

Abstract
Background
Leishmania species belong to the family Trypanosomatidae and cause leishmaniasis, a geographically widespread disease that infects humans and other vertebrates. This disease remains endemic in China. Due to the large geographic area and complex ecological environment, the taxonomic position and phylogenetic relationship of Chinese Leishmania isolates remain uncertain. A recent internal transcribed spacer 1 and cytochrome oxidase II phylogeny of Chinese Leishmania isolates has challenged some aspects of their traditional taxonomy as well as cladistics hypotheses of their phylogeny. The current study was designed to provide further disease background and sequence analysis.
Methods
We systematically analyzed 50 cytochrome b (cyt b) gene sequences of 19 isolates (16 from China, 3 from other countries) sequenced after polymerase chain reaction (PCR) using a special primer for cyt b as well as 31 sequences downloaded from GenBank. After alignment, the data were analyzed using the maximum parsimony, Bayesian and netwok methods.
Results
Sequences of six haplotypes representing 10 Chinese isolates formed a monophyletic group and clustered with Leishmania tarentolae. The isolates GS1, GS7, XJ771 of this study from China clustered with other isolates of Leishmania donovani complex. The isolate JS1 was a sister to Leishmania tropica, which represented an L. tropica complex instead of clustering with L. donovani complex or with the other 10 Chinese isolates. The isolates KXG-2 and GS-GER20 formed a monophyletic group with Leishmania turanica from central Asia. In the different phylogenetic trees, all of the Chinese isolates occurred in at least four groups regardless of geographic distribution.
Conclusions
The undescribed Leishmania species of China, which are clearly causative agents of canine leishmaniasis and human visceral leishmaniasis and are related to Sauroleishmania, may have evolved from a common ancestral parasite that came from the Americas and may have split off earlier than the other old world Leishmania. Our results also suggest the following: the isolates GS7, GS1 and XJ771 occur as part of the L. donovani complex; the JS1 isolate is L. tropica; and the isolate GS-GER20 identified as Leishmania gerbilli is close to KXG-2 which is L. turanica.
Keywords: Leishmania, Phylogeny, cyt b, China, Sauroleishmania
Background
The leishmaniases are a group of vector borne diseases that are caused by flagellate of the genus Leishmania, which is transmitted by the bite of the sandfly, and affect as many as 12 million people worldwide with 1.5–2 million new cases each year in 88 countries [1]. The genus Leishmania consists of nearly 30 species of morphologically similar kinetoplastid protozoa, and approximately 20 of these species are responsible for a spectrum of human diseases that ranges from mild to fatal infections [2,3].
It is well accepted now that the genus Leishmania forms a monophyletic group with three distinct subgenera Leishmania (Leishmania), Leishmania (Viannia), and Leishmania (Sauroleishmania) [4]. Once, the Leishmania-like parasites of reptiles were recognized as a separate genus L. (Sauroleishmania) [5,6]. But the classification of lizard Leishmania as subgenus L. (Sauroleishmania) was proposed by Saf’janova [7], Croan et al.[8], Orlando et al.[9], Zelazny et al.[10] and Fraga et al.[4] on the basis of biological criteria and analysis of different Leishmania gene. According to the absolute chromosomal size difference index (aCSDI) analysis of as many as 31 “conserved” chromosomes, subgenera L. (Leishmania) is divided into clusters: (1) Old world representatives of subgenus Leishmania (OWL), and (2) New world representatives of subgenus Leishmania (NWL) and subgenus Viannia (NWV) [6].
The complexity of the taxonomy and phylogenetic relationships of the Chinese Leishmania was due to the extensive geographic area and complex ecological environment. Identification of species responsible for different leishmaniasis and clinical manifestation remains uncertain. The strains from cutaneous leishmaniasis (CL) in Xinjiang Uygur Autonomous Region (Xinjiang) especially in Karamay is closely related to L. tropica with analysis of SSU rDNA gene[11,12], whereas the pathogen identified as Leishmania infantum[13,14] or Leishmania turanica[15] from the same geographic region Karamay also could cause CL. However, L. turanica is nonpathogenic to humans, according to Strelkova et al.[16]. The parasites of some visceral leishmaniasis (VL) cases in Sichuan and Gansu provinces were L. donovani or undescribed species Leishmania sp. [17-19]. VL and CL have been reported in China to date the species of Leishmania comprises much more than that. The isolates in China were more heterogeneous than previously thought, requiring the reassignment of some isolates into different groups as described by Lu et al.[20].
Over the past few decades, DNA markers including coding and non-coding genes have become additional information for advancing our understanding of evolutionary and phylogenetic relationships and species differentiation. Data pertaining to the ribosomal RNA (rRNA) gene sequences–in particular, the two non-coding, highly variable internal transcribed spacer regions (ITS1 and ITS2) are considered to be acceptable molecular criteria for resolving taxonomic questions and determining the phylogenetic affinities among closely related Leishmania species [17,21-25]. While coding genes are also wildly used for taxonomic studies, such as metabolic enzymes (ICD, ME, MPI, G6PDH, ASAT, GPI, NH1, NH2, PGD and FH) [26,27], heat-shock protein 70 gene (hsp70) [4], cytochrome oxidase II (CO II) [28], the gene encoding the largest subunit of RNA polymerase II (rpoIILS) [6,8], the glycoprotein 63 gene (gp63) [29], cysteine protease B genes (cpb) [30] and cytochrome b (cyt b) [31-35].
In previous ITS1 and CO II study [17,19], we summarized the four endemic Leishmania species in China: L. donovani, L. infantum, Leishmania gerbilli, and L. turanica. We also noted that there might be an undescribed Leishmania species endemic in China and highlighted that the isolate IPHL/CN/77/XJ771 from Bachu County, Xinjiang, is L. donovani instead of L. infantum. To elucidate the phylogeny, evolution and epidemiology of interesting group of strains in China, further studies of more genes are required.
Cyt b is one of the cytochromes involved in the electron transport process of the mitochondrial respiratory chain is considered one of the most useful genes for phylogenetic work [34]. Marco et al. proved that the cyt b gene sequencing can precisely identify the Leishmania spp. for all of the local stocks that are well characterized by multi-locus enzyme electrophoresis (MLEE), the current gold standard [32]. Phylogeny and sequence variation of the genus Leishmania has also been discussed successfully with cyt b sequencing [34,35]. In this paper, the cyt b gene of Leishmania from China was sequenced and analyzed using bioinformatics methods. Moreover, the phylogenetic relationships were reconstructed using cyt b sequences obtained by this study and download from the GenBank database. We then discuss in detail the implications of relationships between strains in China and other locations.
Methods
Leishmania isolates
A population of cloned promastigotes (including 16 Chinese Leishmania isolates and three isolates from other countries) was stored in liquid nitrogen, and kept at the Department of Parasitology, Western China School of Preclinical and Forensic Medicine, Sichuan University. All of the Leishmania isolates used in this study are listed in Table 1. The promastigotes were cultivated in medium 199 supplemented with 15% heat-inactivated fetal bovine serum (HIFBS) at 28°C. Approximately 1–5 × 109 promastigotes were collected at room temperature by centrifugation at 3300 × g for 10 min and washed with phosphate-buffered saline.
Table 1.
List of Leishmania strains, origin, and database accession numbers, including sequences of Leishmania retrieved from GenBank
| Sequence length (bp) | GenBank accession numbers | MLEE-based species assignment | WHO code | Origin | Reference |
|---|---|---|---|---|---|
| 1079 |
HQ908255a |
Leishmania sp. |
MHOM/CN/84/SD1 |
Shandong, China |
This study |
| 1078 |
HQ908260a |
Leishmania sp. |
MHOM/CN/90/SC10H2 |
Sichuan, China |
This study |
| 1079 |
HQ908263a |
Leishmania sp. |
MHOM/CN/89/GS6 |
Gansu, China |
This study |
| 1079 |
HQ908264a |
Leishmania sp. |
MHOM/CN/86/SC6 |
Sichuan, China |
This study |
| 1080 |
HQ908271a |
Leishmania sp. |
MHOM/CN/84/GS3 |
Gansu, China |
This study |
| 1080 |
HQ908273 |
Leishmania sp. |
MHOM/CN/83/GS2 |
Gansu, China |
This study |
| 1079 |
HQ908266 |
Leishmania sp. |
MHOM/CN/80/XJ801 |
Xinjiang, China |
This study |
| 1079 |
HQ908272 |
Leishmania sp. |
MHOM/CN/90/SC11 |
Sichuan, China |
This study |
| 979 |
HQ908268 |
Leishmania sp. |
MCAN/CN/86/SC9 |
Sichuan, China |
This study |
| 1079 |
HQ908269 |
Leishmania sp. |
MHOM/CN/89/GS5 |
Gansu, China |
This study |
| 1080 |
HQ908259 |
L. gerbilli |
MGER/CN/60/GS-GER20 |
Gansu, China |
This study |
| 1080 |
HQ908256 |
L. turanica |
MRHO/CN/88/KXG-2 |
Karamay, China |
This study |
| 1079 |
HQ908262 |
L. donovani complex |
MCAN/CN/60/GS1 |
Gansu, China |
This study |
| 1079 |
HQ908261 |
L. donovani complex |
MHOM/CN/93/GS7 |
Gansu, China |
This study |
| 1079 |
HQ908267 |
L. donovani complex |
IPHL/CN/77/XJ771 |
Xinjiang, China |
This study |
| 1060 |
HQ908265 |
L. tropica |
MHOM/CN/84/JS1 |
China |
This study |
| 1080 |
HQ908270b |
L. tropica |
MHOM/SU/74/K27 |
Soviet Union |
This study |
| 1080 |
HQ908257b |
L. tropica |
- |
- |
This study |
| 1080 |
AB095960b |
L. tropica |
MHOM/SU/58/Strain OD |
Soviet Union |
Luyo-Acero et al., 2004 [35] |
| 1079 |
AB095965c |
L. garnhami |
MHOM/VE/76/JAP78 |
Venezuela |
Luyo-Acero et al., 2004 [35] |
| 1078 |
HQ908258c |
L. mexicana |
- |
- |
This study |
| 1079 |
AB095957d |
L. donovani |
MHOM/SD/62/2S-25M-C2 |
Sudan |
Luyo-Acero et al., 2004 [35] |
| 1079 |
AB095958d |
L. infantum |
MHOM/TN/80/IPT1 |
Tunisia |
Luyo-Acero et al., 2004 [35] |
| 1080 |
AB095959e |
L. chagasi |
MHOM/BR/74/PP75 |
Brazil |
Luyo-Acero et al., 2004 [35] |
| 872 |
EF579896e |
L. donovani |
MHOM/IN/80/DD8 |
India |
Foulet et al., 2007 [33] |
| 1079 |
AB434677 |
L. archibaldi |
MHOM/ET/72/GEBRE1 |
Ethiopia |
Asato et al., 2009 [34] |
| 872 |
EF579897f |
L. chagasi |
MHOM/BR/74/PP75a |
Brazil |
Foulet et al., 2007 [33] |
| 872 |
EF579913f |
L. infantum |
MCAN/GR/94/CRE69 |
Greece |
Foulet et al., 2007 [33] |
| 1080 |
AB095962 |
L. aethiopica |
MHOM/ET/72/L100 |
Ethiopia |
Luyo-Acero et al., 2004 [35] |
| 872 |
EF579908 |
L. aethiopica |
MHOM/ET/72/L100 |
Ethiopia |
Foulet et al., 2007 [33] |
| 1080 |
AB095970 |
L. major |
MHOM/EC/88/PT-115 |
Ecuador |
Luyo-Acero et al., 2004 [35] |
| 1080 |
AB434675 |
L. turanica |
MRHO/SU/80/CLONE3720 |
Soviet Union |
Asato et al., 2009 [34] |
| 1080 |
AB434674 |
L. arabica |
MPSA/SA/83/JISH220 |
Saudi Arabia |
Asato et al., 2009 [34] |
| 1104 |
M97357 |
L. mexicana |
- |
- |
Lee et al., 1992 [36] |
| 1089 |
M92829 |
L. mexicana |
- |
- |
Lee et al., 1992 [36] |
| 1078 |
AB095964 |
L. amazonensis |
MHOM/BR/73/M2269 |
Brazil |
Luyo-Acero et al., 2004 [35] |
| 1079 |
AB095963g |
L. mexicana |
MHYC/BZ/62/M379 |
Belize |
Luyo-Acero et al., 2004 [35] |
| 872 |
EF579909g |
L. amazonensis |
LMAMPRO/BR/72/M1841 |
Brazil |
Foulet et al., 2007 [33] |
| 872 |
EF579902g |
L. amazonensis |
MHOM/BR/73/M2269 |
Brazil |
Foulet et al., 2007 [33] |
| 1078 |
AB434678 |
L. aristidesi |
MORY/PA/69/GML |
Panama |
Asato et al., 2009 [34] |
| 1078 |
M10126 |
L. tarentolae |
- |
- |
de la Cruz et al., 1984 [37] |
| 1078 |
AB095966 |
L. braziliensis |
MHOM/BR/75/M2904 |
Brazil |
Luyo-Acero et al., 2004 [35] |
| 1078 |
AB434682 |
L. braziliensis |
MHOM/BR/75/M2903 |
Brazil |
Asato et al., 2009 [34] |
| 1078 |
AB095967 |
L. braziliensis |
MHOM/EC/88/INH-03 |
Ecuador |
Luyo-Acero et al., 2004 [35] |
| 872 |
EF579905h |
L. guyanensis |
MHOM/GF/79/LEM85 |
French Guiana |
Foulet et al., 2007 [33] |
| 872 |
EF579912h |
L. guyanensis |
MHOM/BR/75/M4147 |
Brazil |
Foulet et al., 2007 [33] |
| 1078 |
AB095968 |
L. panamensis |
MHOM/BR/71/LS94 |
Brazil |
Luyo-Acero et al., 2004 [35] |
| 1078 |
AB434680 |
L. shawi |
MHOM/BR/79/M15065 |
Brazil |
Asato et al., 2009 [34] |
| 1080 |
AB434686 |
L. equatorensis |
MCOH/EC/82/LSP-1 |
Ecuador |
Asato et al., 2009 [34] |
| 1078 | M94286 | Trypanosoma brucei | - | - | Feagin et al., 1987 [38] |
a, b, c, d, e, f, g, and h shared the same haplotype.
DNA extraction and polymerase chain reaction (PCR)
Total genomic DNA of the parasite was extracted by proteinase K digestion and phenol/chloroform/isoamyl alcohol extraction procedures followed by ethanol precipitation to purify the extracted DNA as described by Sambrook and Russell [39]. PCR was performed to generate a fragment spanning cyt b kinetoplast DNA (kDNA) between the forward primer COIIIF (5′-TAATACGACTCACTATAGTTTATATTG ACATTTTGTWGATT-3′) and the reverse primer MURF4R (5′- GGGTTTTCCCAG TCACGACGAATCTCTCTCTCCCTT −3′) [35]. The PCR protocols for amplification were: 94°C for 3 min followed by 35 cycles of 94°C for 30s, 58°C for 30s, and 72°C for 1.5 min, followed by a final elongation step at 72°C for 10 min. The amplified products were purified on a 2.0% agarose gel stained with ethidium bromide, using a commercial DNA purification kit according to the manufacturer’s protocol. The purified PCR product was then sequenced. The DNA sequences of each individual and each species were deposited in the GenBank database under accession numbers (HQ908255-HQ908273).
Sequence alignment and analyses
A set of cyt b sequences of Leishmania were retrieved from GenBank, included 29 sequences of genus Leishmania (AB095957–AB095960, AB095962–AB095968, AB095970, EF579896–EF579897, EF579902, EF579905, EF579908, EF579909, EF579912, EF579913, AB434674, AB434675, AB434677, AB434678, AB434680, AB434682, M97357, M92829, M10126), Leishmania equatorensis (AB434686) and Trypanosoma brucei (M94286) (Table 1). The sequences were first aligned using Clustal X 1.83 [40] with a gap-opening penalty of 5 and gap-extension penalty of 1 following the small gap costs recommendation of Hickson et al.[41]. The aligned matrix from this procedure was verified to have the same length, and minor adjustments were then made manually using SeaView v.4.2.5 [42]. The data matrices are available from the corresponding author. The haplotype analyses were performed to 37 sequences using DAMBE software for 50 sequences [43]. Compositional heterogeneity was evaluated using chi-square (χ2) tests implemented in PAUP* 4.0b10 [44]. Distances from the predicted amino acid sequences were determined with the p-distance models which were computed by MEGA v. 4.1 [45].
Phylogenetic analyses
Phylogenetic hypotheses of Leishmania were generated with cyt b kDNA segments using two types of commonly applied phylogenetic techniques: heuristic searches using maximum parsimony (MP) analyses performed with the program PAUP* program and Bayesian inference (BI) using the MrBayes v.3.2 program [46]. In both MP and BI analyses, gaps were treated as missing data. For heuristic searches under parsimony, invariant characters were removed from the dataset. Each search involved 10 random additional replicates, one tree held at each step, with tree bisection and reconnectin branch swapping, steepest descent on, and a maximum of 10,000 saved trees. Non-parametric bootstrapping was used to generate phylogeny confidence values [47], with 1,000 pseudoreplicates using a heuristic tree search for each pseudoreplicate. Trypanosoma brucei (M94286) was used to root the trees.
Prior to Bayesian analyses, the best-fit model of evolution, TIM3 + G, was selected using jModeltest v. 0.1.1 [48] under the Bayesian information criterion [49], following recent recommendations [50]. We estimated the posterior probability distributions by allowing four incrementally heated Markov chains (default heating values) to proceed to four million generations, and with samples were taken every 200 generations. Analyses were repeated beginning with different starting trees to ensure that the analyses were not restricted from the global optimum [51]. Convergence was first tested by examining the average deviation of the split frequencies of the two runs, in order to determine whether the two runs had converged. MCMC convergence was also explored by examining the potential scale reduction factor (PSRF) convergence diagnostics for all model parameters (provided by the sump and sumt commands). The first one million generations before this chain became stationary were discarded, and the remaining samples from the independent runs were pooled to obtain the final approximation of the posterior tree distribution.
Sequence alignments were additionally inferred from uncorrected p-distances through NJ networks (Neighbor Net) obtained by SplitsTree 4 [52,53]. This software can detect the alternative evolutionary paths supported by the sequence alignments, and as such, they do not enforce the single bifurcating dendrogram. To yield a single phylogeny hypothesis, the posterior distribution was summarized as a 50% majority rule consensus.
Results
Base composition and nucleotide substitution patterns
The size of the newly determined cyt b fragments is shown in the Table 1. Of the 911 aligned characters, 324 were variable, including 225 that were parsimony-informative. Percentage base compositions were as follows: A, 27.8; C, 7.2; G, 15.6; and T, 50.4. The average maximum likelihood estimated Ti/Tv ratio was 1.2.
A base stationarity test showed insignificant differences among the taxa in base composition bias in the data (χ2 = 85.386150, df = 108, p = 0.94687017). The p-distances among the 10 isolates (10 isolates, SD1, SC10H2, GS6, SC6, GS3, GS2, XJ801, SC11, SC9, GS5; the isolates SD1, SC10H1, GS6, SC6 and GS3 share the same sequence) in China were ranged from 0.000 to 0.023 (mean = 0.010), which are smaller than the distances between these isolate and any other known species. These 10 isolates were then classified into the Leishmania sp. group. The divergence between Leishmania sp. and other Leishmania species ranged from 0.051 (Leishmania sp. versus L.tarentolae) to 0.131 (Leishmania sp. vs. L. turanica and L. gerbilli), with an average of 0.096, a value that is larger than that within Leishmania sp. group. The L. donovani complex group contains seven haplotypes: GS1, GS7, XJ771, PP75a (the same sequence with CRE69), 2S-25M-C2 (the same sequence shared with IPT1), PP75 (the same sequence with DD8). The distance within the strains of L. donovani complex ranged from 0.001 (the strain GS7 of China vs. L. chagasi PP75a and L. infantum CRE69) to 0.013 (L. archibaldi GEBRE1 vs. L. chagasi PP75 and L. donovani DD8), which are smaller than the distances between these strains and other known species. The average distance in this group as a whole is 0.006. The p distances among all species except Leishmania sp. and T. brucei were from 0.002 (between L. turanica and L. gerbilli) to 0.136 (between Leishmania arabica and L. equatoresnsis). Most pairwise comparisons mentioned above had divergence values < 0.136, with an average of 0.106 (Table 2).
Table 2.
Pairwise genetic distances for cytb segments among Leishmania species
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
Leishmania sp. |
- |
|
|
|
|
|
|
|
|
|
|
| 2 |
L. tarentolae |
0.051 |
- |
|
|
|
|
|
|
|
|
|
| 3 |
L. braziliensis complex |
0.095 |
0.095 |
- |
|
|
|
|
|
|
|
|
| 4 |
L. mexicana complex |
0.104 |
0.106 |
0.107 |
- |
|
|
|
|
|
|
|
| 5 |
L. donovania complex |
0.110 |
0.113 |
0.102 |
0.095 |
- |
|
|
|
|
|
|
| 6 |
L.turaniaca |
0.131 |
0.131 |
0.122 |
0.107 |
0.092 |
- |
|
|
|
|
|
| 7 |
L. gerbilli |
0.131 |
0.131 |
0.123 |
0.107 |
0.093 |
0.002 |
- |
|
|
|
|
| 8 |
L. arabica |
0.130 |
0.130 |
0.124 |
0.106 |
0.101 |
0.085 |
0.085 |
- |
|
|
|
| 9 |
L. tropica complex |
0.116 |
0.119 |
0.113 |
0.095 |
0.081 |
0.088 |
0.088 |
0.095 |
- |
|
|
| 10 |
L. equatoresnsis |
0.126 |
0.128 |
0.119 |
0.112 |
0.120 |
0.126 |
0.127 |
0.136 |
0.125 |
- |
|
| 11 | Trypanosoma brucei | 0.177 | 0.176 | 0.150 | 0.158 | 0.158 | 0.183 | 0.183 | 0.185 | 0.176 | 0.155 | - |
Pairwise genetic distances between different groups are detected by MEGA4.
Phylogenetic relationships
The heuristic search of the cyt b matrix resulted in 10,000 equally parsimonious trees of 15519 steps with high CI (0.6112) and RI (0.8427) velues. In the strict consensus phylogram (Figure 1), three clades (BP = 88%) were formed. Leishmania sp., L. tarentolae and L. braziliensis complex form a clade (BP = 68%); six haplotypes in China formed a strongly cluster Leishmania sp. (BP = 88%), and clustered with with L. tarentolae (BP = 99%); and next joined by the L. braziliensis complex (BP = 99%) containing three haplotypes of L. braziliensis, L. guyanensis, L. panamensis and L. shawi. The OWL clade (BP = 86%) clustered with the following OWL species: L. donovani, L. major, L. tropica, L. arabica, L. turanica and L. gerbilli. Leishmania chagasi (with the synonymous L. infantum), L. donovani, Leishmania archibaldi (with the synonymous L. donovani) [54], L. infantum, and the isolates GS7, GS1 and XJ771of this study from China formed a monophyletic clade L. dovovani complex. The strain JS1 from Jiangsu province of China clustered with L. tropica from the Soviet Union (BP = 100%) and then clustered with L. aethiopica (BP = 100%), which formed the L. tropica complex except L. major (BP = 89%). L. turanica from the Soviet Union clustered with strains GS-GER20 and KXG-2 (BP = 63%) in China from this study clustered together (BP = 100%), which next joined by L. arabica (BP = 94%) and L. major (BP = 54%). The clade NWL (BP = 100%) is clustered with L. mexicana, L. garnhami, L. amazonensis, and Leishmania aristides. Information about the strains is shown in Table 1.
Figure 1.

Maximum parsimony consensus tree of the cyt b dataset by using PAUP*.Trypanosoma brucei (M94286) is the outgroup. Tree length = 15519, CI = 0.6112, RI = 0.8427. The tree is based on haplotypes (identical haplotypes are presented by one strain). The numbers above the branch represent percent recovery in bootstrap analysis (1,000 pseudoreplicates), only bootstrap values >50% are shown. Strains information is shown in Table 1. The letters (a, b, c, d, e, f, g, and h) indicate sharing of a haplotype.
For the BI analyses, the likelihood value of the 50% majority consensus tree (Figure 2) was ln L = −4132.1156, while the average PSRF was 1.001. The topology of the BI tree is a little different from that of MP tree. Two robust clades were formed. In one robust clade (PP = 0.95), Leishmania sp. (PP = 0.99) was a sister of L. tarentolae (PP = 1.00) and forms one clade with the species of L. (Leishmania) (LL), the other clade was consis of the species of L. (Viannia) (LV) (PP = 1.00) (see Figure 3). The group of Leishmania sp. and L. tarentolae was basal to subgenus Leishmania and L. equatorensis. L. mexicana, L. garnhami, L. amazonensis, and L. aristidesi formed the L. mexicana complex (PP = 1.00). The L. mexicana and L. equatorensis are fundamental to all remaining subgenus Leishmania species. Within the other members of subgenus Leishmania, the L. donovani complex is sister to the clade (PP = 1.00) that clustered with L. tropica (PP = 1.00) plus L. aethiopica (PP = 1.00), and the clade (PP = 0.97) that consists of L. turanica, L. gerbilli and L. arabica, next joined by L. major (PP = 1.00).
Figure 2.

The 50% majority-rule consensus tree inferred from Bayesian inference of cyt b dataset using MrBayes v. 3.2. The numbers at the nodes represent Bayesian posterior probabilities; TIM3 + G was selected using jModeltest v. 0.1.1. Trypanosoma brucei (M94286) is the outgroup. The tree is based on haplotypes (identical haplotypes are presented by one strain). Strain information is shown in Table 1. The letters (a, b, c, d, e, f, g, and h) indicate sharing of a haplotype.
Figure 3.
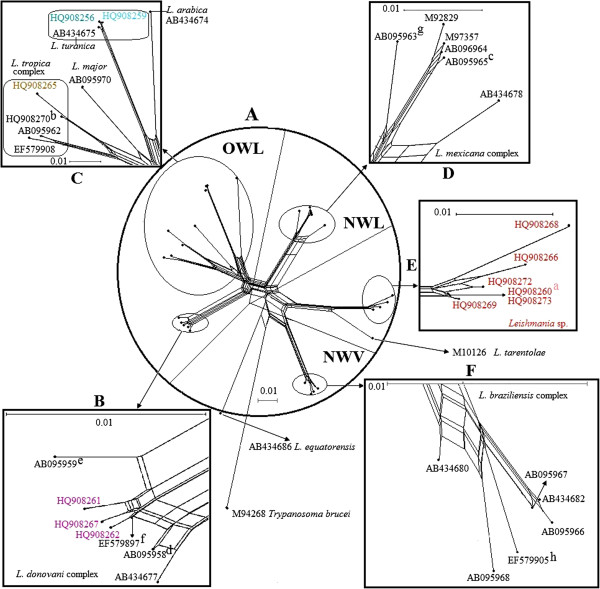
The phylogenetic network of the Leishmania cyt b sequences shown in Table 1 and Figures 1,2 was built with 1000 bootstrap replicates. It was algorithm, excluding all conserved site. Distances were calculated using the Kimura 2-parameter distance. Trypanosoma brucei (M94286) is the outgroup. Each A-F panel is drawn to the scale indicated and expressed as dissimilarity per nucleotide counted over variable sites (Figures 1–2) in cyt b alignment. The dots indicate the sequence position in the network. A: Complete network with representation of the five groups shown in detail in the remaining panels; the whole network excluding the L. equatorensis and T. brucei is divided into four segments: OWL, NWL, the Leishmania sp. and L. tarentolae group, and NWV. B: The L. donovani complex includes three Chinese isolates. C: Includes the species L. tropica, L. aethiopica, L. major, L. turanica, L. gerbilli, and L. arabica.D: The New World Leishmania subgenus. E: The Leishmania sp. of this study. F: The L. (Viannia) subgenus. Strain information is shown in Table 1.
In addition to the common phylogenetic relationships among the different species shown by the MP tree and BI tree, the network (Figure 3) calculated by SplitsTree 4 also indicated a clear evolutionary path with a high value. Leishmania sp. and L. tarentolae share most of their evolutionary paths.
Discussion
As a part of worldwide Leishmania population, the phylogenetics of Chinese isolates with analysis of the cyt b genetic sequences of 16 Leishmania isolates was discussed in this paper which demonstrated similarities and differences compared with previous data [17,19] and keep the genus evolutionary unity and integrity over large geographic ranges and time periods.
Leishmania sp. of China
Most interestingly 10 Chinese strains, representing 6 closely related haplotypes, could not be assigned to any of the so far described species of Leishmania, a finding that is congruent with our earlier ITS1 and COII studies [17,19]. These Leishmania sp. isolates were most closely related to the lizard-infecting L. tarentolae (Figures 1, 2, 3).
It was reported that one of these isolates SC6 was collected from patients with VL in Nanping County of Sichuan Province, was infected successfully 8 dogs (8/12) and its amastigotes were detected in their bone marrow smears [55]. Another isolate SC10H2 was proved that it clustered with the pathogen of canine leishmaniasis in Beichuan County, Sichuan Province, China based on the 17S RNA gene [18]. The non-pathogenic to humans L. tarentolae has been classified as subgenus L. (Sauroleishmania) on the basis of biological criteria and different genes [4,8,10,34]. In such cases, we can conclude that the undescribed Leishmania species which is clearly a causative agent of canine leishmaniasis and human VL do exist in China are related to the Sauroleishmania. However, the more lizard parasites are required to confirm whether Leishmania sp. is assigned to the Sauroleishmania.
The pairwise genetic distance analysis (Table 2) and phylogenetic network (Figure 3) suggest that the cyt b sequences of the Chinese/tarentolae group (Leishmania sp. and L. tarentolae) are closer to the Viannia clade than the older world Leishmania. This finding is in contrast to that of our ITS1 study [17] and other studies: as an OWL species branching from within New World taxa, L. tarentolae (Sauroleishmania) are closer to the Leishmania subgenus than to the Viannia subgenus based on different DNA marks (polA and RNA polymerase II, 7SL RNA, hsp 70) [4,8,10]. It is well knows that different genes can have different evolutionary histories and be influenced by selection and horizontal gene transfer, and the phylogenies are also prone to sampling bias; therefore, more genes of diverse geographic original strains would be needed to elucidate the phylogeny, evolution, and epidemiology of the Chinese/tarentolae group.
The isolates of Leishmania sp. were collected from different foci (plain, desert and hill), and the longest distance between isolates is more than 2000 miles (from Shandong to the Xinjiang) (Figure 4). Meanwhile, different species were found in the same area. The isolate XJ801 of Leishmania sp. is from Kashi city of Xinjiang. The isolate 801 identified as L. donovani based on ITS1 sequences by Wang et al.[16] and Yang et al.[17] is also from Kashi city. Another two isolates MHOM/CN/76/BT013 and MHOM/CN/81/812 which is differs from L. donovani (PHON/CN/77/771), L. turanica (MRHO/CN/88/KXG-2) and L. gerbilli (MRHO/CN/62/1) based on polymorphisms in both kinetoplast (kDNA) and nuclear (nDNA) DNAs that also collected from the same area Kashi [20]. As such, the Leishmania isolates in China were more heterogeneous, further epidemiologic survey and more strains are required in Kashi.
Figure 4.
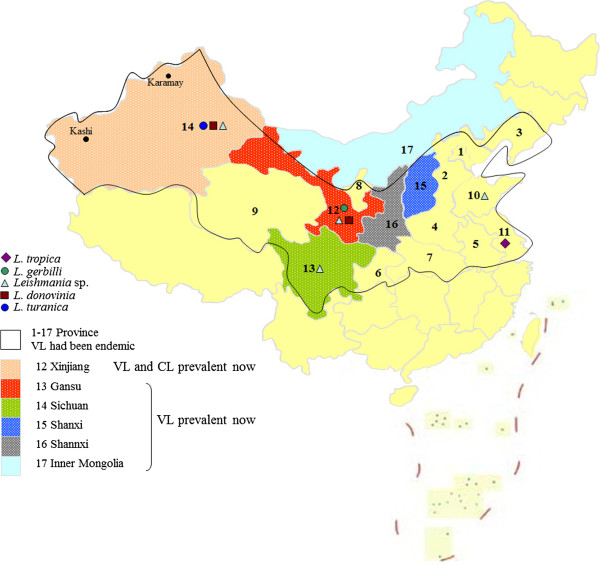
Distribution of leishmaniasis in China and the specimen collection areas. 1: Beijiang and Tianjin; 2: Hebei; 3: Liaoning; 4: Henan; 5: Anhui; 6: Chongqing; 7: Hubei; 8: Ningxia Hui Autonomous Region; 9: Qinghai; 10: Shandong; 11: Jiangshu; 12: Gansu; 13: Sichuan; 14: Xinjiang Uygur Autonomous Region (Xinjiang); 15: Shanxi; 16: Shannxi; 17: Inner Mongolia Autonomous Region (Inner Mongolia). The species of this study from different province are shown as different shapes on the map.
L. donovani complex of China
Analysis in the current study revealed that the cyt b sequences of GS1, GS7 and XJ771 clustered with other species of L. donovani complex (PP = 1.00). On the basis of MLEE of the representative isolates from the plain, hill, and desert regions of China, Xu et al. were first to identify the causative agents responsible of VL as Leishmania donovani sensu lato and L. infantum[56]. The results based on sequences of cyt b, ITS1 [17] and COII [19] sequences clearly proved the existence of L. donovani in China. However, L. donovani or L. infantum standard isolates cannot be distinguished from L. donovani complex isolate using the cyt b gene in the BI and MP trees. These findings aren’t consistent the ITS1 study showing three isolates clustered with L. donovani and a clear classification within subspecies between Leishmania donovani donovani and Leishmania donovani infantum. Therefore, the inter-specific variation of the ribosomal RNA gene ITS1 was inferred to be more suitable than mtDNA segment cyt b for studying the phylogenetic relationships among subspecies. Of course, we can’t exclude the possibility that the different inter-specific variation between ITS1 and cyt b are calculated by choosing the different samples or numbers of the isolates or strains.
L. turanica of China
Our cyt b data demonstrate that the isolates KXG-2 and GS-GER20 clustered with L. turanica (AB434675) from central Asia, findings that are congruent with those of our earlier studies [17,19] and then clustered with L. arabica from western Asia, a finding that agrees with that of Asato et al.[34] (Figures 1, 2, 3). The definitive hosts of L. gerbilli, L. turanica, L. arabica are rodents of the Old World [57]. Using MLEE methods, the isolate KXG-2 was identified as L. turanica[16], and the isolate GS-GER20 was identified as L. gerbilli[58]. In the 1990s, L. turanica and L. gerbilli were identified in rodents or sandflies in Karamay, Xinjiang and L. turanica was proved to be pathogenic in both monkeys and humans in the laboratory, Phlebotomus mongolensis and Phlebotomus andrejevi were its major vectors [16]. We considered the isolates KXG-2 and GS-GER20 to be L. turanica and L. gerbilli, respectively via the cyt b gene sequences.
L. tropica of China
The species of the L. tropica complex cause the urban form of Old World CL. In Iran, Iraq, and India, it is transmitted by Phlebotomus papatasi. This species is rarely reported in China. The fact that the isolate JS1 was collected from Jiangsu Province clustered with L. tropica, which agrees with the results of our earlier study based on the COII gene [19]. Lu et al. used random amplified polymorphic DNA data to suggest a close relationship between the isolate JS1 and L. tropica (K27) [59]. Thus, we infer that the isolate JS1 may be L. tropica. However to further confirm this inference, more data such as host specificity, life cycle, and biochemical analysis will be needed.
Evolution inference and epidemiology of China
In our analysis, Leishmania cyt b sequences are consistent with the genus Leishmania that contains three subgenera: Leishmania, Sauroleishmania and Viannia[4]. Based on the suggestion that mammalian Leishmania did not evolve from those of lizards but vice versa[60,61], Lukeš et al.[54] proposed that the ancestor of the new world Leishmania evolved in South America and then migrated via the Bering land bridge to Asia via multiple independent genetic loci. The Leishmania lineage would have been dispersed throughout central and/or Southeast Asia, where a major diversification gave rise to L. aethiopica, L. major, L. gerbilli, L. turanica, L. tropica, and the L. donovani complex. The isolates from China were absent in this analysis. However Fraga et al. thought this theory puts L. tarentolae (Sauroleishmania) in an illogical position. Our data suggest that Leishmania sp. of the pathogen of VL and CanL clustering with L. tarentolae (Sauroleishmania) was in the same “illogical position”. The maximum parsimony consensus tree (Figure 1) and splitstree (Figure 3) supports the idea of a common origin with the Viannias subgenus, whereas the Bayesian tree (Figure 2) show the Chinese/taretolae group clustered together with species of Leishmania subgenus. This ambiguous position of L. tarentolae had been discussed by Luyo-Acero et al. based on the same DNA marker cyt b[35] that L. tarentolae clustered with Viannia in the NJ tree consisting with the minicircle phylogenetic analysis [62], and clustered with Leishmania in the MP tree supported by ATPase 6 gene [63]. The position of L. equatorensis as falling outside the Leishmania clade in the parsimony tree is supported by the phylogeny suggested by Cupolillo et al.[64]. However the Chinese/tarentolae group which was not described by Lukeš et al.[54], may have evolved from a common ancestral parasite that came from the Americas and may split off earlier than the other OWL.
Leishmaniasis remains endemic in China, especially in the west and northwest frontier regions. The epidemic foci of VL in China were classified into three types according to different geographical origin, infective agent, and clinical evidences, i.e., plain foci, hill foci, and desert foci [20]. Human VL and CL occur in China, most being VL along with rare CL cases [56,65-67]. VL was one of the most important parasitic diseases occurring in over 17 Chinese provinces in 1951[68]. Since the condition has come under control, currently, VL is mainly prevalent in six provinces in northwest China [69] (Figure 4). This study proved that the evolution hypothesis of Tian and Chen related to the Chinese Leishmania isolates from different epidemic foci was limited and lacked integrity [70]. In fact, the Chinese Leishmania species occurs as the multiple species L. donovani (L. donovani donovani, L. donovani infantum), L. turanica, L. tropic, Leishmania sp. and so forth, and some of these such as L. donovani and L. turanica were shared with neighbouring countries including India, Russia, and Uzbekistan.
Conclusions
The current study investigated the Chinese Leishmania parasites using cyt b sequence data. Undescribed Leishmania species which are clearly causative agents of CanL and human VL do exist in China and are related to the Sauroleishmania subgenus, may have evolved from a common ancestral parasite that came from the Americas and split off earlier than the other OWL. Our cyt b results also suggest the following: the isolates GS7, GS1 and XJ771 occur as part of the L. donovani complex; the isolate JS1 is L. tropica; and the isolate KXG-2 is close to the isolate GS-GER20, which is L. turanica and L. gebilli respectively. The results of the current study indicate that the isolates from China may have had a more complex evolutionary history. In the future, we will build upon the currently described data set to gain more insight into the fascinating spectrum of Chinese Leishmania.
Competing interests
The authors have no competing interests to declare.
Authors’ contributions
JPC and BBY conceived, designed and coordinated the field study, while DLC, LL, XSH and JNX participated in the study design and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Bin-Bin Yang, Email: binbinyangbinbin@yahoo.com.cn.
Da-Li Chen, Email: cdl1978119@sina.com.
Jian-Ping Chen, Email: jpchen007@163.com.
Lin Liao, Email: liaolin08@163.com.
Xiao-Su Hu, Email: huxiaosu28@sina.com.
Jia-Nan Xu, Email: xjn420@yahoo.com.cn.
Acknowledgements
This work was supported by the National Natural Science Foundations of China (NO: 30771883, 30800094 and 81171607) and the Chinese Important Scientific Research Project on Infectious Diseases (NO 2008-ZX10004-011).
References
- World Health Organization. Proceedings of a meeting of the WHO Expert Committee, Geneva, 22–26 March 2010. Geneva: World Health Organization; 2010. Control of the leishmaniase; pp. 22–26. [Google Scholar]
- Shaw JJ. Taxonomy of the genus Leishmania: Present and future trends and their implications. Mem Inst Oswaldo Cruz. 1994;89:471–478. doi: 10.1590/s0074-02761994000300033. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Control of the leishmaniases.Proceedings of a meeting of the WHO Expert Committee, Geneva, 1–158 March 1990. Geneva: World Health Organization; 1990. [PubMed] [Google Scholar]
- Fraga J, Montalvo AM, de Doncker S, Dujardin J-C, der Auwera GV. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect Genet Evol. 2010;10:238–245. doi: 10.1016/j.meegid.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Lainson R, Shaw JJ. In: The Leishmaniasis in Biology and Medicine. Killick W, Kendrick R, editor. London Academic Press; 1987. Evolution, classification and geographical distribution of Leishmania; pp. 1–120. [Google Scholar]
- Dujardin JC, Henriksson J, Victoir K, Brisse S, Gamboa D, Arevalo J, Le Ray D. Genomic Rearrangements in Trypanosomatids: an Alternative to the ‘One Gene’ Evolutionary Hypotheses? Mem Inst Oswaldo Cruz. 2000;95:527–534. doi: 10.1590/s0074-02762000000400015. [DOI] [PubMed] [Google Scholar]
- Saf'janova VM. The problem of taxonomy with Leishmania. Ser Protozool Sov Acad Sci Leningr. 1982;7:5–109. [Google Scholar]
- Croan DG, Morrison DA, Ellis JT. Evolution of the genus Leishmania revealed by comparison of DNA and RNA polymerase gene sequences. Mol Biochem Parasitol. 1997;89:149–159. doi: 10.1016/s0166-6851(97)00111-4. [DOI] [PubMed] [Google Scholar]
- Orlando TC, Rubio MA, Sturm NR, Campbell DA, Floeter-Winter LM. Intergenic and external transcribed spacers of ribosomal RNA genes in lizard-infecting Leishmania: molecular structure and phylogenetic relationship to mammal-infecting Leishmania in the subgenus Leishmania(Leishmania) Mem Inst Oswaldo Cruz. 2002;97:695–701. doi: 10.1590/s0074-02762002000500020. [DOI] [PubMed] [Google Scholar]
- Zelazny AM, Fedorko DP, Li L, Neva FA, Fisher SH. Evaluation of 7SL RNA gene sequences for the identification of Leishmania spp. Am J Trop Med Hyg. 2005;72:415–420. [PubMed] [Google Scholar]
- Chen JP, Hu XS, Jing BQ, Yang WT. Studies of SSUrDNA sequence homologies of Leishmania species causing cutaneous leishmanisis in Xinjiang, China by PCR and hybridization. Acta Parasitol Med Entomol Sin. 1996;3:89–92. [Google Scholar]
- Zhang T, Hu XS, Jing BQ, Zhen XL, Ma Y, Li F. PCR Amplification and Cloning of SSU rDNA Variable Region of Cutaneous Leishmaniasis Pathogen from Xinjiang. China. J Fujian Med Univ. 1998;32:1–5. [Google Scholar]
- Ren HY, Guan LR, Yang YQ, Zuo XP, Li F, Xu YX, Abulikemujiang, Deng LQ. Clinicopathologic analysis on ninety cases of cutaneous leishmaniasis in Karamay region of Xinjiang. Chin J Parasitol Parasit Dis. 1996;14:226–230. [Google Scholar]
- Wang JY, Qu JQ, Wang RQ, Guan LR, Ren HY, Chang KP. Analysis on homology in several isolates of Leishmania from Karamay Xinjiang. Chin J Parasitol Parasit Dis. 1996;14:266–269. [Google Scholar]
- Guan LR, Yang YQ, Xu YX, Wu JT. Leishmaniasis in Karamay XI. The development of cutaneous Leishmaniasis in monkey and man experimentally infected with Leishmania from Karamay big gerbil. Bull Dis Control Prev. 1992;10:263–266. [PubMed] [Google Scholar]
- Strelkova MV, Shurkhal AV, Kellina OI, Eliseev LN, Evans DA, Peters W, Chapman CJ, Le Blancq SM, van Eys GJ. A new species of Leishmania isolated from the great gerbil Rhombomys opimus. Parasitology. 1990;101:327–335. doi: 10.1017/s0031182000060510. [DOI] [PubMed] [Google Scholar]
- Yang BB, Guo XG, Hu XS, Zhang JG, Liao L, Chen DL, Chen JP. Species discrimination and phylogenetic inference of 17 Chinese Leishmania isolates based on internal transcribed spacer 1 (ITS1) sequences. Parasitol Res. 2010;107:1049–1065. doi: 10.1007/s00436-010-1969-9. [DOI] [PubMed] [Google Scholar]
- Sun K, Guan W, Zhang JG, Wang YJ, Tian Y, Liao L, Yang BB, Chen DL, Chen JP. Prevalence of canine leishmaniasis in Beichuan County, Sichuan, China and phylogenetic evidence for an undescribed Leishmania sp. in China based on 7SL RNA. Parasit Vectors. 2012;5:75. doi: 10.1186/1756-3305-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DP, Guo XG, Chen DL, Chen JP. Species delimitation and phylogenetic relationships of Chinese Leishmania isolates reexamined using kinetoplast cytochrome oxidase II gene sequences. Parasitol Res. 2011;109:163–173. doi: 10.1007/s00436-010-2239-6. [DOI] [PubMed] [Google Scholar]
- Lu HG, Zhong L, Guan LR, Qu JQ, Hu XS, Chai JC, Xu ZB, Wang CT. Separation of Chinese Leishmania isolates into five genotypes by kinetoplast and chromosomal DNA heterogeneity. Am J Trop Med Hyg. 1994;50:763–770. doi: 10.4269/ajtmh.1994.50.763. [DOI] [PubMed] [Google Scholar]
- Dávila AMR, Memen H. Internaltranscribedspacer (ITS) sequences used to explore phylogenetic relationships within Leishmania. Ann Trop Med Parasitol. 2000;94:651–654. doi: 10.1080/00034980050152085. [DOI] [PubMed] [Google Scholar]
- Schönian G, Akuffo H, Lewin S, Maasho K, Nylén S, Pratlong F, Eisenberger CL, Schnur LF, Presber W. Genetic variability within the species Leishmania aethiopica does not correlate with clinical variations of cutaneous leishmaniasis. Mol Biochem Parasitol. 2000;106:239–248. doi: 10.1016/s0166-6851(99)00216-9. [DOI] [PubMed] [Google Scholar]
- El Tai NO, El Fari M, Mauricio I, Miles MA, Oskam L, El Safi SH, Presber WH, Schönian G. Leishmania donovani: intraspecific polymorphisms of Sudanese isolates revealed by PCRbased analyses and DNA sequencing. Exp Parasitol. 2001;97:35–44. doi: 10.1006/expr.2001.4592. [DOI] [PubMed] [Google Scholar]
- Kuhls K, Mauricio IL, Pratlong F, Presber W, Schönian G. Analysis of ribosomal DNA internal transcribed spacer sequences of the Leishmania donovani complex. Microbes Infect. 2005;7:1224–1234. doi: 10.1016/j.micinf.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Parvizi P, Moradi G, Akbari G, Farahmand M, Ready PD, Piazak N, Assmar M, Amirkhani A. PCR detection and sequencing of parasite ITSrDNA gene from reservoirs host of zoonotic cutaneous leishmaniasis in central Iran. Parasitol Res. 2008;103:1273–1278. doi: 10.1007/s00436-008-1124-z. [DOI] [PubMed] [Google Scholar]
- Mauricio IL, Yeo M, Baghaei M, Doto D, Pratlong F, Zemanova E, Dedet JP, Lukes J, Miles MA. Towards multilocus sequence typing of the Leishmania donovani complex: Resolving genotypes and haplotypes for five polymorphic metabolic enzymes (ASAT, GPI, NH1, NH2, PGD) Int J Parasitol. 2006;36:757–769. doi: 10.1016/j.ijpara.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Zemanová E, Jirkü M, Mauricio IL, Horák A, Miles MA, Lukeš J. The Leishmania donovani complex: Genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int J Parasitol. 2007;37:149–160. doi: 10.1016/j.ijpara.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Ibrahim ME, Barker DC. The origin and evolution of the Leishmania donovani complex as inferred from a mitochondrial cytochrome oxidase II gene sequence. Infect Genet Evol. 2001;1:61–68. doi: 10.1016/s1567-1348(01)00009-0. [DOI] [PubMed] [Google Scholar]
- Mauricio IL, Gaunt MW, Stothard JR, Miles MA. Glycoprotein 63 (gp63) genes show gene conversion and reveal the evolution of Old World Leishmania. Int J Parasitol. 2007;37:565–576. doi: 10.1016/j.ijpara.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Hide M, Bras-Goncalves R, Bañuls AL. Specific cpb copies within the Leishmania donovani complex: evolutionary interpretations and potential clinical implications in humans. Parasitology. 2007;134:379–389. doi: 10.1017/S0031182006001600. [DOI] [PubMed] [Google Scholar]
- Marco JD, Uezato H, Mimori T, Barroso PA, Korenaga M, Nonaka S, Baosombrío MA, Taranto NJ, Hashiguchi Y. Are cytochrome b gene sequencing and polymorasw chain reaction as reliable as multilocus enzyme electrophoresis for identifying Leishmania sp from Argentina? Am J Trop Med Hyg. 2006;75:256–260. [PubMed] [Google Scholar]
- Marco JD, Bhutto AM, Soomro FR, Baloch JH, Barroso PA, Kato H, Uezato H, Katakura K, Korenaga M, Nonaka S, Hashiguchi Y. Multilocus enzyme electrophoresis and cytochrome b gene sequencingbased identification of Leishmania isolates from different foci of cutaneous leishmaniasis in Pakistan. Am J Trop Med Hyg. 2006;75:261–266. [PubMed] [Google Scholar]
- Foulet F, Botterel F, Buffet P, Morizot G, Rivollet D, Deniau M, Pratlong F, Costa JM, Bretagne S. Detection and identification of Leishmania species from clinical specimens by using a realtime PCR assay and sequencing of the cytochrome B gene. J Clin Microbiol. 2007;45:2110–2115. doi: 10.1128/JCM.02555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asato Y, Oshiro M, Myint CK, Yamamoto Y, Kato H, Marco JD, Mimori T, Gomez EA, Hashiguchi Y, Uezato H. Phylogenic analysis of the genus Leishmania by cytochrome b gene sequencing. Exp Parasitol. 2009;121:352–361. doi: 10.1016/j.exppara.2008.12.013. [DOI] [PubMed] [Google Scholar]
- LuyoAcero GE, Uezato H, Oshiro M, Takei K, Kariya K, Katakura K, GomezLandires E, Hashiguchi Y, Nonaka S. Sequence variation of the cytochrome b gene of various human infecting members of the genus Leishmania and their phylogeny. Parasitology. 2004;128:483–491. doi: 10.1017/s0031182004004792. [DOI] [PubMed] [Google Scholar]
- Lee ST, Tarn C, Wang CY. Characterization of sequence changes in kinetoplast DNA maxicircles of drugresistant Leishmania. Mol Biochem Parasitol. 1992;56:197–207. doi: 10.1016/0166-6851(92)90169-k. [DOI] [PubMed] [Google Scholar]
- de la Cruz VF, Neckelmann N, Simpson L. Sequences of six genes and several open reading frames in the kinetoplast maxicircle DNA of Leishmania tarentolae. J Biol Chem. 1984;259:15136–15147. [PubMed] [Google Scholar]
- Feagin JE, Jasmer DP, Stuart K. Developmentally regulated addition of nucleotides within apocytochrome b transcripts in Trypanosoma brucei. Cell. 1987;49:337–345. doi: 10.1016/0092-8674(87)90286-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J. Russell D W: Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson RE, Simon C, Perrey SW. The performance of several multiplesequence alignment programs in relation to secondary structure features for an rRNA sequence. Mol Biol Evol. 2000;17:530–539. doi: 10.1093/oxfordjournals.molbev.a026333. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Xia X, Xie Z. DAMBE: data analysis in molecular biology and evolution. J Hered. 2001;92:371–373. doi: 10.1093/jhered/92.4.371. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP* Phylogenetic analysis using parsimony (* and other methods). version 4 Sinauer. MA: Sunderland; 2002. [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 40. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Felsensten JP. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Posada D. j ModelTest: phylogenetic model averaging. Mol Biol E. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6:461–464. [Google Scholar]
- Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approach. Syst Biol. 2004;53:793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Larget B, Miller RE, Ronquist F. Potential applications and pitfalls of Bayesian inference of phylogeny. Syst Biol. 2002;51:673–688. doi: 10.1080/10635150290102366. [DOI] [PubMed] [Google Scholar]
- Huson DH. SplitTree: analyzing amd visualizing evolutionaty data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryan D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Lukeš J, Mauricio IL, Schönian G, Dujardin JC, Soteriadou K, Dedet JP, Kuhls K, Tintaya KWQ, Jirků M, Chocholová E, Haralambous C, Pratlong F, Oborník M, Horák A, Ayala FJ, Miles MA. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc Natl Acad Sci USA. 2007;104:9375–9380. doi: 10.1073/pnas.0703678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XS, Yang WT, Lu HG, Yan HP, Chen JP, Ma Y, Jing BQ, Zhang T. Sequencing a specific kinetoplast DNA fragment of Leishmania donovani for polymerase chain reaction amplification in diagnosis of Leishmaniasis in bone marrow and blood samples. J Parasito. 2000;84:822–826. doi: 10.1645/0022-3395(2000)086[0822:SASKDF]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Xu ZB, Le Blancq S, Evans DA, Peters W. The characterization by isoenzyme electrophoresis of Leishmania isolated in the People’s Republic of China. Trans R Soc Trop Med Hyg. 1984;78:689–693. doi: 10.1016/0035-9203(84)90243-8. [DOI] [PubMed] [Google Scholar]
- Ashford RW. The leishmaniases as emerging and reemerging zoonoses. Int J Parasitol. 2000;30:1269–1281. doi: 10.1016/s0020-7519(00)00136-3. [DOI] [PubMed] [Google Scholar]
- Guan LR, Yang YQ, Ren HY, Shen WX. Discovery and study of Leishmania turanica for the first time in China. Bull World Health Organ. 1995;69:595–601. [PMC free article] [PubMed] [Google Scholar]
- Lu DM, Hu XS, Qiao ZD. Analysis of Leishmania species and strains from China by RAPD technique. Chin J Parasitol Parasit Dis. 2001;19:290–293. [PubMed] [Google Scholar]
- Shaw JJ. Ecological and evolutionary pressures on leishmanial parasites. Braz J Gen. 1997;20:123–128. [Google Scholar]
- Noyes H. Implications of a Neotropical origin of the genus Leishmania. Memórias do Instituto Oswaldo Cruz. 1998;93:657–661. doi: 10.1590/s0074-02761998000500017. [DOI] [PubMed] [Google Scholar]
- Yurchenko V, Kolesnikov A, Lukes J. Phylogenetic analysis of Trypanosomatina (Protozoa: Kinetoplastida) based on minicircle conserved regions. Folia Parasitol. 2000;47:1–5. doi: 10.14411/fp.2000.001. [DOI] [PubMed] [Google Scholar]
- Brewster S, Barker DC. The ATPase subunit 6 gene sequence predicts that RNA editing is conserved between lizard-and human-infecting Leishmania. Gene. 1999;235:77–84. doi: 10.1016/s0378-1119(99)00207-3. [DOI] [PubMed] [Google Scholar]
- Cupolillo E, Medina-Acosta E, Noyes H, Momen H, Grimaldi G Jr. A revised classification for Leishmania and Endotrypanum. Parasitol Today. 2000;16:142–144. doi: 10.1016/s0169-4758(99)01609-9. [DOI] [PubMed] [Google Scholar]
- Guan LR, Yang YQ, Ren HY, Chai JJ. Eight cutaneous Leishmaniasis cases detected in Karamay, Xinjiang Uygur Autonomous Region, China. Southeast Asian J Trop Med Public Health. 1992;23:803–804. [PubMed] [Google Scholar]
- Zheng CJ, Wang LY, Xu X, Zhu XH, Wu WP. Visceral Leishmaniasis in China during 2004–2007. Chin J Parasitol Parasit Di. 2009;27:344–346. [PubMed] [Google Scholar]
- Chen J, Xu MJ, Zhou DH, Song HQ, Wang CR, Zhu XQ. Canine and feline parasitic zoonoses in China. Parasit Vectors. 2012;5:152. doi: 10.1186/1756-3305-5-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Cui G, Chen HT, Zhou XN, Gao CH, Yang YT. Current epidemiological profile and features of visceral leishmaniasis in People’s Republic of China. Parasit Vectors. 2012;5:31. doi: 10.1186/1756-3305-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XN, Lv S, Yang GJ, Kristensen TK, Bergquist NR, Utzinger J, Malone JB. Spatial epidemiology in zoonotic parasitic diseases: insights gained at the 1st International Symposium on Geospatial Health in Lijiang, China, 2007. Parasit Vectors. 2009;2:10. doi: 10.1186/1756-3305-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Chen JP. Cloning and sequence analysis is of ITS gene of Leishmania donovani isolates from different epidemic Foci in China. J Biomed. 2005;22:540–544. [PubMed] [Google Scholar]