMed] [Google Scholar]
- 7.Szatalowicz VL, Arnold PE, Chaimovitz C, Bichet D, Berl T, Schrier RW. Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure. New England Journal of Medicine. 1981;305:263–266. doi: 10. 1056/NEJM198107303050506. [DOI] [PubMed] [Google Scholar]
- 8.Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. New England Journal of Medicine. 2006;355:2099–2112. doi: 10.1056/NEJMoa065181. [DOI] [PubMed] [Google Scholar]
- 9.Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. American Journal of Medicine. 1967;42:790–806. doi: 10.1016/0002-9343(67)90096-4. [DOI] [PubMed] [Google Scholar]
- 10.Ware JE, Jr, Kosinski M, Bayliss MS, McHorney CA, Rogers WH, Raczek A. Comparison of methods for the scoring and statistical analysis of SF-36 health profile and summary measures: summary of results from the Medical Outcomes Study. Medical Care. 1995;33:AS264–AS279. doi: 10.1097/00005650-199501001-00005. [DOI] [PubMed] [Google Scholar]
- 11.Ware J, Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Medical Care. 1996;34:220–233. doi: 10.1097/ 00005650-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Guyatt GH, Osoba D, Wu AW, Wyrwich KW, Norman GR. Methods to explain the clinical significance of health status measures. Mayo Clinic Proceedings. 2002;77:371–383. doi: 10.4065/ 77.4.371. [DOI] [PubMed] [Google Scholar]
- 13.Saito T, Ishikawa S, Abe K, Kamoi K, Yamada K, Shimizu K, Saruta T, Yoshida S. Acute aquaresis by the nonpeptide arginine vasopressin (AVP) antagonist OPC-31260 improves hyponatremia in patients with syndrome of inappropriate secretion of antidiuretic hormone (SIADH) Journal of Clinical Endocrinology and Metabolism. 1997;82:1054–1057. doi: 10.1210/jc.82.4.1054. [DOI] [PubMed] [Google Scholar]
- 14.Soupart A, Gross P, Legros JJ, Alfoldi S, Annane D, Heshmati HM, Decaux G. Successful long-term treatment of hyponatremia in syndrome of inappropriate antidiuretic hormone secretion with satavaptan (SR121463B), an orally active nonpeptide vasopressin V2-receptor antagonist. Clinical Journal of the American Society of Nephrology. 2006;1:1154–1160. doi: 10.2215/CJN.00160106. [DOI] [PubMed] [Google Scholar]
- 15.Berl T, Quittnat-Pelletier F, Verbalis JG, Schrier RW, Bichet DG, Ouyang J, Czerwiec FS. Oral tolvaptan is safe and effective in chronic hyponatremia. Journal of the American Society of Nephrology. 2010;21:705–712. doi: 10.1681/ASN.2009080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sterns RH, Riggs JE, Schochet SS., Jr Osmotic demyelination syndrome following correction of hyponatremia. New England Journal of Medicine. 1986;314:1535–1542. doi: 10.1056/ NEJM198606123142402. [DOI] [PubMed] [Google Scholar]
- 17.Sterns RH, Silver S, Kleinschmidt-DeMasters BK, Rojiani AM. Current perspectives in the management of hyponatremia: prevention of CPM. Expert Review of Neurotherapeutics. 2007;7:1791–1797. doi: 10.1586/14737175.7.12.1791. [DOI] [PubMed] [Google Scholar]
- 18.Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines: expert panel recommendations. American Journal of Medicine. 2007;120:S1–S21. doi: 10. 1016/j.amjmed.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. Journal of the American Society of Nephrology. 1994;4:1522–1530. doi: 10.1681/ASN.V481522. [DOI] [PubMed] [Google Scholar]
- 20.Boscoe A, Paramore C, Verbalis JG. Cost of illness of hyponatremia in the United States. Cost Effectiveness and Resource Allocation. 2006;4:10–20. doi: 10.1186/1478-7547-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]