

Abstract
Background
Over 90% of adults aged 20 years or older with permanent teeth have suffered from dental caries leading to pain, infection, or even tooth loss. Although caries prevalence has decreased over the past decade, there are still about 23% of dentate adults who have untreated carious lesions in the US. Dental caries is a complex disorder affected by both individual susceptibility and environmental factors. Approximately 35-55% of caries phenotypic variation in the permanent dentition is attributable to genes, though few specific caries genes have been identified. Therefore, we conducted the first genome-wide association study (GWAS) to identify genes affecting susceptibility to caries in adults.
Methods
Five independent cohorts were included in this study, totaling more than 7000 participants. For each participant, dental caries was assessed and genetic markers (single nucleotide polymorphisms, SNPs) were genotyped or imputed across the entire genome. Due to the heterogeneity among the five cohorts regarding age, genotyping platform, quality of dental caries assessment, and study design, we first conducted genome-wide association (GWA) analyses on each of the five independent cohorts separately. We then performed three meta-analyses to combine results for: (i) the comparatively younger, Appalachian cohorts (N = 1483) with well-assessed caries phenotype, (ii) the comparatively older, non-Appalachian cohorts (N = 5960) with inferior caries phenotypes, and (iii) all five cohorts (N = 7443). Top ranking genetic loci within and across meta-analyses were scrutinized for biologically plausible roles on caries.
Results
Different sets of genes were nominated across the three meta-analyses, especially between the younger and older age cohorts. In general, we identified several suggestive loci (P-value ≤ 10E-05) within or near genes with plausible biological roles for dental caries, including RPS6KA2 and PTK2B, involved in p38-depenedent MAPK signaling, and RHOU and FZD1, involved in the Wnt signaling cascade. Both of these pathways have been implicated in dental caries. ADMTS3 and ISL1 are involved in tooth development, and TLR2 is involved in immune response to oral pathogens.
Conclusions
As the first GWAS for dental caries in adults, this study nominated several novel caries genes for future study, which may lead to better understanding of cariogenesis, and ultimately, to improved disease predictions, prevention, and/or treatment.
Keywords: Dental caries, Genetics, Genome wide association, Permanent dentition, Genomics
Background
Dental caries is a common chronic disease that causes pain and disability across all age groups [1]. Untreated caries can lead to pain spread of infection to adjacent tissue, tooth loss, and edentulism (total tooth loss). Caries prevalence increases with age, and by the third decade of life, approximately 91% of dentate adults have experienced dental caries in the US. Although overall caries experience has decreased by about 3.3% over the last decade, this trend is most apparent in younger adults (aged 20–39 years) with higher educational attainment (NHANES surveillance summaries on oral health, 2005). Nevertheless, about 23% of adults have untreated tooth decay, nationwide.
The etiology of dental caries involves a complex interplay of environmental and genetic factors. Heritability analyses have revealed the notable role of genes on caries disease [2-4]. We previously conducted a heritability analysis on dental caries based on 2,600 participants from 740 multi-generational families [5]. For caries in the permanent dentition, we estimated approximately 35-55% of phenotypic variation in disease experience was attributable to genetic factors. Importantly, we also showed that genes affecting susceptibility to caries in the primary dentition partly differ from those in permanent teeth.
Previous studies of the genetics of dental caries have focused mostly on candidate genes. Genes affecting taste preferences (such as taste receptor gene TAS2R38) may affect dietary habits, a major known caries risk factor [6]. Other examples are amelogenin (AMELX) [7,8] and tuftelin (TUFT1) [9], enamel matrix proteins, and CD14 , an innate immune response gene involved in bacterial pattern-recognition during cariogenesis [10]. In the only genome-wide association study (GWAS) conducted to date on caries [11], a few loci (ACTN2, MTR, and EDARADD, MPPED2, and LPO) with possible biological roles in susceptibility to caries, although not genome-wide significant, demonstrated suggestive evidence for association with caries phenotypes.
Despite these efforts, few specific genes for dental caries in the permanent dentition have been identified or replicated. Therefore, our goal was to perform genome-wide association scans (GWAS) to identify genetic variants associated with dental caries in permanent dentition in adults. Identification of caries genes will contribute to our understanding of caries etiology, and may lead to preventative interventions and/or treatment strategies for dental caries.
Methods
Sample recruitment and data collection
As shown in Table 1, five independent samples were included in this study. 1) The first sample (N = 970) was ascertained through the Center for Oral Health Research in Appalachia (COHRA), an initiative to study the causes of oral health disparities in rural Appalachia. In brief, the sample was drawn from largely rural Appalachian communities in Pennsylvania and West Virginia according to a household-based recruitment protocol requiring at least one biological child–parent pair in order to participate [12]. 2) The second cohort of participants (N = 223, DRDR1) was ascertained through the University of Pittsburgh, School of Dental Medicine Dental Registry and DNA Repository (DRDR). In this ongoing project, every individual that comes to the dental school for treatment is invited to be part of the registry [13]. These samples together with the COHRA sample were included as part of GENEVA dental caries project [14]. 3) The third cohort comprises an additional 290 participants subsequently accepted into the DRDR (DRDR2), with similar demographic characteristics as DRDR1. 4) The fourth cohort (N = 4230) was from the Atherosclerosis Risk in Communities (ARIC) Study, which was designed to investigate the etiology and natural history of atherosclerosis [15]. The Dental ARIC, an ancillary project supported by the National Institute of Dental and Craniofacial Research (NIDCR), was conducted at the fourth visit between 1996 and 1998 [16]. 5) The fifth cohort was from a nested case–control of type 2 diabetes samples within the Health Professionals Follow-up Study [17,18] (HPFS; N = 1730), a prospective on-going project targeting male health professionals aged between 40 and 75 years in the US. Parti cipants particularly involved in our project were recruited in the middle or late 1990s for both ARIC and HPFS, whereas for COHRA and the two DRDR cohorts, samples were brought in on or after 2005. Recruiting for all five sample cohorts was not based on participants’ dental caries status. Written informed consent was obtained from all participants at each individual project. All study procedures were reviewed and approved by the Institutional Review Boards at universities at each site (Federal Wide Assurance (FWA) # for GENEVA dental caries project: FWA00006790; ARIC project: FWA00004801 and HPFS-T2D: FWA00000484).
Table 1.
Description of the five cohorts
| Cohort Description* | COHRA | DRDR1 | DRDR2 | ARIC | HPFS |
|---|---|---|---|---|---|
|
PI |
Marazita |
Vieira & Marazita |
Vieira |
Boerwinkle |
Hu |
|
Sample Size† |
970 |
223 |
290 |
4230 |
1730 (male) |
|
Age§ |
34.3 ± 9.4 |
41.9 ± 16.9 |
41.8 ± 17.9 |
63.1 ± 5.6 |
65.2 ± 8.4 |
|
Age Range |
17-67 |
17-84 |
17-89 |
53-75 |
49-83 |
|
Caries Prevalence (%) |
95.3 |
94.5 |
97.7 |
99.5 |
98.7 |
|
Caries Phenotype¶ |
DMFS |
DMFS |
DMFS |
Proportion DFS |
Caries Severity |
|
Genotyping Platform |
Illumina 610-Quad |
Illumina 610-Quad |
Illumina 610-Quad |
Affymetrix 6.0 |
Affymetrix 6.0 |
|
Genotyping Cente** |
CIDR |
CIDR |
PITT-GPCL |
BICGA |
BICGA |
| Imputed data | Y | Y | N | Y | N |
* COHRA: Center for Oral Health Research in Appalachia cohort.
DRDR1: Dental Registry and DNA Repository cohort phase 1.
DRDR2: Dental Registry and DNA Repository cohort phase 2.
ARIC: Atherosclerosis Risk in Community cohort.
HPFS: Health Professionals Follow-Up Study cohort.
† All summary statistics and subsequent analyses only include genotyped non-Hispanic Whites.
§ Mean ± SD.
¶ DMFS: Decayed, Missing due to Decay, or Filled tooth surfaces.
Proportion DFS: Decayed or Filled tooth surfaces / total surfaces at risk.
Caries severity: total # of caries coded as 0, 1; 2–4; 5–9 and 10 or more.
** CIDR: the Johns-Hopkins Center for Inherited Disease Research;
PITT-GPCL: Genomics and Proteomics Core Laboratories at University of Pittsburgh.
BICGA: Broad Institute Center for Genotyping and Analysis at Harvard and MIT.
Caries Phenotype assessment
For COHRA, dental caries of permanent teeth was assessed by dentists or dental hygienists via visual inspection. Data for DRDR1 and DRDR2 were extracted from evaluations done by dentists. Examiners across all sites were calibrated periodically. Each tooth surface was scored as sound, decayed, filled, missing due to decay, or missing due to reasons other than decay, in accordance with the World Health Organization recommended scale and in accordance with the NIH/NIDCR-approved protocol for assessing dental caries for research purposes [12,19]. This method of caries assessment is compatible with the Phen-X Toolkit ( http://www.phenxtoolkit.org) to facilitate combining data across studies, and the National Center for Health Statistics Dental Examiners Procedures Manual (See Section 4.9.1.3). Third molars were excluded from caries assessment. Edentulous individuals were recruited into the study but were excluded from caries assessment and follow-up analysis. The phenotype, DMFS, used in GWAS analysis represents the count of decayed, missing due to decay, or filled (restored) tooth surfaces across an individual’s permanent dentition.
Caries assessment in the ARIC cohort was similar to the approach indicated above, except that no distinction was made between teeth that were missing due to decay or missing due to another reason. Thus, the DFS (decayed or filled tooth surface) phenotype was available for this dataset. In order to account for the variation of total number of teeth at risk among this older sample of individuals, we created a new phenotype where the proportion of DFS equals to the original DFS counts divided by the total number of tooth surfaces at risk.
In the HPFS cohort, caries was assessed by self-reported questionnaires. Baseline caries measurement collected in 1996 was used in our analysis. In general, data was collected on the total number of cavities in permanent teeth. The response to this question was an ordered categorical variable representing different levels of caries severity (no cavity, 1 affected tooth, 2–4, 5–9, and 10 or more affected teeth).
As reported previously [6,12], both inter- and intra-examiner concordances of caries assessments were high in the COHRA cohort. However this calibration process was not available for other cohorts, either because such design was not part of the original study (DRDR1 and DRDR2), or the caries phenotype collection was of a side interest (ARIC), or the caries assessment was simply based on self-reported information from questionnaire (HPFS).
Genotyping, quality assurance, and imputation
As part of GENEVA dental caries project, genotyping for COHRA and DRDR1 samples was carried out on behalf of the GENEVA consortium by the Johns-Hopkins Center for Inherited Disease Research (CIDR) through a National Institutes of Health contract. Genotyping of these cohorts was performed using the Illumina Human610-Quadv1_B BeadChip (Illumina, San Diego, CA, USA). Additional details are available at the National Center for Biotechnology Information database of Genotype and Phenotypes (dbGaP, http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap, study accession designation phs000095.v1.p1). The DRDR2 cohort was genotyped at the University of Pittsburgh Genomics and Proteomics Core Laboratory using the same Illumina Human610-Quad chip. Genotyping for both ARIC and HPFS cohorts was performed at the Broad Institute of MIT and Harvard’s Center for Genotyping and Analysis using the Affymetrix 6.0 SNP array (Affymetrix, Santa Clara, CA, USA) and the Birdseed calling algorithm. Additional details are available at dbGaP (study accession designations phs000090.v1.p1 for ARIC and phs000091.v2.p1 for HPFS)
Genotype data for all cohorts except DRDR2 went through an extensive process of cleaning, imputation, and quality assurance, performed by the GENEVA consortium Coordinating Center at the University of Washington [14,20,21]. The entire cleaning procedure included but was not limited to, checks for gender identity, chromosomal anomalies, sample relatedness, population structure, missing call rates, plate effects, Mendelian errors, duplicate discordance, etc. Detailed cleaning reports are publicly available for each study at the above referenced dbGaP resource. The data cleaning and quality control for DRDR2 genotypes were conducted by our own team using similar procedures as above.
Genotype imputation (i.e., inferring unobserved genotypes based on observed ones from a reference sample with similar genetic background) was performed by the GENEVA coordinating center for three cohorts (COHRA, DRDR1 and ARIC). Imputed data were released for all successfully imputed SNPs (approximately 1.4 million) using subjects from a HapMap Phase III reference panel (genetically-determined European ancestry, CEU sample) and BEAGLE software [22]. Quality metrics were provided for each imputed SNP that were further used in analysis for filtering imputation results on a per-SNP level. Imputed genotypes are provided as the probability of each of the three genotype states, reflecting the level of certainty in the genotype prediction. These probabilities were directly incorporated into downstream statistical analyses within PLINK, rather than taking the most likely imputed genotype. For detailed description of this imputation procedure and follow-up quality control, please refer to the report available on dbGaP.
Statistical analysis
Genome-wide association scans were limited to self-reported non-Hispanic Whites, which comprised the majority of samples in our study. This was to minimize the risk of inflated type I error caused by population stratification and to avoid reduction in power due to possible genetic heterogeneity. Before analysis, principal component analysis (PCA) based on independent autosomal SNPs was applied to verify the self-reported race variable against the DNA evidence. Hapmap controls (CEU, YRI, CHB, JPT) were used as reference. High concordance between self-reported race and genetically-determined ancestry was observed across all cohorts. The very rare outliers were excluded in further analysis. For the COHRA sample, which included participants of all ages, statistical analysis was limited to permanent teeth in individuals 17 years or older. All participants in the other cohorts were adults, and therefore were included in analysis.
All GWAS scans were performed in PLINK ( http://pngu.mgh.harvard.edu/~purcell/plink) [23] using linear regression (−−linear option) while adjusting for age and sex as covariates. The above analyses were performed separately in each cohort with genotyped data and imputed data if available (COHRA, DRDR1 and HPFS). Before analysis, HWE (P-value ≤ 10E-4) and minor allele frequency (MAF ≤ 0.02) filters were applied to exclude outlier or rare SNPs. Next, we combined the GWAS association results from each study by performing meta-analysis in METAL ( http://www.sph.umich.edu/csg/abecasis/Metal/) [24] using its weighted Z-score method based on sample size, P-value and direction of effect in each study (fixed effect model). Due to the differences in age, birth cohort, demography, genotyping platform, and quality of dental caries assessment, as well as possible genetic heterogeneity among our cohorts, we performed three meta-analyses: 1) Meta 1 (COHRA, DRDR1, and DRDR2): we combined these three cohorts because they were each comprised of comparatively younger individuals from Appalachia. In addition, they were genotyped on the same Illumina chip, and have the most informative caries DMFS phenotype; 2) Meta 2 (ARIC and HPFS): we combined these two cohorts because they were both genotyped using Affymerix 6.0 chip and they both included comparatively older participants (all samples ≥49 years) with poorer quality dental caries assessments; 3) Meta 3 (all five cohorts combined).
We explored all signals with “suggestive significance” (P-value ≤ 10E-5) using several online bioinformatics tools and databases, such as SCAN ( http://www.scandb.org/) [25], and WGAViewer ( http://compute1.lsrc.duke.edu/softwares/WGAViewer/) [26]. This step was crucial and based on the assumption that associated SNPs, which may not themselves be causal, were in LD with the causal variant nearby. Moreover, it is currently unknown where a causal variant may be located with respect to the gene it affects, although cis-acting (i.e., physically proximal) variants are widely believed to be important. Therefore, for every SNP meeting suggestive significance, we explored whether any nearby genes had known biological functions relevant to cariogenesis. The calculation of genomic inflation factor, lambda, and the generation of Quantile-Quantile plots were conducted in the R statistical package (R Foundation for Statistical Computing, Vienna, AU). Manhattan plots were created using Haploview [27]. Regional visualization of GWAS top signals were produced using LocusZoom ( http://csg.sph.umich.edu/locuszoom/) [28]. We also generated genotype intensity plots (i.e. cluster plots) for genotyped SNPs within top signals to verify high-quality genotype calling. Because over 95% of our samples were unrelated individuals, we did not adjust analysis for family relatedness, but closely monitored evidence of genomic inflation.
Results
Table 1 shows descriptive characteristics of the five cohorts used in our study. ARIC and HPFS were the two largest cohorts containing comparatively older participants aged 49 years or greater. The mean ages of these cohorts were more than 20 years greater than those from the other three cohorts. The difference of birth year is even larger between two older and three younger cohorts because subjects in ARIC and HPFS were ascertained almost 10 years earlier. The HPFS cohort included only males. The DRDR1 and DRDR2 cohorts were similar. Caries prevalence was extremely high (94.5-99.5%) for all of our five cohorts, substantially higher than that reported by NHANES in 2005 (86.8-96.3%) for corresponding age groups.
Different methods of caries assessment were performed across the five cohorts (Table 1). Tooth surface-level caries assessment was performed for COHRA, DRDR1 and DRDR2, by intra-oral examination, from which DMFS index was generated. DMFS index is the count of carious surfaces across the dentition, and is the most widely used measure of dental caries experience along with DMFT (index by tooth). Caries measurements in the other two cohorts were different and presumably less complete from above. In ARIC, data on teeth missing due to decay were not collected, and therefore the DMFS index could not be generated. Instead we used the proportion DFS as our caries phenotype, which measures caries experience with respect to the number of tooth surfaces for which we have data (as opposed to the full permanent dentition, as in DMFS). In HPFS, dental caries was assessed as a self-reported categorical variable representing approximate number of carious lesions at tooth level.
Figure 1 shows Manhattan plots for the three meta-analyses. No association signals passed the genome-wide significance threshold (i.e., marginal P-value ≤ 5.0 × 10-8). The genomic inflation factor, λ, was 1.0345, 1.0055 and 1.0125 for three meta-analyses, respectively, indicating negligible P-value inflation. We investigated the genes (and possible biological functions) at or near SNPs with suggestive P-values (i.e., P-value ≤ 10E-5) in each meta-analysis, and compared common genetic signals across meta-analyses.
Figure 1.

GWAS results in three Meta-Analyses: Manhattan and Q-Q plots. All P-values are negative log10 transformed. Each point represents a genotyped or imputed (whenever available) SNP marker.
Top Signals within each meta-analysis (P-values ≤ 10E-7)
Altogether, there were 5 regions identified in our study where at least one SNP achieved this level of significance: three from Meta 1 and one each from Meta 2 and 3 (Table 2). The SNP exhibiting the strongest evidence of association in Meta 1 was rs635808 on chromosome 6 (P-value = 1.06 × 10-7) located in the intronic region of RPS6KA2 (Figure 2A, Additional file 1: Table S1). This gene encodes an enzyme from the RSK (ribosomal S6 kinase) family, which is capable of phosphorylating various substrates, including members of the mitogen-activated kinase (MAPK) signaling pathway. It has been previously reported that the activation of MAPK pathway (through p38 phosphorylation) plays pivotal role in inflammatory cytokine and chemokine gene regulation and thus it is involved in oral-related diseases such as dental caries [29], caries-induced pulpitis [30], chronic oral pain and periodontal disease.
Table 2.
Effect size and P-values for top SNPs in three meta-analyses*
| Gene/SNPs | Chr | Base Pair position | Data Status† |
Effect Size§ |
P-value (Meta 1) | P-value (Meta 2) | P-value (Meta 3) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| COHRA | DRDR1 | DRDR2 | ARIC | HPFS | |||||||
|
RHOU |
|
|
|
|
|
|
|
|
|
|
|
| rs3936161 |
1 |
227336163 |
Illumina |
−1.95 |
7.17 |
-- |
−0.70 |
-- |
0.721 |
1.55E-05 |
6.76E-05 |
| rs12072775 |
1 |
227339176 |
Affymetrix |
−1.94 |
7.17 |
-- |
−0.71 |
0.007 |
0.725 |
4.23E-05 |
0.001 |
| rs9287022 |
1 |
227344972 |
Imputed |
−2.09 |
7.19 |
-- |
−0.61 |
-- |
0.673 |
3.79E-06 |
1.86E-05 |
| rs9793739 |
1 |
227352481 |
Imputed |
−2.08 |
7.88 |
-- |
−0.75 |
-- |
0.721 |
5.27E-07 |
4.28E-06 |
| rs2988738 |
1 |
227427128 |
Affymetrix |
0.67 |
9.55 |
-- |
−1.75 |
−0.15 |
0.567 |
2.02E-05 |
0.002 |
|
ADAMTS3 |
|
|
|
|
|
|
|
|
|
|
|
| rs788919 |
4 |
73572758 |
Illumina |
−1.40 |
−4.14 |
−3.56 |
−0.47 |
-- |
0.026 |
1.36E-04 |
1.02E-05 |
| rs4694123 |
4 |
73606652 |
Illumina |
−1.31 |
−2.94 |
−3.89 |
−0.46 |
-- |
0.038 |
1.18E-04 |
1.26E-05 |
| rs10805050 |
4 |
73612147 |
Illumina |
1.01 |
2.93 |
2.50 |
0.42 |
-- |
0.093 |
4.88E-06 |
1.68E-06 |
| rs788911 |
4 |
73632087 |
Illumina |
0.99 |
2.54 |
3.29 |
0.38 |
-- |
0.084 |
4.77E-06 |
1.46E-06 |
| rs1383934 |
4 |
73636388 |
Illumina |
1.19 |
3.20 |
3.70 |
0.64 |
-- |
0.046 |
1.77E-06 |
2.96E-07 |
|
RPS6K2 |
|
|
|
|
|
|
|
|
|
|
|
| rs505982 |
6 |
167095386 |
Imputed |
3.66 |
8.52 |
-- |
−0.35 |
-- |
8.93E-06 |
0.859 |
0.025 |
| rs635808 |
6 |
167097412 |
Illumina |
−4.21 |
−7.53 |
−8.30 |
0.44 |
-- |
1.06E-07 |
0.898 |
0.010 |
|
PTK2B |
|
|
|
|
|
|
|
|
|
|
|
| rs17057381 |
8 |
27416801 |
Affymetrix |
16.39 |
28.98 |
-- |
0.47 |
−0.03 |
4.02E-07 |
0.267 |
0.764 |
|
CNIH |
|
|
|
|
|
|
|
|
|
|
|
| rs1953743 |
14 |
53722229 |
Both |
−3.08 |
−6.55 |
−6.87 |
0.01 |
0.04 |
1.98E-06 |
0.371 |
0.027 |
| rs4251631 |
14 |
53945934 |
Illumina |
−3.78 |
−6.32 |
−10.02 |
−0.35 |
-- |
2.13E-07 |
0.013 |
1.80E-06 |
| rs11850320 |
14 |
53990173 |
Illumina |
−4.57 |
−7.93 |
−6.78 |
−0.33 |
-- |
9.92E-07 |
0.177 |
0.0003 |
| rs7150062 |
14 |
53997400 |
Both |
4.52 |
7.93 |
6.78 |
0.40 |
0.01 |
1.15E-06 |
0.295 |
0.001 |
| rs7143579 | 14 | 54010435 | Illumina | −4.42 | −7.15 | −7.89 | −0.42 | -- | 1.16E-06 | 0.137 | 0.0002 |
* Summarizes genes/regions containing at least one SNP with significant P-values ≤ 10E-7 (bolded); Listed are the first five most significant SNPs if more than five SNPs observed at the corresponding region;
† Illumina/Affymetrix/Both: SNP was genotyped in Illumina 610Quad/Affymetrix 6.0/both chips respectively;
Imputed: SNP data was generated by imputation only. “--" indicating the corresponding SNP was not genotyped in DRDR2 or
HPFS;
§ Effect size can be directly compared ONLY among Meta 1 cohorts (COHRA, DRDR1 and DRDR2).
Figure 2.
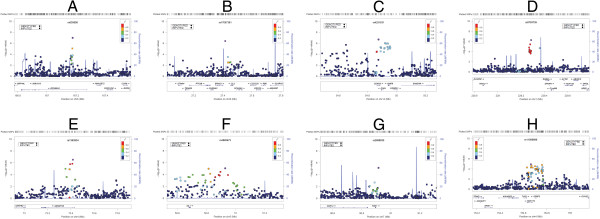
Regional plots of P-values at top loci in meta-analyses. Negative log10 transformed P-values and physical positions for SNPs in the region are shown. Colors indicate linkage disequilibrium between the index SNP (colored in purple) and other SNPs based on HapMap CEU data. The rug plot indicates regional SNP density. The recombination rate overlay is based on HapMap CEU data. Gene positions and directions of transcription are annotated based on hg19/1000 Genomes Nov 2010 release.
Another suggestive signal observed in Meta 1 was rs17057381 (P-value = 4.02 × 10-7) on chromosome 8. Within a ±100 kb region, there are five genes including PTK2B. No direct evidence implicates these genes in cariogenesis; however, previous studies have shown that PTK2B mediates the p38-dependent MAPK pathway [31,32] and is important for oral disorders including dental caries. (Figure 2B)
The third suggestive signal observed in Meta 1 was a broad region of association on chromosome 14 (Figure 2C; top SNP was rs4251631, P-value = 2.13 × 10-7). Multiple low LD SNPs (in reference to rs4251631) demonstrated suggestive significance and four of them were among the top SNPs in Meta 3 (P-values between 8.17 × 10-5 and 1.80 × 10-6). The association signal is centered over a region of low recombination harboring 4 genes, CDKN3, CNIH, GMFB and CGRRF1 (none of which have known or biologically plausible roles in dental caries). The association signal extends 500 kb upstream to the 5’ untranslated region of BMP4 gene. Bone morphogenetic proteins are important for regeneration/repair of the dentin-pulp complex after cariogenic injury [33], and BMP4, in particular, has been shown to initiate and regulate repair of carious tissue [34,35].
In Meta 2 we observed a suggestive signal on chromosome 1 (rs9793739, P-value = 5.27 × 10-7). No relevant information with caries was found for genes near this SNP except that about 400 kb upstream of the top hit, was the RHOU gene (the closest hit, Figure 2D), a member of the Rho family of GTPases. Evidence suggests that GTPases act as key mediators of the Wnt signaling cascade [36], a pathway that is well-known for its role in regulating tooth morphology during tooth development [37]. In 2001, Tao et al. showed in mice the possible role of RHOU in the regulation of cell morphology and proliferation through the Wnt1 signaling pathway [38]. Though biologically plausible, it is currently unknown whether RHOU is involved in genetic susceptibility to dental caries.
In Meta 3 we observed a suggestive association with rs1383934 (P-value = 2.96 × 10-7). This SNP is located on chromosome 4 in the intronic region of ADAMTS3 (Figure 2E), which is highly expressed during tooth development in the dental papilla in mice [39]. The role of ADAMTS3 in cariogenesis is unknown; however, given its role in tooth development in mouse, it is plausible that this gene affects susceptibility to dental caries.
Other interesting signals (P-values ≤ 10E-5)
In Meta 1 we also observed suggestive association for a 400 kb region on chromosome 5 including the ISL1 gene (rs4865673, P-value = 8.73 × 10-6, Figure 2F). In mice, this gene is exclusively expressed in epithelial cells of developing incisors, and is a crucial regulator of jaw and tooth development [40], suggesting a possible mechanism through which ISL1 may affect susceptibility to dental caries.
For Meta 2, we also observed suggestive association with the gene FZD1 on chromosome 7 (rs2888830, P-value = 7.01 × 10-6, Figure 2G). As receptor of Wnt family signaling molecules, FZD1 is responsible for activating intracellular signals for Wnt pathways for tooth initiation (eruption) [41].
In Meta 3, we observed suggestive association with the gene TLR2 on chromosome 4 (rs11099896, P-value = 1.24 × 10-5, Figure 2H). TLR2 is involved in the immune response against cariogenesis; the gene-coded receptor is expressed on the cell surface of odontoblasts. During cariogenesis, the receptor recognizes oral bacterial and triggers the immune defense system [42]. In both dentin [43] and dental pulp [44], similar mechanisms were observed.
Cross-Meta-analysis signals
Shared signals were observed across meta-analyses including associations of common SNPs and common regions (i.e., within 100 kb) in two or more meta-cohorts. There were 29 loci that exhibited suggestive association across meta-analyses (See Figure 3 and Additional file 1: Table S3). Besides genes (such as RHOU, ADAMTS3, CDKN3/CNIH/GMFB, FZD, etc.) which had been highlighted in individual meta-analysis, this list also includes ZNF160 on chromosome 19 (rs10405102, P-value = 3.02 × 10-5 in Meta 1; rs9967593 and rs1650966, P-value = 2.23 × 10-5 and 2.22 × 10-5 respectively in Meta 2; rs2288421, P-value = 5.96 × 10-5 in Meta 3), which represses TLR4 [45], another odontoblast cell-surface receptor that recognizes oral pathogens to mediate immune response [46].
Figure 3.

Venn diagram summarizing common Genes (on or near SNPs with P-value ≤ 10E-5) cross meta-analyses.
Discussion
We performed the first GWAS for dental caries in the permanent dentition in adults, which complements earlier scans for childhood caries [11], tooth eruption [47] and the whole genome linage scans for caries using family data [48]. Though we did not observe any genetic associations meeting genome-wide significance, we did nominate several statistically suggestive loci with plausible biological roles in dental caries. Specifically we nominated RPS6KA2 and PTK2B involved in p38-dependent MAPK signaling; RHOU and FZD1 involved in Wnt signaling cascade. Both of these pathways have been implicated in dental caries. ADMTS3 and ISL1 are involved in tooth development; and TLR2 is involved in immune response to oral pathogens.
Our study investigated the genetics of dental caries separately in our younger Appalachian cohorts and comparatively older non-Appalachia cohorts. Comparing the ARIC and HPFS cohorts versus the other three Appalachian ones, the mean age difference is over 20 years and the participants in older cohorts were ascertained about 10 years earlier. In other words, subjects were born 30 years earlier, on average, in ARIC and HPFS. We speculate that this birth cohort effect may serve as a surrogate for unmeasured life history variables that differ between the Appalachian and non-Appalachian cohorts. For instance, water and tooth paste fluoridation was introduced between the 1950s and 1970s in the US. For participants in ARIC and HPFS studies, the majority had little exposure to sources of fluoride in their first 20 to 30 years of life. In comparison, the majority of COHRA, DRDR1, and DRDR2 participants had fluoride exposure throughout their entire lives. Given the protective role of fluoride on dental caries, and the likely involvement of gene-by-fluoride interactions, we speculate that fluoride exposure may account for some of the genetic heterogeneity between Meta 1 and Meta 2. Other unknown factors that differ between cohorts may have a similar effect.
This study benefits from several strengths including a large sample size of 7,200 participants, quality genotyping and imputation data generated by CIDR, Broad CGA and the GENEVA coordinating center, and carefully-designed meta-analyses assessing genetic effects within and across multiple cohorts. However, several limitations warrant further discussion. First, we did not replicate genetic association with any genes implicated in the previous GWAS of childhood dental caries. This is perhaps because the current analysis studied a different dentition type (permanent vs. primary teeth). In addition, we achieved lower performances in larger cohorts. For example, although Meta 2 had four times larger sample size than Meta 1, in Meta 2 we observed fewer suggestive genetic signals than analysis in Meta 1 (141 vs. 222 and 10 vs. 41 SNPs of P-values ≤ 10E-5 and 10E-6 respectively). Possible explanations include the poorer quality assessment of caries, the imbalance in the sex ratio, and the advanced age of participants for whom the cumulative environmental assault across decades may have greatly overshadowed genetic effects. Furthermore, during the analysis on HPFS case–control cohort of type 2 diabetes, we failed to adjust the diabetes status variable due to the IRB restriction. There existed evidence showing that individuals with type 2 diabetes may exhibit poorer oral health [49]. However, the definite answer for association between dental caries and type 2 diabetic status remains uncertain [50,51].
Conclusions
We designed and performed the first genome-wide association study for dental caries in the permanent dentition in adults. The GWAS analyses were first conducted in each of five independent cohorts; three meta-analyses were subsequently performed on part or all data from over 7000 combined samples. Although we did not observe any genetic associations meeting genome-wide significance, we identified a few loci that demonstrated both the suggestive P-values and the biologically relevant functions for dental caries. Of note, several of these nominated genes may be involved in common signaling pathways.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XW, JRS, EF, DEW, MLM conceived and designed this study; XW analyzed the data; XW and JRS wrote the manuscript; XW, JRS, ZZ, FB, EF, DEW and MLM managed, cleaned and quality checked the data together with other co-authors from the coordinating center at University of West Virginia, University of Washington, CIDR, ARIC and HPFS collaborators ; XW, JRS, EF, KTC, MKL, DEW and MLM interpreted the results; XW, JRS, ZZ, FB, ARV, KTC, MKL, DEP, RJW, DEW and MLM read, revised and approved the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
SNPs with P-value ≤ 10E-5 in Meta 1, Meta 2 and Meta 3. This files contains 3 tables (Supplement Table 1A, 1B and 1C), each of which shows the top-hit SNPs (P-value ≤ 10E-5 as cut-off) and other corresponding information from the three meta-analyses (meta 1, meta2 and meta 3) respectively.
Contributor Information
Xiaojing Wang, Email: timanwang@gmail.com.
John R Shaffer, Email: jrs51@pitt.edu.
Zhen Zeng, Email: zhz43@pitt.edu.
Ferdouse Begum, Email: ferdouse@pitt.edu.
Alexandre R Vieira, Email: arv11@pitt.edu.
Jacqueline Noel, Email: jbn9@pitt.edu.
Ida Anjomshoaa, Email: ida2@pitt.edu.
Karen T Cuenco, Email: ktc14@pitt.edu.
Myoung-Keun Lee, Email: myl2@pitt.edu.
James Beck, Email: jim_beck@dentistry.unc.edu.
Eric Boerwinkle, Email: Eric.Boerwinkle@uth.tmc.edu.
Marilyn C Cornelis, Email: mcorneli@hsph.harvard.edu.
Frank B Hu, Email: frank.hu@channing.harvard.edu.
David R Crosslin, Email: davidcr@u.washington.edu.
Cathy C Laurie, Email: cclaurie@u.washington.edu.
Sarah C Nelson, Email: sarahcn@u.washington.edu.
Kimberly F Doheny, Email: kdoheny@cidr.jhmi.edu.
Elizabeth W Pugh, Email: epugh1@jhmi.edu.
Deborah E Polk, Email: dpolk@pitt.edu.
Robert J Weyant, Email: rjw1@pitt.edu.
Richard Crout, Email: rcrout@hsc.wvu.edu.
Daniel W McNeil, Email: Daniel.McNeil@mail.wvu.edu.
Daniel E Weeks, Email: weeks@pitt.edu.
Eleanor Feingold, Email: feingold@pitt.edu.
Mary L Marazita, Email: marazita@pitt.edu.
Acknowledgments
(1) Funding support for the study entitled “Dental Caries: Whole Genome Association and Gene x Environment Studies” was provided by the National Institutes of Dental and Craniofacial Research (NIDCR) as part of the trans-NIH Genes, Environment and Health Initiative [GEI] (U01-DE018903). This study is one of the genome-wide association studies funded as part of the Gene Environment Association Studies (GENEVA) program of the GEI. Genotyping was done by the Johns Hopkins University (JHU) Center for Inherited Disease Research (CIDR), with funding from the National Institute of Dental and Craniofacial Research (NIDCR), through the National Institutes of Health (NIH) contract to JHU, contract number HHSN268200782096C. Funds for this project’s genotyping were provided by the NIDCR through CIDR’s NIH contract. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center (U01-HG004446) and by NCBI. Data and samples were provided by the Center for Oral Health Research in Appalachia (a collaboration of the University of Pittsburgh and West Virginia University funded by NIDCR R01-DE 014899); (2) the University of Pittsburgh School of Dental Medicine Dental Registry and DNA Repository (DRDR). The DRDR is supported by the School of Dental Medicine and NIH Grant 5TL1RR024155. I. Anjomshoaa was supported by the CTSI START UP program, the short-term pre-doctoral award through the Clinical and Translational Science Institute and the Institute for Clinical Research Education at the University of Pittsburgh (NIH Grant 5TL1RR024155-02). Financial support for A.R. Vieira was provided by NIH Grant R01-DE018914. (3) Additional support was provided by R03-DE021425, and UL1RR024153 .
Dental data from two other GENEVA projects ARIC and HPFS were also included. ARIC dental data collection was funded by R01-DE11551 from NIDCR. Data collection for HPFS T2D cohort included in this project was funded by U01-HG004399 from NIH.
The datasets used for the analyses described in this manuscript are available from dbGaP [ http://www.ncbi.nlm.nih.gov/gap]; specifically dbGaP accession number phs000095.v1.p1 for the GENEVA dental caries data, accession number phs000090.v1.p1 for ARIC and phs000091.v2.p1 for HPFS.
References
- Beltran-Aguilar ED, Barker LK, Canto MT, Dye BA, Gooch BF, Griffin SO, Hyman J, Jaramillo F, Kingman A, Nowjack-Raymer R. et al. Surveillance for dental caries, dental sealants, tooth retention, edentulism, and enamel fluorosis–United States, 1988–1994 and 1999–2002. Morb Mortal Wkly Rep Surveill Summ. 2005;54(3):1–43. [PubMed] [Google Scholar]
- Bretz WA, Corby PM, Melo MR, Coelho MQ, Costa SM, Robinson M, Schork NJ, Drewnowski A, Hart TC. Heritability estimates for dental caries and sucrose sweetness preference.[see comment] Arch Oral Biol. 2006;51(12):1156–1160. doi: 10.1016/j.archoralbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Bretz WA, Corby PM, Schork NJ, Robinson MT, Coelho M, Costa S, Melo Filho MR, Weyant RJ, Hart TC. Longitudinal analysis of heritability for dental caries traits. J Dent Res. 2005;84(11):1047–1051. doi: 10.1177/154405910508401115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraas JC, Messer LB, Till MJ. A genetic contribution to dental caries, occlusion, and morphology as demonstrated by twins reared apart. J Dent Res. 1988;67(9):1150–1155. doi: 10.1177/00220345880670090201. [DOI] [PubMed] [Google Scholar]
- Wang X, Shaffer JR, Weyant RJ, Cuenco KT, DeSensi RS, Crout R, McNeil DW, Marazita ML. Genes and their effects on dental caries may differ between primary and permanent dentitions. Caries Res. 2010;44(3):277–284. doi: 10.1159/000314676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendell S, Wang X, Brown M, Cooper ME, DeSensi RS, Weyant RJ, Crout R, McNeil DW, Marazita ML. Taste genes associated with dental caries. J Dent Res. 2010;89(11):1198–1202. doi: 10.1177/0022034510381502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SW, Yoon I, Lee HW, Cho J. Association between AMELX polymorphisms and dental caries in Koreans. Oral Dis. 2011;17(4):399–406. doi: 10.1111/j.1601-0825.2010.01766.x. [DOI] [PubMed] [Google Scholar]
- Deeley K, Letra A, Rose EK, Brandon CA, Resick JM, Marazita ML, Vieira AR. Possible association of amelogenin to high caries experience in a Guatemalan-Mayan population. Caries Res. 2008;42(1):8–13. doi: 10.1159/000111744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slayton RL, Cooper ME, Marazita ML. Tuftelin, mutans streptococci, and dental caries susceptibility. J Dent Res. 2005;84(8):711–714. doi: 10.1177/154405910508400805. [DOI] [PubMed] [Google Scholar]
- De Soet JJ, van Gemert-Schriks MC, Laine ML, van Amerongen WE, Morre SA, van Winkelhoff AJ. Host and microbiological factors related to dental caries development. Caries Res. 2008;42(5):340–347. doi: 10.1159/000151329. [DOI] [PubMed] [Google Scholar]
- Shaffer JR, Wang X, Feingold E, Lee M, Begum F, Weeks DE, Cuenco KT, Barmada MM, Wendell SK, Crosslin DR. et al. Genome-wide association scan for childhood caries implicates novel genes. J Dent Res. 2011;90(12):1457–1462. doi: 10.1177/0022034511422910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk DE, Weyant RJ, Crout RJ, McNeil DW, Tarter RE, Thomas JG, Marazita ML. Study protocol of the Center for Oral Health Research in Appalachia (COHRA) etiology study. BMC Oral Health. 2008;6(3):8–18. doi: 10.1186/1472-6831-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjomshoaa I, Cooper ME, Vieira AR. Caries is Associated with Asthma and Epilepsy. Eur J Dentistry. 2009;3(4):297–303. [PMC free article] [PubMed] [Google Scholar]
- Cornelis MC, Agrawal A, Cole JW, Hansel NN, Barnes KC, Beaty TH, Bennett SN, Bierut LJ, Boerwinkle E, Doheny KF. et al. The Gene, Environment Association Studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010;34(4):364–372. doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JD, Elter JR, Heiss G, Couper D, Mauriello SM, Offenbacher S. Relationship of periodontal disease to carotid artery intima-media wall thickness: the atherosclerosis risk in communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2001;21(11):1816–1822. doi: 10.1161/hq1101.097803. [DOI] [PubMed] [Google Scholar]
- Borrell LN, Beck JD, Heiss G. Socioeconomic disadvantage and periodontal disease: the Dental Atherosclerosis Risk in Communities study. Am J Public Health. 2006;96(2):332–339. doi: 10.2105/AJPH.2004.055277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud DS, Liu Y, Meyer M, Giovannucci E, Joshipura K. Periodontal disease, tooth loss, and cancer risk in male health professionals: a prospective cohort study. Lancet Oncol. 2008;9(6):550–558. doi: 10.1016/S1470-2045(08)70106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Cornelis MC, Kraft P, Stanya KJ, Linda Kao WH, Pankow JS, Dupuis J, Florez JC, Fox CS, Pare G. et al. Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum Mol Genet. 2010;19(13):2706–2715. doi: 10.1093/hmg/ddq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury TF, Horowitz AM, Ismail AI, Maertens MP, Rozier RG, Selwitz RH. Diagnosing and reporting early childhood caries for research purposes. A report of a workshop sponsored by the National Institute of Dental and Craniofacial Research, the Health Resources and Services Administration, and the Health Care Financing Administration. J Public Health Dent. 1999;59(3):192–197. doi: 10.1111/j.1752-7325.1999.tb03268.x. [DOI] [PubMed] [Google Scholar]
- Bennett SN, Caporaso N, Fitzpatrick AL, Agrawal A, Barnes K, Boyd HA, Cornelis MC, Hansel NN, Heiss G, Heit JA. et al. Phenotype harmonization and cross-study collaboration in GWAS consortia: the GENEVA experience. Genet Epidemiol. 2011;35(3):159–173. doi: 10.1002/gepi.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, Boehm F, Caporaso NE, Cornelis MC, Edenberg HJ. et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol. 2010;34(6):591–602. doi: 10.1002/gepi.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26(2):259–262. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Zhang K, Need AC, Martin O, Fellay J, Urban TJ, Telenti A, Goldstein DB. WGAViewer: software for genomic annotation of whole genome association studies. Genome Res. 2008;18(4):640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC. Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb Protoc. 2009;2009(10):pdb ip71. doi: 10.1101/pdb.ip71. [DOI] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S, Smith AJ, Berdal A, Lumley PJ, Cooper PR. The MAP kinase pathway is involved in odontoblast stimulation via p38 phosphorylation. J Endod. 2010;36(2):256–259. doi: 10.1016/j.joen.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Botero TM, Son JS, Vodopyanov D, Hasegawa M, Shelburne CE, Nor JE. MAPK signaling is required for LPS-induced VEGF in pulp stem cells. J Dent Res. 2010;89(3):264–269. doi: 10.1177/0022034509357556. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Tanaka N, Mitani Y, Miyazaki T, Fujii H, Sato M, Kovarik P, Decker T, Schlessinger J, Taniguchi T. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-gamma, but not IFN-alpha, signaling. EMBO J. 1999;18(9):2480–2488. doi: 10.1093/emboj/18.9.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P, Avraham S, Kumar S, Nakazawa A, Place A, Ghanem L, Rana A, Kumar V, Majumder PK, Avraham H. et al. Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. J Biol Chem. 1999;274(15):10140–10144. doi: 10.1074/jbc.274.15.10140. [DOI] [PubMed] [Google Scholar]
- Nakashima M. Bone morphogenetic proteins in dentin regeneration for potential use in endodontic therapy. Cytokine Growth Factor Rev. 2005;16(3):369–376. doi: 10.1016/j.cytogfr.2005.02.011. [DOI] [PubMed] [Google Scholar]
- About I, Laurent-Maquin D, Lendahl U, Mitsiadis TA. Nestin expression in embryonic and adult human teeth under normal and pathological conditions. Am J Pathol. 2000;157(1):287–295. doi: 10.1016/S0002-9440(10)64539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung IH, Choung PH, Ryu HJ, Kang YH, Choung HW, Chung JH, Choung YH. Regulating the role of bone morphogenetic protein 4 in tooth bioengineering. J Oral Maxillofac Surg. 2007;65(3):501–507. doi: 10.1016/j.joms.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23(3):265–277. doi: 10.1101/gad.1760809. [DOI] [PubMed] [Google Scholar]
- Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, Andl T, Yang SH, Lu MM, Piccolo S, Schmidt-Ullrich R. et al. Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol. 2008;313(1):210–224. doi: 10.1016/j.ydbio.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Pennica D, Xu L, Kalejta RF, Levine AJ. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes Dev. 2001;15(14):1796–1807. doi: 10.1101/gad.894301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133(8):1587–1596. doi: 10.1242/dev.02308. [DOI] [PubMed] [Google Scholar]
- Mitsiadis TA, Angeli I, James C, Lendahl U, Sharpe PT. Role of Islet1 in the patterning of murine dentition. Development. 2003;130(18):4451–4460. doi: 10.1242/dev.00631. [DOI] [PubMed] [Google Scholar]
- Kouskoura T, Fragou N, Alexiou M, John N, Sommer L, Graf D, Katsaros C, Mitsiadis TA. The genetic basis of craniofacial and dental abnormalities. Schweiz Monatsschr Zahnmed. 2011;121(7–8):636–646. [PubMed] [Google Scholar]
- Horst OV, Horst JA, Samudrala R, Dale BA. Caries induced cytokine network in the odontoblast layer of human teeth. BMC Immunol. 2011;12:9. doi: 10.1186/1471-2172-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerayutthwilai O, Byers MR, Pham TT, Darveau RP, Dale BA. Differential regulation of immune responses by odontoblasts. Oral Microbiol Immunol. 2007;22(1):5–13. doi: 10.1111/j.1399-302X.2007.00310.x. [DOI] [PubMed] [Google Scholar]
- Mutoh N, Tani-Ishii N, Tsukinoki K, Chieda K, Watanabe K. Expression of toll-like receptor 2 and 4 in dental pulp. J Endod. 2007;33(10):1183–1186. doi: 10.1016/j.joen.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol. 2009;183(10):6522–6529. doi: 10.4049/jimmunol.0901271. [DOI] [PubMed] [Google Scholar]
- Horst OV, Tompkins KA, Coats SR, Braham PH, Darveau RP, Dale BA. TGF-beta1 Inhibits TLR-mediated odontoblast responses to oral bacteria. J Dent Res. 2009;88(4):333–338. doi: 10.1177/0022034509334846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller F, Feenstra B, Zhang H, Shaffer JR, Hansen T, Esserlind AL, Boyd HA, Nohr EA, Timpson NJ, Fatemifar G. et al. Genome-wide association study identifies four loci associated with eruption of permanent teeth. PLoS Genet. 2011;7(9):e1002275. doi: 10.1371/journal.pgen.1002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR, Marazita ML, Goldstein-McHenry T. Genome-wide scan finds suggestive caries loci. J Dent Res. 2008;87(5):435–439. doi: 10.1177/154405910808700506. [DOI] [PubMed] [Google Scholar]
- Sandberg GE, Sundberg HE, Fjellstrom CA, Wikblad KF. Type 2 diabetes and oral health: a comparison between diabetic and non-diabetic subjects. Diabetes Res Clin Pract. 2000;50(1):27–34. doi: 10.1016/S0168-8227(00)00159-5. [DOI] [PubMed] [Google Scholar]
- Collin HL, Uusitupa M, Niskanen L, Koivisto AM, Markkanen H, Meurman JH. Caries in patients with non-insulin-dependent diabetes mellitus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85(6):680–685. doi: 10.1016/S1079-2104(98)90035-X. [DOI] [PubMed] [Google Scholar]
- Hintao J, Teanpaisan R, Chongsuvivatwong V, Dahlen G, Rattarasarn C. Root surface and coronal caries in adults with type 2 diabetes mellitus. Community Dent Oral Epidemiol. 2007;35(4):302–309. doi: 10.1111/j.1600-0528.2007.00325.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SNPs with P-value ≤ 10E-5 in Meta 1, Meta 2 and Meta 3. This files contains 3 tables (Supplement Table 1A, 1B and 1C), each of which shows the top-hit SNPs (P-value ≤ 10E-5 as cut-off) and other corresponding information from the three meta-analyses (meta 1, meta2 and meta 3) respectively.