

Abstract
Background
The size of the protein sequence database has been exponentially increasing due to advances in genome sequencing. However, experimentally characterized proteins only constitute a small portion of the database, such that the majority of sequences have been annotated by computational approaches. Current automatic annotation pipelines inevitably introduce errors, making the annotations unreliable. Instead of such error-prone automatic annotations, functional interpretation should rely on annotations of ‘reference proteins’ that have been experimentally characterized or manually curated.
Results
The Seq2Ref server uses BLAST to detect proteins homologous to a query sequence and identifies the reference proteins among them. Seq2Ref then reports publications with experimental characterizations of the identified reference proteins that might be relevant to the query. Furthermore, a plurality-based rating system is developed to evaluate the homologous relationships and rank the reference proteins by their relevance to the query.
Conclusions
The reference proteins detected by our server will lend insight into proteins of unknown function and provide extensive information to develop in-depth understanding of uncharacterized proteins. Seq2Ref is available at: http://prodata.swmed.edu/seq2ref.
Keywords: Web server, Functional interpretation, Sequence homology, Reference protein, PubMed literature
Background
Due to the avalanche of protein sequences made available by high-throughput genome sequencing, complete manual annotation is unfeasible, leaving a large fraction of protein functions to be predicted by automatic functional annotation pipelines [1]. However, without experimental characterization, the quality of annotation is often questionable, owing to errors in automatic annotation transfer and lack of updates from new findings. In spite of recent advances in highly integrative functional prediction methods [2], a recent investigation [3] into the annotation quality of well-characterized enzyme families revealed that the average percentage of misannotation for the haloacid dehalogenase (HAD) superfamily in the three largest public databases, i.e. non-redundant (nr) [4], TrEMBL [5] and KEGG [6], is over 60%. The possible causes of such annotation errors include multi-domain problems [7], experimental data misinterpretations, threshold relativity problems, and paralog-ortholog misclassifications [8-12]. Moreover, the simplified descriptions recorded in protein sequence and protein family databases are usually inadequate for understanding the precise function of a protein [1].
Such errors and omissions make database annotations insufficient for complete functional interpretation of a protein. A more accurate source of annotations is the ‘reference proteins’ closely related to the protein of interest. We define ‘reference, proteins’ as proteins that have been experimentally studied, manually curated, and reported in the literature. Information about reference proteins is essential for accurate functional interpretation and experimental design. The cross-links between proteins, genes, and associated literature available from National Center for Biotechnology Information (NCBI) provide a basis for reference protein identification. However, it is not trivial to identify a good set of reference proteins and supporting literature because such reference proteins constitute only a small portion of protein databases. Additionally, many proteins linked to large-scale studies (such as genome sequencing) do not provide sufficient functional information.
We have developed a web server named Seq2Ref to assist the identification of applicable reference proteins. Seq2Ref employs BLAST [13] to perform homology searches and exploits crosslinks created by NCBI between proteins and literature to detect reference proteins. Homologs from the Protein Data Bank (PDB) [14] and Swiss-Prot (SP) [15] databases are detected as well, as these databases contain experimental data on 3D protein structures and manually curated annotation on sequence records, respectively. Moreover, we developed a plurality-based rating system integrating reciprocal BLAST and Multiple Sequence Comparisons (MSC) to rank the reference proteins. By retrieving homologous reference proteins, Seq2Ref can contribute to precisely inferring unknown protein function and developing detailed functional interpretation.
Results and discussion
Server interface
The input and output interfaces are shown in Figure 1. An email address and the query protein are the minimal requirements to initiate a job. Options for BLAST search parameters and selection of server modes (fast/slow) are available in the PARAMETERS panel. We recommend manually selecting the organism of the input sequence for reciprocal BLAST if the input sequence is not in the nr database. The total run time is usually 5 to 15 min for fast mode and 1 hour or more for slow mode. When the job completes, an email notification will be sent to the address provided by the user.
Figure 1.
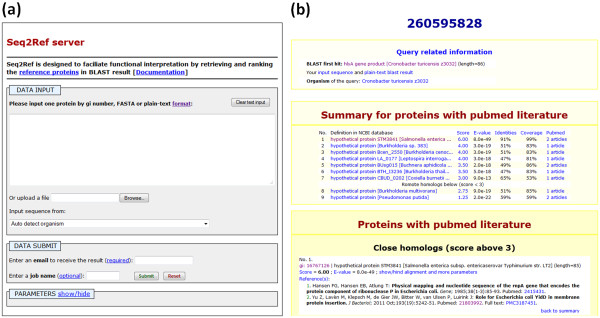
The Seq2Ref webpage interfaces (a) A screenshot of the Seq2Ref submission interface shows the regions for the query input. The query can be submitted by typing in the protein sequence or uploading a file containing the sequence in three formats (gene index (gi) number, FASTA or plain-text). An email address is required for receipt of results. (b) A screenshot shows the top region of the output interface. Query related information (top) is reported. Each section of the results contains a summary table and a detailed panel displaying reference proteins and their relevant information. Full result is in the link: http://prodata.swmed.edu/wenlin/server/user_data/seq2ref/S2RGsaCMG/result.html.
The results page (shown in Figure 1) lists the reference proteins and relevant information in a ranked order. Reference proteins from three sources are shown, respectively, as: (1) a summary table containing protein definition, rating score and BLAST statistics (expectation value, sequence identity and coverage); (2) and a detailed description panel with the rating records, BLAST statistics and scores, and relevant database information. Reference proteins are ranked first by the rating score and second by the expectation value; the publications associated with each protein are sorted by the publication date. As functional studies of remote homologs may not be applicable to the query protein, by default we do not display reference proteins with rating scores lower than 3 in the detailed description panel.
Benchmark
To assess the performance of the Seq2Ref server, especially the ability of our plurality-based rating system to sort out the most relevant references, we applied our algorithm to the enolase superfamily, which has been thoroughly characterized and recorded in the Structure-Function Linkage Database (SFLD) [16-18]. The enolase superfamily contains seven subgroups, which are further divided into 20 families. Proteins within one family share the same substrate specificity and can be considered orthologs; proteins within one subgroup share the same general base(s) in the active site and have the similar catalytic mechanism [19]. For each family, we selected one representative sequence, usually the one with an available 3D structure (Additional file 1: Table S1), as the input for benchmark.
At each rating score cutoff, coverage and average accuracy were used as parameters to evaluate the performance of Seq2Ref (Table 1). The coverage is defined as the percentage of tested sequences that detect reference proteins above the score cutoff. The accuracy is defined as the average of the true positive rates among tested sequences above the score cutoff. Two criteria were used to define true positives: (1) in a stringent (family) context, a true positive hit must be from the same family as the query; and (2) in a broader (subgroup) context, hits from the same subgroup but from different families are also considered true. As shown in Table 1, the accuracy is always 100% with score cutoffs no less than 4; when the cutoff drops to 3, Seq2Ref reaches 100% coverage but starts to include those hits from the same subgroup but different families. Although the biased dataset from only one family might cause overfitting of the statistics, the benchmark suggests that accurate functional interpretation at family level should be achieved by utilizing the reference proteins with a score no less than 4. The information for marginal hits with scores between 3 and 4 is valuable to understand the broad function of the protein subgroup. However, one should not directly transfer the specific functions of marginal hits, such as substrate specificity, to the query.
Table 1.
The accuracy and coverage of the rating system
| Score | Subgroup accuracy1 | Family accuracy1 | Coverage2 |
|---|---|---|---|
| 6 |
100% |
100% |
80% |
| >=5 |
100% |
100% |
85% |
| >=4 |
100% |
100% |
95% |
| >=3 | 100% | 78.5% | 100% |
1: accuracy calculated by averaging the family/subgroup true positive rates.
2: coverage calculated by taking the ratio of testing sequences that detect reference proteins above the score cutoff.
Case study and examples
Due to its ability to retrieve reference proteins and their relevant information in a ranked order, the Seq2Ref server is useful for finding PubMed references relevant to proteins of unknown function, as well as obtaining a deeper understanding of proteins than that revealed by short annotations, as illustrated by the following examples.
Organizing new information for proteins of unknown function
Hypothetical proteins of unknown function constitute a remarkably large portion of the database [20]. Novel studies on uncharacterized proteins and their orthologs provide new insights about their functions, but sequence databases often do not incorporate this information in a timely manner. By retrieving literature, Seq2Ref helps to obtain the most recent information about proteins.
The Macaca mulatta protein, corresponding to gi|355567738, is annotated as a hypothetical protein EGK_07670 in the NCBI Protein database (Seq2Ref results: http://prodata.swmed.edu/wenlin/server/user_data/seq2ref/S2Rnv4cun/result.html). This hypothetical protein contains three conserved domains of unknown function (two DUF3730 and one DUF3028). Our server detects one close homolog, a hypothetical protein (gi|23345097) in human, which has been experimentally studied. The highly confident statistics in BLAST (e-value around 0; 98% identity, 100% coverage) and similar protein domain composition support an orthologous relationship between these two proteins. The human protein was recently (in 2012) reported to be a tumor suppressor in gliomas. It was named ‘focadhesin’, due to its cellular localization at the focal adhesion of the cell membrane [21]. As a likely ortholog, the M. mulatta protein might also be a tumor suppressor and localized at the focal adhesion. Thus, by finding a homolog with the latest experimental publication not yet incorporated in sequence databases, Seq2Ref can serve as a basis for reliable functional prediction of unknown proteins.
Providing detailed information about a protein’s function
Although conserved domains in proteins usually suggest their functions, overly broad descriptions of domain functions are less informative than more specific descriptions. By presenting reference proteins and associated literature, Seq2Ref can offer more definitive and reliable information about protein functions.
One example is the hlyA gene product in Cronobacter turicensis z3032 (gi|260595828, Seq2Ref results: http://prodata.swmed.edu/wenlin/server/user_data/seq2ref/S2RGsaCMG/result.html). A search of the Conserved Domain Database (CDD) merely suggests that this protein contains a ‘haemolytic domain’, with the most similar hit (lowest expectation value) annotated as a ‘hypothetical protein’ and one possible informative hit as ‘conserved hypothetical protein YidD’. The ‘conserved hypothetical protein YidD’ domain (TIGR00278) shows neither functional studies nor a detailed functional description. The publication [22] associated with a Pfam domain record (pfam01809) in the CDD search result suggests that the name ‘haemolytic domain’ originated because one protein (ytjA from Bacillus subtilis) containing this domain can cause cells to lyse in culture. Unfortunately, this study failed to suggest a specific molecular function. Seq2Ref provided more information by detecting (e-value 8.0e-49; 90% identity; reciprocal best hit) the experimentally studied protein YidD from E. coli (gi|67476547), which is identical to the NCBI nr database representative protein (gi|16767126) from Salmonella enterica. This orthology is reinforced by the common conserved genomic context [23] (Additional file 2: Table S2) and the CLANS [24] protein similarity network, in which E. coli YidD and C. turicensis hlyA cluster tightly together among their homologs (Additional file 3: Figure S1). The reference [23] associated with E. coli YidD detected by our server suggests that YidD assists YidC, the protein insertase, in insertion of inner membrane proteins. As a confident ortholog of E. coli YidD, the hlyA gene from C. turicensis very likely shares the same function. Thus, the homologous reference protein, detected by Seq2Ref, contributes to understanding the protein function more specifically.
Limitations
As shown in the examples, the Seq2Ref server detects reference proteins, which can facilitate deeper understanding of the protein function. However, we should keep in mind the limitations. The main concern regards the quality of cross-links between the NCBI Protein and PubMed databases. Missing or wrong links defined by NCBI would result in the loss of or the inappropriate assignment of relevant literature. Another concern is that although the top ranked reference proteins are very likely functionally similar to the query proteins, one should still be careful in directly transferring the information from the hit to the query, as verification of orthology requires additional diligent analysis. To come to the best conclusions about a protein’s function, one should critically inspect the relevance of the publications and the homology of the reference proteins to the query.
Conclusions
Seq2Ref is a homology-based tool to identify reference proteins from PubMed, PDB and SP databases. We have developed a plurality-based rating system that evaluates homologous relationships to indicate the degree of confidence one should have in transferring annotations from a well-studied reference protein to a similar new protein. Thus, by retrieving both experimental studies and high-quality functional annotations of reference proteins, our server provides a solid basis for correct function interpretation of novel proteins.
Methods
Detection of homologs and identification of reference proteins
The workflow of the Seq2Ref server is available in Additional file 4: Figure S2. Seq2Ref performs the BLAST search against the NCBI nr database to detect homologs of the query protein. Based on BLAST search results, reference proteins are identified as: (1) the hits linked to PubMed literature by NCBI (those publications associated with more than 100 protein records are excluded); (2) the hits from PDB; (3) and the hits from SP. Reference proteins from PDB and SP databases are obtained by parsing the protein descriptions recorded in nr. Retrieval from PubMed requires fast but thorough searching of cross-links between NCBI databases. To implement this search, Seq2Ref has two modes: a “fast mode” based on searching a pre-processed local database (updated every 6 months) that consists of the reference proteins in nr, and a “slow mode” in which the most updated reference proteins are retrieved in real-time via NCBI Entrez [25].
Analysis of homologous relationship
We assign orthology firstly by the approximate method of reciprocal best hits [26]. In this method, it is necessary to know the the source-organism of the query protein. To automatically detect the species, Seq2Ref identifies the taxon of the first BLAST hit with at least 97% identity and 90% coverage. Alternatively, the user can manually specify the organism of the input sequence. To avoid possible false negatives caused by variants of the same gene in reciprocal BLAST, such as alleles containing a single nucleotide polymorphism, we pre-cluster the proteins from each genome using CD-HIT [27] (identity cutoff: 97%; coverage cutoff: 90%).
Reference proteins are further analyzed by the method of multiple sequence comparison (MSC) shown in Figure 2. Specifically, we retrieve the sequences most closely related to the query, and then compare the reference proteins to those closely related sequences. Such multiple comparisons allow us to obtain more robust statistics in evaluating homology compared to simple pairwise comparison.
Figure 2.
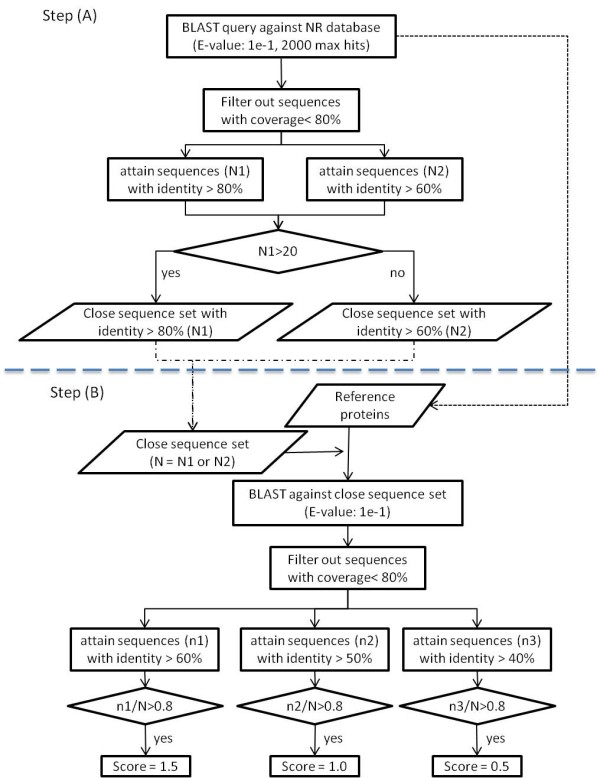
Flow chart of multiple sequence comparison (MSC) method. In step (A), MSC uses BLAST with filters as indicated to define a group of closely related sequences to the query (close sequence set, N); in Step (B), the Reference protein set consists of protein sequences identified without filters by BLAST in step A that are 1) from the Swiss-Prot database, 2) from the Protein Data Bank (PDB), and 3) linked to PubMed articles in the NCBI. Reference protein sequences are then used as queries against a database consisting of the close sequence set. Hits are filtered as indicated. If the filtered hit ratio is larger than 0.8, then a score is assigned to the reference protein.
Rank reference proteins by relevance to the query
We developed a plurality-based rating system with scores ranging from 1 to 6, with 6 indicating the most relevant hits (shown as Table 2). To note, our rating system aims to provide intuitive indicators for the level of similarity, but not act as a statistical predictor of functionality. Four features are considered: reciprocal BLAST, MSC, pairwise comparison between the query and the hit, and whether the hit protein is a “reference protein”, e.g. if there are PubMed citations linked to the protein in the current version of NCBI databases. The maximal rating score for each aspect is 2, 1.5, 1.5 and 1, respectively. A higher total rating score indicates the query protein is closer to the hit and is more likely to function similarly. Proteins with scores lower than 3 would be considered more distant homologs whose functions may have diverged, because they are neither reciprocal BLAST best hits nor with confident statistics in MSC and pairwise comparison.
Table 2.
The rating system
|
Feature |
Criterion |
Points |
||
|---|---|---|---|---|
| True | False | NA | ||
| Reciprocal BLAST |
Query-to-hit-genome1 best hit |
+1 |
0 |
+0.253 |
| |
Hit-to-query-genome2 best hit |
+1 |
0 |
+0.253 |
| Multiple Sequence Comparison (MSC) Method4 |
Accept with identity cutoff 60% |
+0.5 |
0 |
\ |
| Accept with identity cutoff 50% |
+0.5 |
0 |
\ |
|
| Accept with identity cutoff 40% |
+0.5 |
0 |
\ |
|
| Pairwise comparison to the query4 |
Identity>60% |
+0.5 |
0 |
\ |
| Identity>50% |
+0.5 |
0 |
\ |
|
| Identity>40% |
+0.5 |
0 |
\ |
|
| others | Reference proteins | +1 | 0 | \ |
1: use the input sequence to BLAST against the genome of the hit.
2: use the hit to BLAST against the genome of the input sequence.
3: the whole genome sequences of the protein are unavailable.
4: coverage in both the input sequence and the hits must larger than 80%.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WL carried out most of the work and drafted the manuscript. QC and LNK participated in the server design and helped draft the manuscript. NVG conceived the study, participated in its design, and helped draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
The benchmark result links of enolase superfamily. The table shows the selected representative proteins for families in enolase superfamily and the webpage links of the server results.
Pairwise BLAST results for proteins located in the operon containing YidD. Five proteins from Escherichia coli are compared with corresponding proteins in Cronobacter turicensis.
The protein similarity network of YidD homologs produced by CLANS program. Each black dot represents one protein sequence. Red circle and green asterisk represent the query (Cronobacter turicensis hlyA) protein and the experimental studied hit (E. coli YidD), respectively. Edges (lines) show BLAST connections between sequences that have an E-value at least as good as 10−33. Lengths of edges indicate that sequences in tightly clustered groups are relatively more similar to each other than sequences with few and distant connections.
The workflow of Seq2Ref. The whole process can be divided into homologous reference protein detection (Step 1) and homology evaluation (Step 2). Starting from a query sequence, a BLAST/PSI-BLAST search of the NCBI non-redundant database (NR) is performed (Step 1–1) to detect homologous proteins. Seq2Ref detects the reference protein among these homologous proteins by retrieving and checking the information in NCBI databases (Step 1–2). Orange lines represent the reference proteins among the BLAST result. Sequentially, Reciprocal BLAST (RB) and multiple sequence comparison (MSC) will be performed to evaluate the homologous relationships (Step 2–1). Integrating the statistics calculated above, a rating system will assign scores and rank the reference proteins (Step 2–2).
Contributor Information
Wenlin Li, Email: wenlin.li@utsouthwestern.edu.
Qian Cong, Email: qian.cong@utsouthwestern.edu.
Lisa N Kinch, Email: lkinch@chop.swmed.edu.
Nick V Grishin, Email: grishin@chop.swmed.edu.
Acknowledgement
The authors thank Dustin Schaeffer and Jeremy Semeiks for critical reading of the manuscript, and Qing Lan for helping edit the documentation of the server. This work was supported by National Institutes of Health (GM094575 to NVG) and the Welch Foundation (I-1505 to NVG).
References
- Valencia A. Automatic annotation of protein function. Curr Opin Struct Biol. 2005;15(3):267–274. doi: 10.1016/j.sbi.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Rentzsch R, Orengo CA. Protein function prediction–the power of multiplicity. Trends Biotechnol. 2009;27(4):210–219. doi: 10.1016/j.tibtech.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5(12):e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2010;38(Database issue):D46–51. doi: 10.1093/nar/gkp1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(Database issue):D142–148. doi: 10.1093/nar/gkp846. 2808944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BH, Cong Q, Grishin NV. HangOut: generating clean PSI-BLAST profiles for domains with long insertions. Bioinformatics. 2010;26(12):1564–1565. doi: 10.1093/bioinformatics/btq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol. 1998;1(1):55–67. [PubMed] [Google Scholar]
- Sasson O, Kaplan N, Linial M. Functional annotation prediction: all for one and one for all. Protein science: a publication of the Protein Society. 2006;15(6):1557–1562. doi: 10.1110/ps.062185706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Bairoch A. Go hunting in sequence databases but watch out for the traps. Trends in genetics: TIG. 1996;12(10):425–427. doi: 10.1016/0168-9525(96)60040-7. [DOI] [PubMed] [Google Scholar]
- Doerks T, Bairoch A, Bork P. Protein annotation: detective work for function prediction. Trends in genetics: TIG. 1998;14(6):248–250. doi: 10.1016/S0168-9525(98)01486-3. [DOI] [PubMed] [Google Scholar]
- Smith TF, Zhang X. The challenges of genome sequence annotation or “the devil is in the details”. Nat Biotechnol. 1997;15(12):1222–1223. doi: 10.1038/nbt1197-1222. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Boeckmann B, Ferro S, Gasteiger E. Swiss-Prot: juggling between evolution and stability. Brief Bioinform. 2004;5(1):39–55. doi: 10.1093/bib/5.1.39. [DOI] [PubMed] [Google Scholar]
- Brown SD, Gerlt JA, Seffernick JL, Babbitt PC. A gold standard set of mechanistically diverse enzyme superfamilies. Genome Biol. 2006;7(1):R8. doi: 10.1186/gb-2006-7-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg SC, Brown S, Ojha S, Huang CC, Ferrin TE, Babbitt PC. Representing structure-function relationships in mechanistically diverse enzyme superfamilies. 2005. pp. 358–369. (Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing). [PubMed]
- Pegg SC, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry. 2006;45(8):2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, Reed GH, Rayment I, Ringe D, Kenyon GL, Gerlt JA. The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylic acids. Biochemistry. 1996;35(51):16489–16501. doi: 10.1021/bi9616413. [DOI] [PubMed] [Google Scholar]
- Jaroszewski L, Li Z, Krishna SS, Bakolitsa C, Wooley J, Deacon AM, Wilson IA, Godzik A. Exploration of uncharted regions of the protein universe. PLoS Biol. 2009;7(9):e1000205. doi: 10.1371/journal.pbio.1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockschmidt A, Trost D, Peterziel H, Zimmermann K, Ehrler M, Grassmann H, Pfenning PN, Waha A, Wohlleber D, Brockschmidt FF. et al. KIAA1797/FOCAD encodes a novel focal adhesion protein with tumour suppressor function in gliomas. Brain: a journal of neurology. 2012;135(Pt 4):1027–1041. doi: 10.1093/brain/aws045. [DOI] [PubMed] [Google Scholar]
- Liu J, Fang C, Jiang Y, Yan R. Characterization of a hemolysin gene ytjA from Bacillus subtilis. Curr Microbiol. 2009;58(6):642–647. doi: 10.1007/s00284-009-9383-1. [DOI] [PubMed] [Google Scholar]
- Yu Z, Laven M, Klepsch M, de Gier JW, Bitter W, van Ulsen P, Luirink J. Role for escherichia coli YidD in membrane protein insertion. J Bacteriol. 2011;193(19):5242–5251. doi: 10.1128/JB.05429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20(18):3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2012;40(Database issue):D13–25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24(3):319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The benchmark result links of enolase superfamily. The table shows the selected representative proteins for families in enolase superfamily and the webpage links of the server results.
Pairwise BLAST results for proteins located in the operon containing YidD. Five proteins from Escherichia coli are compared with corresponding proteins in Cronobacter turicensis.
The protein similarity network of YidD homologs produced by CLANS program. Each black dot represents one protein sequence. Red circle and green asterisk represent the query (Cronobacter turicensis hlyA) protein and the experimental studied hit (E. coli YidD), respectively. Edges (lines) show BLAST connections between sequences that have an E-value at least as good as 10−33. Lengths of edges indicate that sequences in tightly clustered groups are relatively more similar to each other than sequences with few and distant connections.
The workflow of Seq2Ref. The whole process can be divided into homologous reference protein detection (Step 1) and homology evaluation (Step 2). Starting from a query sequence, a BLAST/PSI-BLAST search of the NCBI non-redundant database (NR) is performed (Step 1–1) to detect homologous proteins. Seq2Ref detects the reference protein among these homologous proteins by retrieving and checking the information in NCBI databases (Step 1–2). Orange lines represent the reference proteins among the BLAST result. Sequentially, Reciprocal BLAST (RB) and multiple sequence comparison (MSC) will be performed to evaluate the homologous relationships (Step 2–1). Integrating the statistics calculated above, a rating system will assign scores and rank the reference proteins (Step 2–2).