

Abstract
The original glucocorticoid (GC) hypothesis of brain aging and Alzheimer’s disease proposed that chronic exposure to GCs promotes hippocampal aging and AD. This proposition arose from a study correlating increasing plasma corticosterone with hippocampal astrocyte reactivity in aging rats. Numerous subsequent studies have found evidence consistent with this hypothesis, in animal models and in humans. However, several results emerged that were inconsistent with the hypothesis, highlighting the need for a more definitive test with a broader panel of biomarkers. We used microarray analyses to identify a panel of hippocampal gene expression changes that were aging-dependent, and also corticosterone-dependent. These data enabled us to test a key prediction of the GC hypothesis, namely, that the expression of most target biomarkers of brain aging should be regulated in the same direction (increased or decreased) by both GCs and aging. This prediction was decisively contradicted, as a majority of biomarker genes were regulated in opposite directions by aging and GCs, particularly inflammatory and astrocyte-specific genes. Thus, the initial hypothesis of simple positive cooperativity between GCs and aging must be rejected. Instead, our microarray data suggest that in the brain GCs and aging interact in more complex ways that depend on the cell type. Therefore, we propose a new version of the GC-brain aging hypothesis; its main premise is that aging selectively increases GC efficacy in some cell types (e.g., neurons), enhancing catabolic processes, whereas aging selectively decreases GC efficacy in other cell types (e.g., astrocytes), weakening GC anti-inflammatory activity. We also propose that changes in GC efficacy might be mediated in part by cell type specific shifts in the antagonistic balance between GC and insulin actions, which may be of relevance for Alzheimer’s disease pathogenesis.
Keywords: Corticosterone, Hippocampus, Insulin, Inflammation, Astrocyte, Microarray, Cognition, Alzheimer’s
1. Background: The Initial Glucocorticoid Hypothesis of Brain Aging and its Predictions
The hypothesis that the chronic actions of glucocorticoids (GCs) promote aging of the brain, particularly of hippocampal neurons [1, 2], originally grew out of a study showing a positive correlation between biomarkers of hippocampal aging and plasma corticosterone in aging rats [3]. The hypothesis proposed that cumulative exposure even to normal concentrations of GCs could promote brain aging, and that elevation of GCs, as in chronic stress, could further accelerate the progression of brain aging. In addition, because it had been reported that hippocampal neurons participated in the negative feedback inhibition of the hypothalamic-pituitary-adrenal axis [4, see 5 for review], it was postulated that as the hippocampus deteriorated with aging it lost some negative feedback control, resulting in elevated plasma corticosterone and adrenal activity [1, 3].
Prior to these initial studies in brain, it had already been observed that elevated GCs in salmon and mammals, including humans, induced degenerative changes in peripheral tissues that in some ways mimicked aging changes [6, 7]. Further, the extensive studies of Selye on the stress response had led to the proposal that stress could accelerate certain aspects of aging [8]. At that point, little was known about normal aging of the brain and about whether and how GCs acted on the brain. Nevertheless, the established role of the hippocampus in memory, the vulnerability of the hippocampus to aging-related neurodegenerative disease, and the discovery that the brain expressed corticosteroid receptors, which were especially dense in the hippocampus [9], warranted a test of GC effects on hippocampal aging [1]. However, a key to performing such a study was the need for a suitable endpoint biomarker that could provide quantitative estimates of brain aging. Astrocyte reactivity, a reliable indicator of neuronal damage, was selected as the aging biomarker because such reactivity was quantifiable, prominent in the hippocampus, and detectable by mid-life [10, 11]. The study measured astrocyte reactivity in the hippocampus of F344 rats and found that it increased steadily with aging, being apparent even by mid-life. The increasing astrogliosis was correlated quantitatively with plasma corticosterone and adrenal weight, which also increased by mid-life. Thus, the results supported the basic GC hypothesis (illustrated schematically in Fig (1)) [1, 3], in which chronic exposure to adrenal glucocorticoids facilitates aging processes in neurons, notably in the hippocampus (astrocytes are not shown, but presumably are activated by the neuronal changes). It should be noted, however, that there can be several variations on the general hypothesis that GCs promote brain aging [2]. As shown in Fig. (2): i) GCs might act to drive or facilitate aging processes (Hypothesis 1); ii) GCs might act on some targets in parallel with aging processes, to accelerate functional endpoints (Hypothesis 2); iii) aging might increase the efficacy of GC effects, resulting in accelerated progression of deleterious consequences of chronic GC actions (Hypothesis 3); iv) conceivably, an extreme version of Hypothesis 3 might be operative, in which the spectrum of brain aging endpoints primarily reflects the sum of cumulative GC actions (Hypothesis 4). Although very different mechanisms could underlie these alternatives, all have in common the proposition that GCs and aging interact cooperatively. Therefore, all versions predict that both GCs and aging should regulate endpoint biomarkers in the same direction (increase or decrease).
Figure 1. Initial glucocorticoid (GC) hypothesis of hippocampal aging.

Long-term exposure to adrenal GCs/stress hormones (red arrows) facilitates aging-related functional decline and vulnerability in hippocampal neurons. Affected cells show heightened sensitivity to extrinsic injuries and disease, causing hippocampal deterioration and eventual cognitive decline. Not shown are activated astrocytes which presumably react to the neuronal changes. See discussion in text. (Redrawn from [2] with permission.)
Figure 2. Possible variations on the interaction between aging and glucocorticoids.
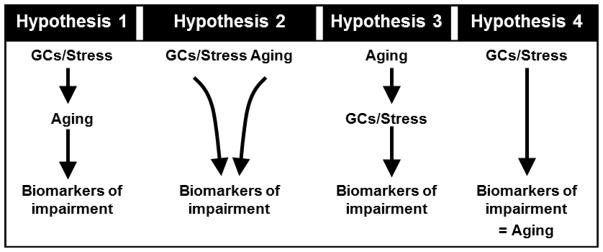
Four sub-hypotheses on the mode of positive cooperativity between aging and GCs. See discussion in text. (Redrawn from [2] with permission.)
2. Additional Studies Supporting the Hypothesis
The initial study was correlative, but we soon followed it with a long-term intervention study. Mid-aged rats were adrenalectomized (ADX) and maintained on very low replacement corticosterone in the drinking water for 9 months, at which point they were placed on a full replacement dose and tested for maze reversal learning. The hippocampus was then processed and several morphometric biomarkers of hippocampal aging (neuronal density, glial reactivity, lipofuscin) were quantified in semi-thin sections. In comparison to similarly-maintained sham, age-matched controls, the ADX rats showed significantly less development of several biomarkers of brain aging, and a strong trend toward better maze performance, thus more closely resembling young animals [12].
These early studies provided initial support for the view that chronic exposure to GCs, particularly if elevated as in stress, promotes aging of the brain. However, multiple studies from other labs also began to accumulate substantial evidence consistent with the hypothesis. Notably, Sapolsky, McEwen and colleagues conducted a series of animal studies that provided extensive support for the view that GCs were elevated with aging and could damage the hippocampus or increase its vulnerability. Their work focused on GC receptors and led to a version of the hypothesis that emphasized the role of downregulation of hippocampal GC receptors during aging, with loss of negative feedback and consequent elevation and damaging effects of circulating GCs [13]. Sapolsky et al [14] also found that GCs could increase vulnerability to toxicity in cultured neurons, possibly by inhibiting glucose uptake. Other work by Meaney and colleagues showed strong correlations between circulating corticosterone and cognitive impairment and hippocampal damage in aged rats [15, 16] and other labs also found inverse relationships between GC exposure and cognitive function. Further, several groups found that chronic stress in rats also could accelerate biomarkers of hippocampal aging [17, 18]. Additionally, it has been found that, similarly to the effects of aging, excess GCs induce hippocampal dendritic atrophy and retard neurogenesis [19, 20]. Most studies employed morphometric and/or behavioral biomarkers to test the effects of GCs on brain aging processes. However, several studies showed that, over periods of minutes to hours, GCs could also increase a number of Ca2+-mediated electrophysiological biomarkers of hippocampal aging [21–23].
Generally analogous findings began to be observed in human studies of aging and AD. A number of imaging studies found that human hippocampal volume decreases with aging [24]. In addition, aging human subjects exhibit higher mean diurnal values of cortisol [25]. Moreover, in an extensive series of longitudinal studies, Lupien and her colleagues have shown that elevated plasma cortisol correlates with reduced hippocampal volume and memory impairment in aging human subjects. In fact, some subjects with higher cortisol developed Alzheimer’s disease (AD) [26]. Further, Cushing’s disease patients, exhibiting very elevated GC values, are also characterized by memory impairment and reduced hippocampal volume [27]. Importantly, it is also well-established that AD subjects have substantially elevated cortisol levels [28]. Although cause-effect relationships are not clear, recent studies show that chronic GC treatment of aged monkeys [29] or a transgenic mouse model of AD [30] can increase amyloid-beta pathology. Additionally, mouse models of AD exhibit higher corticosterone values, which may contribute to insulin resistance and memory impairment [31]. Chronic GC treatment also worsened cognition in AD subjects [32]. Together, these studies suggest that GCs may contribute to AD pathogenesis.
Thus, many findings continued to be consistent with the basic GC hypothesis, supporting the conclusion that excess GCs, and even chronic exposure to normal values, directly drive important components of declining brain function with aging, and perhaps, with AD. Unfortunately, unraveling fundamental biological processes is rarely, if ever, that simple, and some results that contradicted the basic tenets of the hypothesis began to appear.
3. Flies in the Ointment: Emergence of Results Inconsistent with the Hypothesis
In the early 1990s several findings emerged that were not easily reconciled with the predictions of the hypothesis. For one, it was found that corticosterone strongly downregulated gene/protein expression of GFAP, a major astrocytic structural protein for which increased expression is a consistent correlate of astrocyte reactivity and a reliable biomarker of hippocampal aging [33]. This observation appeared to imply an inverse relationship between astrocyte reactivity and corticosterone should develop with aging, yet in our initial study, we had found a positive correlation [3].
Second, a series of studies began to appear showing that for some kinds of learning, such as emotional learning, GCs were facilitative and enhanced memory consolidation (although apparently impairing retrieval) [34]. Certainly it seems reasonable that GC hormones released with stress would facilitate rather than impair memory of the biologically important stressful events. However, because memory capacity declines with aging, this result appeared to be another example of anti-aging rather than pro-aging effects of GCs.
Third, investigations of the caloric restriction model of increased longevity found that GCs of restricted animals were elevated above those of controls not only at feeding times, but across the 24-hr diurnal cycle [35, 36]. Further, several groups found that, in addition to peripheral markers, CR reduced some (but not all) brain biomarkers of aging [37–39]. Consequently, it appeared that in essentially the only established model for retarding multiple markers of aging, circulating GCs were not reduced, and in fact, might be increased.
Thus, these several lines of evidence were not consistent with the prediction of the basic GC hypothesis, noted above (Fig. (2))[2], that both GCs and aging should modulate endpoint markers of aging in the same direction. In fact, the studies cited above found that, under some conditions, GCs modulate the expression of GFAP, memory consolidation and other markers of aging in the direction opposite to that in which they normally change with aging. That is, for these specific markers and conditions, elevated GCs were associated with effects that would tend to oppose, rather than enhance, aging changes.
In addition to the contradictory findings above, some of the biomarkers employed and some of the basic findings from supporting studies were also becoming controversial. A few studies in different strains of rats reported inability to replicate the reduced number or density of hippocampal pyramidal neurons with aging [40], one of the biomarkers used to assess GC effects in several studies. Other reports did not find elevated corticosterone in aged rats [41], and evidence accumulated that it was primarily the mineralocorticoid receptor rather than the glucocorticoid receptor that declined in hippocampus with aging [42]. The emerging contradictions and controversies noted above began to raise some significant doubts, making it clear that at the least some critical reevaluation was necessary [43, 44] (see reviews in [45]).
4. The Need for a Definitive Test of the GC Hypothesis Using Improved Biomarkers
Many of these controversies could apparently be attributed to strain differences and differing rates of brain aging, genetic drift in long-maintained colonies, or to methodological differences [43, 44, 46]. Furthermore, a large body of animal and human data continued to support the basic GC hypothesis. Nevertheless, the three main results that were inconsistent with the key prediction that aging and GCs should alter biomarker endpoints in the same direction, noted above, highlighted the need for a more definitive test of the basic GC hypothesis.
However, a more definitive test required a much broader range of biomarkers, such that anomalies could be detected and the main direction of changes could be assessed. Further, although many prior studies have used one or a few biomarkers to test predictions of this and other aging hypotheses, a panel of multiple biomarkers is likely to provide a more accurate index [47]. Given these considerations, the recent advent of gene microarray technology for the study of brain aging and AD may have been particularly fortunate.
5. Genomic Biomarkers Determined by Microarray Analyses of Brain Aging and AD
For reasons partly outlined above, the use of gene microarray technology appears to hold great promise for the development of biomarkers of brain aging and for a definitive test of the GC hypothesis. That is, microarray expression profiling, which permits parallel measurement of the activity of thousands of genes, is uniquely positioned to find hundreds of markers (genes) that change consistently with aging and can be measured simultaneously. As a corollary, the effects of GCs can be compared to the effects of aging on each of these biomarkers, thereby allowing a large-scale test of predictions of the GC hypothesis.
However, before that test could be performed we had to define a panel of reliable aging-dependent gene expression changes. To date, we and others have employed microarray technology in several studies of brain aging [48–51] and in studies of Alzheimer’s disease using human tissue or animal models of AD [52–59]. These have yielded substantial information on selected genes and related pathways, but results for marker genes have not been highly consistent across studies (reviewed in [60]).
Along with its powerful potential, microarray analysis poses formidable bioinformatics and resource problems. The massive data sets generated and the thousands of comparisons involved create major problems of false positives, false negatives and functional interpretation. In our own microarray studies of hippocampal aging [51], incipient Alzheimer’s disease [59] and aging-dependent cognitive impairment we have attempted to address these problems with statistical rigor, well-powered groups, expression correlations with function, and systematic pathway analysis [60]. In particular, the use of relatively large n’s and well-powered groups has enabled us to ameliorate type II error (false negatives) and to generate extensive lists of genes and related pathways that change in the hippocampus with aging and/or with cognitive impairment. These age-dependent changes in expression appear highly reliable, as many have been detected in both of our large hippocampal aging studies ([51]; and manuscript in preparation1). Notably, nearly all of the genes identified in our aging studies had already changed substantially by mid-life (12–14 months-old) [51]. The majority (but not all) of downregulated genes appeared to reflect neuronal processes, whereas the majority of upregulated genes appeared to reflect glial processes. The specific genes found to change with aging in these studies also elucidated larger functional pathways and categories (as organized in the Gene Ontology database) that were statistically over-represented by coregulated genes.
6. Microarray Test of the Glucocorticoid Hypothesis of Hippocampal Aging
With this extensive list of genes and processes that were down- or up-regulated reliably with hippocampal aging available for use as biomarkers, it was now possible to test the prediction that GCs should modulate targets in the same direction as they change with aging. Moreover, it was also possible to test another prediction of the hypothesis, namely that more genes and pathways should be commonly affected by both GCs and aging than expected by chance.
To determine which genes in the hippocampus are regulated by GCs, we adrenalectomized (ADX) two groups of mid-aged 15-month-old F344 rats and sham operated another (n=10/group). One ADX group was implanted with corticosterone-releasing pellets that supplied a very low dose, and the other group was implanted with pellets that released a high dose of corticosterone (essentially to Sham levels). The groups were maintained for 3 months (to 18 months of age), at which point their hippocampi were dissected and processed for microarray analysis (one hippocampus per array). The data were then analyzed statistically to identify the genes that differed as a function of corticosterone (Cort) levels. This set of Cort-sensitive genes was then compared to the sets of aging-dependent genes, identified in our other studies [51], for detection of common genes and for similarity or difference in direction of change among common genes.
7. Paradoxical Results: Increased and Decreased Efficacy of GCs
Fig. (3) shows a summary of the results from the experiment described above (detailed results will be published elsewhere). The prediction that the number of genes regulated both by GCs and by aging should be greater than expected by chance was confirmed, although many genes regulated by aging were not regulated by Cort, and vice versa. However, the prediction that genes regulated by both Cort and aging should be regulated predominately in the same direction was definitively rejected. As shown in Fig. (3), among those genes downregulated with aging, only approximately half were also downregulated by Cort. Even more striking was the effect for genes upregulated with aging; among these, almost two-thirds were downregulated by Cort. Consequently, the view of simple positive cooperativity between GCs and aging processes, as implied by any of the four sub-hypotheses in Fig. (2), is clearly untenable.
Figure 3. Overlap of aging-related and corticosterone (Cort)-sensitive genes.
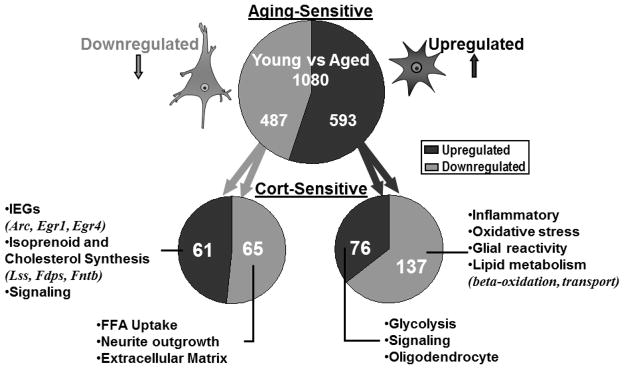
Using microarray technology, the genes that were aging-sensitive (upper) and also Cort- sensitive (lower) were identified. Numbers shown represent the number of genes in each category of directional change (down or up). Most genes downregulated with aging are expressed primarily in neurons, whereas upregulated genes are primarily glial. More genes were altered in opposite directions by Cort and aging than in the same direction, contravening a key prediction of the original GC hypothesis. Selected processes (and some genes from which they were identified) are shown for each directional combination of Cort and aging changes.
Thus, after many years of testing, and despite much evidence consistent with its predictions, the basic GC hypothesis of brain aging apparently must be rejected. Nevertheless, aspects of the findings here suggest that aging and GCs interact in substantial fashion in the hippocampus, and that many genes are commonly regulated by aging and GCs. Instead of only positive interactions between aging and GCs, however, the results indicate that some genes may be regulated in opposite directions (negative cooperativity) and some in the same direction (positive cooperativity). That is, the number of genes that are regulated in opposite directions by Cort and aging is much higher than expected by chance (Fig. (3)). Furthermore, genes regulated in the same direction by Cort and aging appeared to be particularly concentrated in a few pathways/categories critical for brain aging. In addition, an analysis of overrepresented pathways/categories for each of the four combinations of directional effects on genes that were regulated by both Cort and aging (eg, Cort-down/Aging-down; Cort-up/Aging-down; Cort-up/Aging-up; Cort-down/Aging-up) revealed profiles that appeared to reflect distinct functions and potentially specific cell-type localization, for each combination.
Based on these observations, we suggest that processes regulated in the same direction by Cort and aging reflect an increasing efficacy of Cort with aging. Conversely, processes regulated in opposite directions by Cort and aging reflect a declining efficacy of Cort with age. For example, genes exhibiting Cort-down/Aging-down patterns (primarily neuronal) were highly represented for pathways of free fatty acid uptake, neurite outgrowth and extracellular matrix formation (Fig. (3)). These results may indicate that, in neurons, an increasing efficacy of GCs’ catabolic actions with aging acts to downregulate energy-consuming processes supporting growth and structural synthesis. On the other hand, many probable glial genes exhibited Cort-down/Aging-up directional combinations and were highly represented for inflammatory, lysosomal, glial activation (including GFAP), oxidative stress, and cholesterol transport processes, among others (Fig. (3). These processes likely reflect astrocyte activation [61–64], and may indicate declining efficacy of Cort in astrocytes with aging, allowing suppressed inflammatory pathways to escape from Cort’s normally negative regulation.
These observations have several important implications: First, they show that aging mechanisms appear able to promote the expression of some key biomarkers of aging either by selectively increasing or by selectively decreasing the effect of GC actions on those biomarker targets, in a cell type-specific manner; second, they indicate that GCs suppress inflammatory processes in the brain, much as they do in the periphery; and third, declining efficacy of Cort likely accounts for some of the inconsistent results noted above, including why a positive correlation was found between Cort and astrocyte reactivity in aging [3], despite Cort’s ability to suppress GFAP [33], and why GCs facilitate some memory processes [34], despite the decline in memory functions with aging..
8. Shifting the GC Balance with Insulin Signaling
If GCs gain efficacy with aging in some brain cell types and lose efficacy in others, or perhaps even gain and lose efficacy on different processes within the same cell, then the question arises of what mechanisms might underlie such specific targeting. One possibility, of course, is that cells might differentially regulate glucocorticoid receptor content. As yet, however, we have not seen major aging changes in GR content of neurons or glia in the hippocampus (unpublished data). A second possibility is that the intracellular concentration of GCs might be regulated by differential expression of 11-beta hydroxysteroid dehydrogenase 1 (11beta -HSD1), the enzyme responsible for the converson of inactive GC to active GC [65]. For example, decreased 11beta-HSD1 in astrocytes with aging could account for the declining Cort efficacy in astrocytes. Yet another possibility is that GC co-factors or modulatory elements might be differentially regulated with aging.
In addition, one intriguing possibility is that GC efficacy might be inversely proportional to insulin signaling. It is well established that the anabolic actions of insulin and the catabolic actions of GCs counteract each other on many critical peripheral metabolic functions, from glucose utilization to lipid storage. Further, increased GC action is a major candidate causal mechanism in aging-related development of insulin resistance and central obesity [65, 66]. Thus, if an analogous counterbalance operated in brain cells, and insulin signaling were decreased in some cell types with aging, the catabolic actions of GCs might be strengthened. Conversely, if insulin pathway signaling were increased in other cell types, the anti-inflammatory/catabolic actions of GCs might be weakened.
Consistent with the view that insulin signaling might vary with aging, we recently found a significant downregulation of a number of hippocampal insulin pathway genes that was specific for aged, cognitively impaired rats1. Given these considerations, it is suggested that GC catabolic action may increase with age in hippocampal neurons as a cause or a consequence of a decrease in opposing insulin signaling. On the other hand, in reactive astrocytes, insulin/IGF signaling may be upregulated, possibly by calcineurin [62], resulting in declining GC efficacy and the escape of astrocytic inflammatory processes from GC-mediated suppression. However, not all GC actions are catabolic, and GCs and insulin work cooperatively on some non-catabolic targets (e.g., on processes activated rather than suppressed by GCs). These targets, presumably, would not be subject to chronic regulation by the antagonistic actions of GCs and insulin, but rather, might be more sensitive to other, shorter-term effects of GCs and cell type-specific co-factors. Clearly, however, a mechanism based on the balance between GC and insulin action is only one of multiple possibilities.
9. Implications for Alzheimer’s Disease
There is also evidence that altered GC efficacy may play a role in AD. That is, recent findings link peripheral insulin resistance to cognitive decline with aging and AD [67] and, concomitantly, GC action is a candidate mechanism in insulin resistance. In addition, as noted earlier, chronic GC treatment may increase some AD-related pathology, possibly associated with a form of “brain diabetes” [29–31, 68]. Moreover, we recently found microarray evidence for increased efficacy of GCs in hippocampus of early-stage AD subjects. Both downregulation of pyruvate dehydrogenase (PDH) and upregulation of pyruvate carboxylase gene expression were seen in incipient Alzheimer’s disease [59]. Similar effects were found with normal hippocampal aging in rats [51]. These changes suggest that reduced pyruvate flux through PDH and decreased oxidative metabolism of glucose may develop early in AD. Interestingly, the inactivation of PDH is also a major pathway through which GC activity acts to conserve glucose, and apparently, to induce insulin resistance [65, 66]. Thus, our data are consistent with the possibility that GC effects on this and other important target pathways in brain are enhanced in both aging and AD. If so, such alterations in GC efficacy may have implications for AD pathogenesis as well as for the increased risk of AD associated with normal aging.
10. A New Version of the GC Hypothesis of Brain Aging and AD
Based on the results summarized in Fig. (3), it is apparent that the original unidirectional GC/brain aging hypothesis (Figs. 1, 2) is no longer viable. Some intriguing patterns in our microarray data continue to support the idea that GCs play important roles in brain aging and in AD. However, a more sophisticated model is necessary to account for the complexities of these and other results, and a new cell-type-specific version of the GC hypothesis is shown in Fig. (4). This model, on the left, retains the process of increasing GC efficacy of the original model (Fig. (2.3)), while on the right, also incorporates brain aging changes arising from decreasing GC efficacy. In this view, different cell types regulate GC efficacy differentially with aging. Thus, the directional combinations of GC and aging effects on target genes may well reflect specific signatures of various cell types. Nevertheless, it is clear that much additional research will be needed before we fully understand how, why and in which cell types the effects of GCs are altered with normal brain aging and the pathological brain aging characteristic of AD.
Figure 4. New GC hypothesis of hippocampal aging.
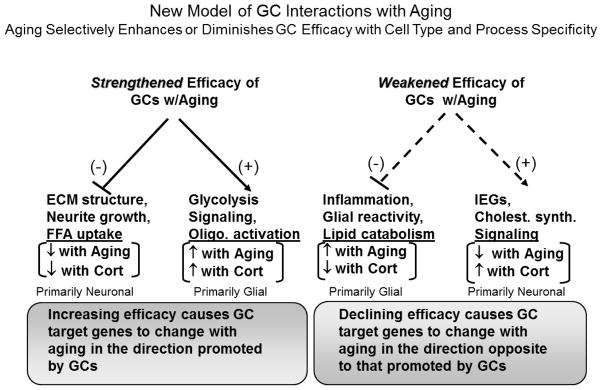
Model incorporating aging-dependent decreases as well as increases in GC efficacy, in different cell type-specific compartments. Target processes (from Fig. (3)) altered during aging by strengthening Cort efficacy (left) should shift with aging in the direction promoted by GCs, whereas targets altered by weakening Cort efficacy (right) should shift with aging in the direction opposite to that normally promoted by GCs. Therefore, in target cells where Cort and aging effects are in same direction (left), Cort efficacy is proposed to increase with aging, and in cells where Cort and aging effects are in opposite directions (right), Cort efficacy is proposed to decrease. Target processes that are regulated by either increased or decreased Cort efficacy may be normally suppressed (−) or activated (+) by GCs. Putative neuronal or glial loci of the effects are based on direction (down or up) of change with aging.
References
- 1.Landfield PW. An endocrine hypothesis of brain aging and studies on brain-endocrine correlations and monosynaptic neurophysiology during aging. Adv Exp Med Biol. 1978;113:179–99. doi: 10.1007/978-1-4684-8893-7_11. [DOI] [PubMed] [Google Scholar]
- 2.Porter NM, Landfield PW. Stress hormones and brain aging: adding injury to insult? Nat Neurosci. 1998 May;1(1):3–4. doi: 10.1038/196. [DOI] [PubMed] [Google Scholar]
- 3.Landfield PW, Waymire JC, Lynch G. Hippocampal aging and adrenocorticoids: quantitative correlations. Science. 1978 Dec 8;202(4372):1098–102. doi: 10.1126/science.715460. [DOI] [PubMed] [Google Scholar]
- 4.Feldman S, Conforti N. Feedback effects of dexamethasone on adrenocortical responses in rats with fornix section. Horm Res. 1976;7(1):56–60. doi: 10.1159/000178709. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991 May;12(2):118–34. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- 6.Finch CE. Enzyme activities, gene function and ageing in mammals. (Review) Exp Gerontol. 1972 Feb;7(1):53–67. doi: 10.1016/0531-5565(72)90035-6. [DOI] [PubMed] [Google Scholar]
- 7.Wexler BC, McMurtry JP. Cushingoid pathophysiology of old, massively obese, spontaneously hypertensive rats (SHR) J Gerontol. 1983 Mar;38(2):148–54. doi: 10.1093/geronj/38.2.148. [DOI] [PubMed] [Google Scholar]
- 8.Selye H. Stress and aging. J Am Geriatr Soc. 1970 Sep;18(9):669–80. doi: 10.1111/j.1532-5415.1970.tb02813.x. [DOI] [PubMed] [Google Scholar]
- 9.McEwen BS. Influences of adrenocortical hormones on pituitary and brain function. Monogr Endocrinol. 1979;12:467–92. doi: 10.1007/978-3-642-81265-1_25. [DOI] [PubMed] [Google Scholar]
- 10.Landfield PW, Rose G, Sandles L, Wohlstadter TC, Lynch G. Patterns of astroglial hypertrophy and neuronal degeneration in the hippocampus of ages, memory-deficient rats. J Gerontol. 1977 Jan;32(1):3–12. doi: 10.1093/geronj/32.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Lindsey JD, Landfield PW, Lynch G. Early onset and topographical distribution of hypertrophied astrocytes in hippocampus of aging rats: a quantitative study. J Gerontol. 1979 Sep;34( 5):661–71. doi: 10.1093/geronj/34.5.661. [DOI] [PubMed] [Google Scholar]
- 12.Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981 Oct 30;214(4520):581–4. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- 13.Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986 Aug;7(3):284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 14.Sapolsky RM, Packan DR, Vale WW. Glucocorticoid toxicity in the hippocampus: in vitro demonstration. Brain Res. 1988 Jun 21;453(1–2):367–71. doi: 10.1016/0006-8993(88)90180-1. [DOI] [PubMed] [Google Scholar]
- 15.Issa AM, Rowe W, Gauthier S, Meaney MJ. Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J Neurosci. 1990 Oct;10(10):3247–54. doi: 10.1523/JNEUROSCI.10-10-03247.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meaney MJ, O’Donnell D, Rowe W, Tannenbaum B, Steverman A, Walker M, et al. Individual differences in hypothalamic-pituitary-adrenal activity in later life and hippocampal aging. Exp Gerontol. 1995 May-Aug;30(3–4):229–51. doi: 10.1016/0531-5565(94)00065-b. [DOI] [PubMed] [Google Scholar]
- 17.Kerr DS, Campbell LW, Applegate MD, Brodish A, Landfield PW. Chronic stress-induced acceleration of electrophysiologic and morphometric biomarkers of hippocampal aging. J Neurosci. 1991 May;11(5):1316–24. doi: 10.1523/JNEUROSCI.11-05-01316.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994 Sep;14(9):5373–80. doi: 10.1523/JNEUROSCI.14-09-05373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McEwen BS. Gonadal and adrenal steroids regulate neurochemical and structural plasticity of the hippocampus via cellular mechanisms involving NMDA receptors. Cell Mol Neurobiol. 1996 Apr;16(2):103–16. doi: 10.1007/BF02088170. [DOI] [PubMed] [Google Scholar]
- 20.Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus. 2006;16(3):233–8. doi: 10.1002/hipo.20155. [DOI] [PubMed] [Google Scholar]
- 21.Kerr DS, Campbell LW, Thibault O, Landfield PW. Hippocampal glucocorticoid receptor activation enhances voltage-dependent Ca2+ conductances: relevance to brain aging. Proc Natl Acad Sci U S A. 1992 Sep 15;89(18):8527–31. doi: 10.1073/pnas.89.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerr DS, Campbell LW, Hao SY, Landfield PW. Corticosteroid modulation of hippocampal potentials: increased effect with aging. Science. 1989 Sep 29;245(4925):1505–9. doi: 10.1126/science.2781293. [DOI] [PubMed] [Google Scholar]
- 23.Joels M, de Kloet ER. Corticosteroid actions on amino acid-mediated transmission in rat CA1 hippocampal cells. J Neurosci. 1993 Sep;13(9):4082–90. doi: 10.1523/JNEUROSCI.13-09-04082.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Leon MJ, Convit A, George AE, Golomb J, de Santi S, Tarshish C, et al. In vivo structural studies of the hippocampus in normal aging and in incipient Alzheimer’s disease. Ann N Y Acad Sci. 1996 Jan 17;777:1–13. doi: 10.1111/j.1749-6632.1996.tb34395.x. [DOI] [PubMed] [Google Scholar]
- 25.Raskind MA, Peskind ER, Wilkinson CW. Hypothalamic-pituitary-adrenal axis regulation and human aging. Ann N Y Acad Sci. 1994 Nov 30;746:327–35. doi: 10.1111/j.1749-6632.1994.tb39251.x. [DOI] [PubMed] [Google Scholar]
- 26.Lupien SJ, Schwartz G, Ng YK, Fiocco A, Wan N, Pruessner JC, et al. The Douglas Hospital Longitudinal Study of Normal and Pathological Aging: summary of findings. J Psychiatry Neurosci. 2005 Sep;30(5):328–34. [PMC free article] [PubMed] [Google Scholar]
- 27.Forget H, Lacroix A, Somma M, Cohen H. Cognitive decline in patients with Cushing’s syndrome. J Int Neuropsychol Soc. 2000 Jan;6(1):20–9. doi: 10.1017/s1355617700611037. [DOI] [PubMed] [Google Scholar]
- 28.Raskind M, Peskind E, Rivard MF, Veith R, Barnes R. Dexamethasone suppression test and cortisol circadian rhythm in primary degenerative dementia. Am J Psychiatry. 1982 Nov;139(11):1468–71. doi: 10.1176/ajp.139.11.1468. [DOI] [PubMed] [Google Scholar]
- 29.Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, et al. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol. 2005 Feb;64(2):139–46. doi: 10.1093/jnen/64.2.139. [DOI] [PubMed] [Google Scholar]
- 30.Green KN, Billings LM, Roozendaal B, McGaugh JL, Laferla FM. Glucocorticoids Increase Amyloid-beta and Tau Pathology in a Mouse Model of Alzheimer’s Disease. J Neurosci. 2006 Aug 30;26(35):9047–56. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006 Jun;199(2):265–73. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology. 2000 Feb 8;54(3):588–93. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 33.Nichols NR, Masters JN, Finch CE. Changes in gene expression in hippocampus in response to glucocorticoids and stress. Brain Res Bull. 1990 May;24(5):659–62. doi: 10.1016/0361-9230(90)90004-j. [DOI] [PubMed] [Google Scholar]
- 34.McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002 Apr;12(2):205–10. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- 35.Masoro EJ. Antiaging action of caloric restriction: endocrine and metabolic aspects. Obes Res. 1995 Sep;3( Suppl 2):241s–7s. doi: 10.1002/j.1550-8528.1995.tb00470.x. [DOI] [PubMed] [Google Scholar]
- 36.Nelson JF, Karelus K, Bergman MD, Felicio LS. Neuroendocrine involvement in aging: evidence from studies of reproductive aging and caloric restriction. Neurobiol Aging. 1995 Sep-Oct;16(5):837–43. doi: 10.1016/0197-4580(95)00072-m. discussion 55–6. [DOI] [PubMed] [Google Scholar]
- 37.Patel NV, Finch CE. The glucocorticoid paradox of caloric restriction in slowing brain aging. Neurobiol Aging. 2002 Sep-Oct;23(5):707–17. doi: 10.1016/s0197-4580(02)00017-9. [DOI] [PubMed] [Google Scholar]
- 38.Weindruch R, Kayo T, Lee CK, Prolla TA. Microarray profiling of gene expression in aging and its alteration by caloric restriction in mice. J Nutr. 2001 Mar;131(3):918S–23S. doi: 10.1093/jn/131.3.918S. [DOI] [PubMed] [Google Scholar]
- 39.Ingram DK, Chefer S, Matochik J, Moscrip TD, Weed J, Roth GS, et al. Aging and caloric restriction in nonhuman primates: behavioral and in vivo brain imaging studies. Ann N Y Acad Sci. 2001 Apr;928:316–26. doi: 10.1111/j.1749-6632.2001.tb05661.x. [DOI] [PubMed] [Google Scholar]
- 40.Rapp PR, Stack EC, Gallagher M. Morphometric studies of the aged hippocampus: I. Volumetric analysis in behaviorally characterized rats. J Comp Neurol. 1999 Jan 25;403(4):459–70. [PubMed] [Google Scholar]
- 41.Sonntag WE, Goliszek AG, Brodish A, Eldridge JC. Diminished diurnal secretion of adrenocorticotropin (ACTH), but not corticosterone, in old male rats: possible relation to increased adrenal sensitivity to ACTH in vivo. Endocrinology. 1987 Jun;120(6):2308–15. doi: 10.1210/endo-120-6-2308. [DOI] [PubMed] [Google Scholar]
- 42.De Kloet ER, Sutanto W, Rots N, van Haarst A, van den Berg D, Oitzl M, et al. Plasticity and function of brain corticosteroid receptors during aging. Acta Endocrinol (Copenh) 1991;125( Suppl 1):65–72. [PubMed] [Google Scholar]
- 43.Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997 Aug;13(3):149–67. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- 44.Landfield PW, Eldridge JC. Evolving aspects of the glucocorticoid hypothesis of brain aging: hormonal modulation of neuronal calcium homeostasis. Neurobiol Aging. 1994 Jul-Aug;15(4):579–88. doi: 10.1016/0197-4580(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 45.DeKloet ER, Azmitia EC, Landfield PW. Brain corticosteroid receptors: studies on the mechanism of action and neurotoxicity of corticosteroid action. Ann N Y Acad Sci. 1994;746:8–21. [PubMed] [Google Scholar]
- 46.Sapolsky RM. Do glucocorticoid concentrations rise with age in the rat? Neurobiol Aging. 1992 Jan-Feb;13(1):171–4. doi: 10.1016/0197-4580(92)90025-s. [DOI] [PubMed] [Google Scholar]
- 47.Landfield PW. Commentary on biomarkers of aging in the nervous system. Exp Gerontol. 1988;23(4–5):413–6. doi: 10.1016/0531-5565(88)90046-0. [DOI] [PubMed] [Google Scholar]
- 48.Jiang CH, Tsien JZ, Schultz PG, Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci U S A. 2001 Feb 13;98(4):1930–4. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000 Jul;25(3):294–7. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 50.Burger C, Cecilia Lopez M, Feller JA, Baker HV, Muzyczka N, Mandel RJ. Changes in transcription within the CA1 field of the hippocampus are associated with age-related spatial learning impairments. Neurobiol Learn Mem. 2006 Jul 10; doi: 10.1016/j.nlm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, et al. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003 May 1;23(9):3807–19. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002 Nov 1;70(3):462–73. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 53.Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol. 2000 Jul;48(1):77–87. [PubMed] [Google Scholar]
- 54.Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer’s disease. DNA Cell Biol. 2001 Nov;20(11):683–95. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- 55.Mufson EJ, Counts SE, Ginsberg SD. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem Res. 2002 Oct;27(10):1035–48. doi: 10.1023/a:1020952704398. [DOI] [PubMed] [Google Scholar]
- 56.Pasinetti GM. Use of cDNA microarray in the search for molecular markers involved in the onset of Alzheimer’s disease dementia. J Neurosci Res. 2001 Sep 15;65(6):471–6. doi: 10.1002/jnr.1176. [DOI] [PubMed] [Google Scholar]
- 57.Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003 Jun 15;23(12):5219–26. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer’s disease. Neurobiol Dis. 2003 Mar;12(2):97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 59.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004 Feb 17;101(7):2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, et al. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer’s disease: statistical reliability and functional correlation. Ageing Res Rev. 2005 Nov;4(4):481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 61.Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006 Feb;83( 2):470S–4S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- 62.Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, et al. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci. 2005 May 4;25(18):4649–58. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rozovsky I, Wei M, Morgan TE, Finch CE. Reversible age impairments in neurite outgrowth by manipulations of astrocytic GFAP. Neurobiol Aging. 2005 May;26(5):705–15. doi: 10.1016/j.neurobiolaging.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 64.Petanceska SS, DeRosa S, Sharma A, Diaz N, Duff K, Tint SG, et al. Changes in apolipoprotein E expression in response to dietary and pharmacological modulation of cholesterol. J Mol Neurosci. 2003;20(3):395–406. doi: 10.1385/JMN:20:3:395. [DOI] [PubMed] [Google Scholar]
- 65.Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 1 as a modulator of glucocorticoid action: from metabolism to memory. Trends Endocrinol Metab. 2004 Nov;15(9):418–24. doi: 10.1016/j.tem.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 66.Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes. 2002 Feb;51(2):276–83. doi: 10.2337/diabetes.51.2.276. [DOI] [PubMed] [Google Scholar]
- 67.Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging. 2005 Dec;26( Suppl 1):65–9. doi: 10.1016/j.neurobiolaging.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 68.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005 Feb;7(1):45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]