

Abstract
Diverse communities of bacteria inhabit plant leaves and roots and those bacteria play a crucial role for plant health and growth. Arabidopsis thaliana is an important model to study plant pathogen interactions, but little is known about its associated bacterial community under natural conditions. We used 454 pyrosequencing to characterize the bacterial communities associated with the roots and the leaves of wild A. thaliana collected at 4 sites; we further compared communities on the outside of the plants with communities in the endophytic compartments. We found that the most heavily sequenced bacteria in A. thaliana associated community are related to culturable species. Proteobacteria, Actinobacteria, and Bacteroidetes are the most abundant phyla in both leaf and root samples. At the genus level, sequences of Massilia and Flavobacterium are prevalent in both samples. Organ (leaf vs root) and habitat (epiphytes vs endophytes) structure the community. In the roots, richness is higher in the epiphytic communities compared to the endophytic compartment (P = 0.024), while the reverse is true for the leaves (P = 0.032). Interestingly, leaf and root endophytic compartments do not differ in richness, diversity and evenness, while they differ in community composition (P = 0.001). The results show that although the communities associated with leaves and roots share many bacterial species, the associated communities differ in structure.
Introduction
Biotic factors such as host species [1]–[3], genotype [4]–[6], and leaf age [7], [8] can all impact the structure of microbial communities associated with plants, as can the many abiotic factors that influence the physiology of host plants (e.g. [9], [10]). One might expect the importance of particular abiotic factors to vary, depending upon the location of the microbial community within the plant, and this may have repercussions for the structure of microbial communities. For example, the microbial community residing in the phyllosphere (the aerial parts of plants) is faced with a nutrient poor and variable environment that is characterized by fluctuating temperature, humidity and UV radiation [11]. The microbial community in the rhizosphere (the soil directly in contact with the roots), on the other hand, resides within a stable environment that is rich in nutrients due to the chemicals exuded by plants to attract beneficial microorganisms and combat pathogens [12]. If environmental variability promotes diversity, as has been suggested [13], [14], then the microbial community within the phyllosphere would be predicted to be more diverse than that within the rhizosphere. There is a little evidence that touches on this hypothesis; however, there are few direct comparisons of rhizosphere and phyllosphere bacterial communities, especially comparisons using material from the same plants [15], [16].
Microbial communities colonizing plants may protect them against pathogen infection [15], [17], and Arabidopsis thaliana is an important model to study plant defense against pathogens. A characterization of the bacterial communities colonizing Arabidopsis thaliana in the field is therefore valuable, not only for exploring how ecological factors shape communities but also for its applied relevance. To date, the bacterial community of the rhizosphere soil associated with A. thaliana has been studied using fingerprinting methods [18], and the bacterial community of the phyllosphere has been characterized with DGGE and clone libraries [19]. However, for both studies, plants were grown in growth cabinets, and therefore, lack potential colonizers. Of more relevance to naturally occurring bacterial communities, Delmotte et al. [20] found that Methylobacterium, Sphingomonas and Pseudomonas proteins were the most abundant in the phyllosphere of A. thaliana grown in the field at one site. None of the three studies above compared communities on the surface (epiphytic) and within the plants (endophytic). Two studies that did compare bacterial communities associated with the inside and the outside of A. thaliana roots used 454 pyrosequencing to find that the endophytic compartment harbors a less diverse community than the rhizosphere [21], [22]. In this study, we also characterized bacterial communities using 454 pyrosequencing of the 16S rRNA gene. We obtained more than 4000 sequences per sample. Our main objective was to describe and compare epiphytic and endopyhtic bacterial communities associated with the roots and leaves of A. thaliana growing under natural conditions.
Materials and Methods
Site Description and Sampling
Arabidopsis thaliana samples were collected at 4 sites: on April 15 2008 at Route Marker 166 (41°21'17.41"N, 86°44'14.47"W), on April 16 2008 at Lake Michigan College (42° 5'24.41"N, 86°23'36.27"W), on April 25 2008 at Michigan Extension (42°5'33.72"N, 86°21'22.76"W), and on April 30 2008 at North Liberty (41°32'24.88"N, 86°25'32.86"W). All of these sites are disturbed. The described field study did not require specific permits and did not involve endangered or protected species. The locations are not privately-owned or protected in any way. In the Midwest, A. thaliana germinates at the end of the summer and overwinters as a rosette. Samples were harvested a few weeks after the snow melt. The plants were healthy-looking, although reddish, indicating that the plants were stressed, potentially from the cold. They were also smaller than A. thaliana grown in growth chambers, which is typical of field collected A. thaliana. Root sizes varied, with plants from Root Marker 166 having the largest roots.
At each site, we collected approximately 20 plants with sterile gloves. The roots were cut from the rosettes in the field using a sterile razor blade and both sample types were stored in sterile 50 ml polystyrene tubes. Plants from one site were bulked into one sample. Samples were brought to the lab on ice, and then stored at −80°C before processing.
Microbes living on the plant surface (epiphytes) were separated from microbes living within the plant (endophytes) using a modified protocol [23]. For the rosette samples: samples were weighed and for each gram of leaf material, 10 ml of 0.1 M potassium phosphate buffer, pH 8.0 was added to the tubes. Tubes were sonicated 1 min and vortexed for 10 sec; this procedure was repeated twice. The wash steps were repeated once. The leaf wash was then filtered using a 115-ml (0.2 um) nitrocellulose filter unit (Nalgene, NY, USA). The roots were washed in a similar way, except the samples (roots and the most tightly associated sand, comprising <1 mm of sand surrounding the roots) were first placed in the filter unit and washed twice with 10 ml phosphate buffer. Roots were then removed from the filter unit and placed in 50-ml vials with 20 ml phosphate buffer, sonicated, and vortexed 6 times every 5 minutes. The root wash was then filtered using the filter unit. Root and leaves were washed twice with 70% ethanol, then stored at −80°C for later extraction of the endophytic fraction.
DNA Extraction, PCR Amplification, and Sample Pooling
Altogether, there are 16 DNA samples: plants were collected from 4 sites, for each site, there are both root and leaves, and for each organ, there are epiphytic and endophytic fractions. For the epiphytic fraction, DNA was extracted from half of the filter using the Power Soil DNA kit (MoBio Laboratories). For the endophytic fraction, each sample was pulverized in liquid nitrogen with a mortar and pestle. An aliquot (150 mg) of each sample was added to the bead tubes from the Power Soil DNA kit (MoBio Laboratories), followed by extraction with the standard MoBio protocol. DNA concentration was determined using PicoGreen (Invitrogen). DNA concentration was adjusted to 10 ng/µl for the endophytic samples and 1 ng/µl for the epiphytic samples; a lower concentration is used for the epiphytic samples because the DNA is mostly microbial DNA. The DNA of the endophytic samples on the other hand also includes plant DNA.
Primer 799F (5'-AACMGGATTAGATACCCKG-3'), which minimizes contamination from plastid DNA [24] and a primer designed for this study, 1193r (5'-ACGTCATCCCCACCTTCC-3'), were used to amplify V5, V6 and V7 of the 16S rRNA gene. The forward primer was fused to the 454 Life Sciences primer B and the reverse primer was fused to the adapter A and a barcode in order to sequence the hypervariable regions V6 and V7.
Each 25 µl PCR reaction contained 10 ng (for the endophytic fraction) or 1 ng (for the epiphytic fraction) of DNA, Mg2+ free PCR buffer (TaKaRa), 3 mM MgCl2 (TaKaRa), 200 µM dNTP, 200 nM forward primer, 200 nM reverse primer, 12.5 µg ultrapure BSA (Ambion), and 1 unit Ex Taq HotStart polymerase (TaKaRa). Cycling conditions were 94°C for 2 min, followed by 25 cycles of 94°C for 30 sec, 55°C for 30 sec, 72°C for 1 min, with a final extension of 72°C for 10 min. All samples were amplified in quadruplicates, which were combined before purification. Primer 799f and 1193r amplify a mitochondrial product of about 800 bp and a bacterial product of about 500 bp. We isolated the bacterial product by separating the PCR products on a 3% low melt agarose gel (2% agarose for root samples) and excising a band of agarose with size 400 bp to 700 bp. DNA was extracted from the gel using the QIAquick gel extraction kit (Qiagen). After purification, DNA was quantified using the PicoGreen assay (Invitrogen) and the quality was checked using a Bioanalyzer (Agilent). DNA concentration was adjusted to 1 ng/µl. The amplicon libraries were prepared by pooling 10 ng of each PCR.
The amplicon libraries were sent to the High-throughput Genome Analysis Core at Joint Institute for Genomics & Systems Biology (University of Chicago/Argonne National Laboratory) for pyrosequencing on a 454 Life Sciences FL (Roche) machine. One region of the 454 run was used for DNA from the rosette samples and the other region was used for the root samples. The sequencing data have been deposited in the NCBI Sequence Read Archive (SRP018030).
Sequence Analysis
The software package mothur (version 1.27.0) was used for sequence analysis [25] while following the Standard Operating Procedure outlined on http://www.mothur.org/wiki/Schloss_SOP. Briefly, sequencing error was reduced using shhh.flows (mothur implementation of the AmpliconNoise algorithm). Then, each unique sequence was aligned with align.seqs using the SILVA reference alignment. A distance matrix was calculated with default parameters. Chimeric sequences were identified using chimera.uchime and removed. Sequences matching “Cyanobacteria_Chloroplast" and "Mitochondria” were also removed. Next, sequences were clustered using the furthest neighbor clustering algorithm to build OTUs (operational taxonomic unit). The resulting file was parsed to separate the data for each sample. OTUs were assigned a taxonomic group with classify.seqs using the RDP reference file and a cutoff of 80% of the bootstrap value. For the description of the community, OTUs with the same taxonomy were binned together at the phylum, class and genus level.
Statistical Analysis
Abundance tables were analyzed using the package vegan [26] within the R statistical environment (R Development Core Team; http://www.R-project.org). To test the hypothesis that none of the taxa co-occur more often than by chance [27], we transformed the species matrix using the Hellinger-transformation [28] and then calculated Kendall’s Coefficient of Concordance on the 50 most heavily sequenced taxa in each community. To estimate diversity, we minimized the impact of sequencing artifacts by restricting our analyses to all OTUs present in at least 2 samples. Percentages of sequences belonging to singletons were arcsine square root transformed before calculating the Student’s t-test. ANOVA was used to test the effect of ‘habitat’ (epiphyte vs. endophyte) and ‘organ’ (root vs. leaf) on the relative abundance of the members of the core community. Correspondence analysis was performed with the function cca. To calculate diversity indices while controlling for sampling effort, 2000 sequences were subsampled 500 times for each biological sample. For each subsampling of 2000 sequences, three diversity indices were calculated and plotted: richness, diversity and evenness. Richness (S) is the number of OTUs. Shannon-Weaver index is H = -sum pi *ln pi, where pi is the proportional abundance of species i. From the Shannon-Weaver index, one can calculate diversity: D = exp(H) [29]. Evenness was calculated with Sheldon’s evenness: E = exp(H)/S) [30]. ANOVA was used to test the effect of ‘habitat’ (epiphyte vs. endophyte) and ‘organ’ (root vs. leaf) on richness, diversity and evenness using the mean of the 500 permutations for each sample. Normality of the standardized residuals was investigated with a qqplot; furthermore, the Shapiro-Wilk test confirmed that the standardized residuals were normally distributed. Paired student’s t-tests were calculated for all pairwise comparisons, P values were adjusted using the fdr correction for multiple testing. For analysis of community composition, pairwise dissimilarities between samples were calculated based on the Bray–Curtis index with the vegan function vegdist. To assess the effect of ‘habitat’ and ‘organ’ on community composition, we used the vegan functions adonis, which is a non-parametric multivariate analysis of variance [31] as well as the functions mrpp [32] and anosim [33].
Results
Analysis of Pyrosequencing Data
We used the standard operating procedure from the software package mothur that includes a denoising step [25]. After removing chimeras, we obtained 135,540 sequences. We found that primer 799f amplifies both bacterial and plant chloroplast DNA under our PCR conditions; the proportion of reads assigned to a plant taxonomic identification ranges from 0 to 23% for each sample. After removing reads assigned to the taxonomic Kingdom Plantae, 129,445 sequences remained.
Sequences were clustered into operational taxonomic units (OTUs) at the 0.05 distance cutoff, which is typically the genus level [34]. Rarefaction curves were starting to level off, suggesting that the plant associated communities were reasonably well characterized with our sampling effort (Figure 1). Interestingly, the rarefaction curves of the epiphytic samples are higher than the endophytic samples for the root samples while the reverse is true for the leaf samples.
Figure 1. Rarefaction curves at the 5% distance cutoff.
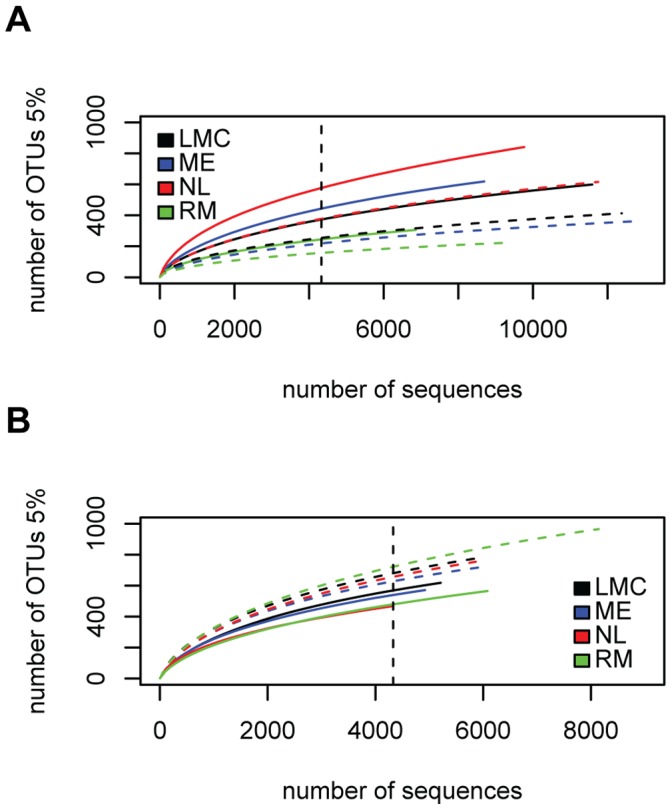
(A) Leaf-associated communities; (B) Root-associated communities. Samples were collected at 4 sites (RM, Route Marker; NL, North Liberty; ME, Michigan Extension; LMC, Lake Michigan College). Continuous lines represent endophytic samples and dashed lines represent epiphytic samples. The dashed vertical line indicates the numbers of sequences subsampled from each sample (4329 sequences).
Description of the Community
To compare samples, the number of sequences per sample was standardized to the minimum number of sequences in a single sample (4329 sequences). First, taxonomy of the sequences was examined at the phylum level on the basis of the RDP Bayesian classifier. The most heavily-sequenced phyla associated with both roots and leaves were Proteobacteria, Actinobacteria, and Bacteroidetes; sequences assigned to the Firmicutes were additionally present in the rhizosphere (Figure 2). Sequences assigned to Actinobacteria were more abundant in the root-associated communities (28.4% of the epiphytic, 30.9% of the endophytic community) compared to the leaf-associated communities (12.3% of the epiphytic, 14.5% of the endophytic community), while sequences assigned to the class Gammaproteobacteria were more abundant in the leaf epiphytic samples (34.9%) compared to the leaf endophytic community (13.5%) and root-associated communities (5.7% of the epiphytic, 6.2% of the endophytic community).
Figure 2. Relative abundance of bacterial phyla associated with Arabidopsis thaliana roots and leaves.

The Proteobacteria OTU has been replaced by 4 OTUs at the subclass level (alpha, beta, gamma, delta). Only OTUs with at least 100 sequences are represented.
We notice that predominance at the phylum and class level is driven by the high abundance of one or two OTUs (Table 1). For example, Gammaproteobacteria were dominant in the leaf epiphytic community due to the large number of sequences of Pseudomonas; similarly, in the roots, Actinobacteria were mostly represented by the OTUs Actinomycetales and Actinoplanes. The phylum Bacteroidetes is mainly represented by sequences belonging to 3 OTUs: Flavobacterium, Chitinophagaceae and, Flavobacteriaceae.
Table 1. The core community associated with roots and leaves of Arabidopsis thaliana.
| OTU number | Genus (or higher) | Phylum | Leaf-associated | Root-associated | Result of ANOVA test | |||||||||||
| endophyte | epiphyte | endophyte | epiphyte | organ | habitat | interaction | ||||||||||
| average propotion | average propotion | average propotion | average propotion | F | P | F | P | F | P | |||||||
| Otu006 | Flavobacteriaceae | Bacteroidetes | 2.56% | 1.77% | 2.25% | 2.60% | 0.21 | 0.657 | 0.80 | 0.389 | 0.87 | 0.370 | ||||
| Otu007 | Methylobacterium | Proteobacteria | 3.24% | a | 2.56% | a | 0.91% | b | 0.62% | b | 35.11 | 6.97E−05 | 1.33 | 0.270 | 0.00 | 0.975 |
| Otu008 | Otu008 | Proteobacteria | 3.10% | 2.37% | 1.99% | 2.10% | 3.18 | 0.100 | 0.56 | 0.468 | 1.45 | 0.252 | ||||
| Otu009 | Otu009 | Otu009 | 3.55% | 6.77% | 7.09% | 9.32% | 6.26 | 0.028 | 2.20 | 0.164 | 0.15 | 0.710 | ||||
| Otu023 | Pseudomonas | Proteobacteria | 10.90% | b | 31.40% | a | 2.02% | c | 2.31% | c | 56.67 | 6.97E−06 | 6.28 | 0.0276 | 3.21 | 0.0986 |
| Otu027 | Actinomycetales | Actinobacteria | 2.67% | ab | 0.86% | a | 2.06% | ab | 3.41% | b | 5.50 | 0.037 | 0.45 | 0.514 | 9.53 | 0.00942 |
| Otu030 | Oxalobacteraceae | Proteobacteria | 4.16% | 4.13% | 4.53% | 3.73% | 0.02 | 0.901 | 0.24 | 0.634 | 0.09 | 0.769 | ||||
| Otu031 | Chitinophagaceae | Bacteroidetes | 0.95% | ab | 0.86% | a | 3.54% | bc | 4.90% | c | 23.07 | 4.31E−04 | 0.16 | 0.696 | 0.42 | 0.530 |
| Otu034 | Sphingomonas | Proteobacteria | 8.26% | a | 9.16% | a | 1.90% | b | 2.04% | b | 100.39 | 3.51E−07 | 0.70 | 0.418 | 0.06 | 0.816 |
| Otu042 | Burkholderiales | Proteobacteria | 1.81% | a | 0.63% | b | 1.80% | a | 2.43% | a | 10.19 | 0.00775 | 2.49 | 0.141 | 9.98 | 0.00824 |
| Otu045 | Massilia | Proteobacteria | 3.09% | 3.39% | 5.60% | 4.32% | 1.43 | 0.256 | 0.28 | 0.608 | 0.01 | 0.942 | ||||
| Otu047 | Flavobacterium | Bacteroidetes | 9.77% | 3.93% | 10.15% | 5.89% | 1.03 | 0.331 | 7.94 | 0.0155 | 0.31 | 0.590 | ||||
| Otu051 | Otu051 | Proteobacteria | 1.25% | 1.84% | 2.47% | 1.68% | 2.10 | 0.173 | 0.10 | 0.753 | 1.69 | 0.218 | ||||
| Otu055 | Microbacteriaceae | Actinobacteria | 1.15% | 2.67% | 1.65% | 1.68% | 0.66 | 0.434 | 1.62 | 0.228 | 0.50 | 0.493 | ||||
| Otu059 | Comamonadaceae | Proteobacteria | 2.69% | 2.40% | 1.90% | 1.80% | 1.71 | 0.215 | 0.03 | 0.869 | 0.03 | 0.873 | ||||
| Otu061 | Actinoplanes | Actinobacteria | 1.21% | ab | 0.25% | b | 3.58% | a | 1.05% | ab | 6.13 | 0.0292 | 6.05 | 0.0301 | 0.52 | 0.485 |
| Otu088 | Rhizobium | Proteobacteria | 2.14% | 2.78% | 1.08% | 0.94% | 4.20 | 0.0630 | 0.14 | 0.716 | 0.56 | 0.467 | ||||
| Otu090 | Arthrobacter | Actinobacteria | 0.29% | a | 0.31% | a | 0.97% | ab | 2.44% | b | 10.22 | 0.0077 | 1.89 | 0.194 | 1.79 | 0.205 |
| Otu119 | Variovorax | Proteobacteria | 3.19% | ns | 2.06% | ns | 1.36% | ns | 1.32% | ns | 8.61 | 0.0125 | 0.79 | 0.391 | 1.04 | 0.329 |
| Otu142 | Kineosporia | Actinobacteria | 0.06% | a | 0.14% | ab | 5.91% | b | 0.91% | ab | 8.54 | 0.0128 | 1.23 | 0.289 | 1.74 | 0.212 |
| Otu151 | Duganella | Proteobacteria | 2.73% | 0.05% | 0.12% | 0.02% | 1.38 | 0.263 | 1.81 | 0.203 | 1.13 | 0.309 | ||||
Letters show result from Tukey's ‘Honest Significant Difference’ test.
We define the core community as the 10 most abundant OTUs of each of the 4 habitats (root endophytes, root epiphytes, rosette endophytes, rosette epiphytes), resulting in 21 OTUs altogether (Table 1); these OTUs constitute 67% of the total sequences. ANOVA was used to test the effect of ‘habitat’ (epiphyte vs. endophyte) and ‘organ’ (root vs. leaf) on the relative abundance of the members of the core community. The Tukey's ‘Honest Significant Difference’ method was performed to compare average proportions. Relative abundances of 3 OTUs were found to be higher in the leaves: Pseudomonas, Sphingomonas and Methlybacterium. Furthermore, Pseudomonas was found to be more abundant in the leaf epiphytic community compared to the leaf endophytic community. Relative abundance of one OTU, Chitinophagaceae, was found to be higher in the roots. In addition, relative abundance of Burkholderiales was lower in the leaf epiphytic community compared to the other three communities, Arthrobacter was higher on the root surface compared to the leaf-associated communities and the relative abundance of Kineosporia was higher in the inside of the root compared to the leaf endophytic community. The other OTUs in the core community were generalist OTUs, for example Flavobacterium and Massilia. In addition, 2 OTUs that could not be classified below the phylum level, OTU8 and OTU9, were very ubiquitous genera, comprising more than 2% sequences in each sample.
We compared ranks of the most heavily-sequenced genera in the leaf and root associated communities (Figure S1). We used Kendall’s coefficient of concordance [27] to test for independence of rankings of the genera in each habitat. The top 50 genera in the leaf communities are concordant (Kendall's W = 0.0848, Friedman's chi-square = 4.54, P = 7.56e−5, 999 permutations) but not the top 50 genera within the root communities, indicating that OTUs in the phyllosphere are found together.
We performed correspondence analysis to analyze whether certain species occur at certain sites. The first axis separates samples based on organ type while the second axis separates samples based on site (Figure 3). Most samples cluster closely together, indicating that the communities are similar; however, communities in the inside of the roots of LMC and RM are relatively distinct from others, and this is correlated with more sequences assigned to Kinesporia. Similarly, leaf epiphytic communities of ME and RM were relatively distinct and this was correlated with more sequences assigned to Pseudomonas.
Figure 3. Correspondence analysis illustrates differences between bacterial communities in roots and leaves at 4 sites.

The two axes represent 41% of the inertia. OTUs are represented in gray; OTUs from the core community are highlighted in three colors: red OTUs are more abundant in the leaf-associated community while purple OTUs are more abundant in the root-associated community, generalist OTUs are labeled in blue.
Organ and Habitat Type Differentiate Communities
One of our goals was to compare bacterial communities associated with leaf and root. First, we examined differences in alpha diversity, which measures the diversity within each sample [35], focusing in particular on richness, diversity and evenness. Singletons, OTUs with only 1 sequence, were removed before calculating these indices, because singletons could be due to sequencing artifacts. 3514 singletons were removed, leaving 3160 OTUs (126841 sequences). We observed that the percent of the sequences belonging to singletons is higher for the root-associated community than for the leaf-associated community (3.22% versus 1.32% respectively; paired t-test, t = −3.8967, P = 0.00592). Tables of OTUs at 97% identity were subsampled 500 times for each sample and diversity indices were calculated for each permutation (Figure 4). We compared the diversity indices using pairwise t-tests (Figure S2). For this analysis, the average of the 500 subsamples was considered. Paired t-tests showed that richness is lower in the leaf epiphytic samples compared to the leaf endophytic samples (P = 0.032) and lower compared to both root communities (P = 0.024); by contrast, richness is higher in the root epiphytic samples compared to the root endophytic samples (P = 0.024). For diversity, paired t-tests showed that both root communities are more diverse than the leaf epiphytic communities (P = 0.024 for both tests). For evenness, paired t-tests showed that evenness is lower in leaf epiphytic communities compared to the root epiphytic community (P = 0.019).
Figure 4. Alpha diversity of the bacterial communities in the leaves and roots of Arabidopsis thaliana.
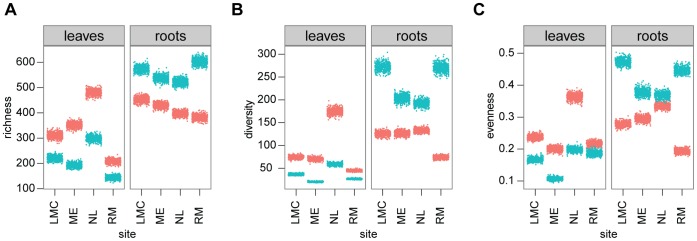
(A) Richness; (B) Diversity; (C) Evenness. Color represents habitat (blue, epiphytic community; red, endophytic community). Dots show the distribution of results of 500 subsampling (2000 sequences subsampled from each sample).
Next, we compared beta diversity [35],which is the variation in species composition. We tested the effect of ‘organ’ (leaf vs root) and ‘habitat’ (epiphyte vs. endophyte) on beta diversity. The adonis test found a significant effect of ‘organ’ (F 1,12 = 5.64, P = 0.001), with pairwise dissimilarities between root and leaf samples higher than within root or within leaf samples. Similarly, the mrpp and anosim tests also found a significant effect of organ (both tests: P = 0.001) but no significant effect of site or habitat.
Discussion
The most heavily sequenced members of the bacterial communities associated with Arabidopsis thaliana roots and leaves were related to described species; 60% of our sequences could be assigned at the genus level. Our ability to assign sequences at the genus level is lower than results obtained in a pyrosequencing study of the potato rhizosphere (75%) [36] but higher than a pyrosequencing study of the spinach phyllosphere (54%) [37]. In general, our study – and these other pyrosequencing studies - find lower representation of culturable species than clone library studies, which have found that 85 to 95% of sequences can be attributed to known genera [20], [38], [39]. This difference is due to the much higher sequencing depth of pyrosequencing. Of course, both clone libraries and 16S rRNA pyrosequencing underestimate true diversity due to primer bias. The primer used in this study (799f), for example, was designed to avoid amplification of chloroplast and therefore excludes Cyanobacteria. In fact, primer 799f matches only about 62% of the sequences in the RDP database (using Probe Match tool on RDPII).
Proteobacteria, Actinobacteria, and Bacteroidetes were the most abundant phyla associated with A. thaliana, all phyla that are typical of the phyllosphere [20], [40] and the rhizosphere [21], [22], [39], [41], suggesting substantial overlap in the key community members across host species. That said, there are many bacterial groups common on other hosts that we did not observe on A. thaliana. For example, the tree phyllosphere is heavily populated by Deinococcus-Thermus and TM7 [40]; all of these are rare in our samples. Moreover, we did not observe many sequences for the Enterobactericeae, which dominate the spinach phyllosphere [37] or for Bacillus and Pantoea, which dominate the lettuce phyllosphere [42], nor did we find any Rheinheimera sequences (and very few sequences for the genera Dyadobacter, Devosia and Pedobacter), which are abundant in the potato root communities [36]. On the other hand, Rathayibacter, found in the Arabidopsis phyllosphere, is not present in spinach, lettuce, or potato.
Comparison of the communities associated with the leaves and roots reveals both ubiquitous and organ specific groups. Flavobacterium (from 4% to 10% sequences) and Sphingomonas (from 2 to 9% sequences) are two abundant genera in both root and leaf associated communities which have potentially beneficial effects for plant growth and health. The abundance of Flavobacterium, a common soil and water bacterium, was positively correlated with potato biomass [36]. Sphingomonas has been isolated from a variety of environmental and plant samples [43] and some strains have a protective effect against plant pathogens [17]. Pseudomonas sequences are common in all four sample types, but they are most abundant in the leaf epiphytic community (31% sequences). In addition, sequences of Methylobacterium, a common phyllosphere colonizer [20], were also found in root samples (0.6 to 0.9% sequences). All these groups were previously known to be abundant on A. thaliana as based on culture-dependent surveys [44], culture-independent studies [36] and proteomic screens [20]. Overall, leaves and roots of A. thaliana are colonized by many of the same genera, albeit in different proportions. This suggests that many of the taxa found in the leaves and roots of A. thaliana may come from similar sources. Since Arabidopsis leaves are close to the ground, bacteria in the leaves may come from rain splashing off the soil. In fact, some soil particles could be observed on the leaves at the time of sampling. Conversely wind and rain, thought to be a source of bacteria in the phyllosphere, also bring bacteria to the soil. A third explanation is that seeds are colonized from the soil, and as the plant grows, bacteria colonize the expanding leaves.
An important caveat applies to interpretation of community studies that utilize pyrosequencing. The genus with the highest number of sequences is not necessarily the most abundant in the community due to several factors, including primer bias and 16S rRNA operon copy numbers. 16S rRNA copies range from 1 to 15 depending on the bacterial species [45]. The proportion of Sphingomonas, which has two copies of 16S rRNA, would thus be underestimated relative to Pseudomonas, which has five copies (rrnDB, Lee et al. 2009). Estimation of community composition based on 16S libraries should ideally take into account the copy number [46]; however, this is not yet feasible with 454 pyrosequencing, because it would be necessary to assign sequences to the species level and the number of species in the database is low (as of Oct 4 2012, 1411 species in the rrnDB).
Environmental variability promotes diversity [13], [14], and for this reason we expected the phyllosphere, generally thought to be quite variable, to be more diverse than the rhizosphere. However, we found the opposite: the root epiphytic community was richer and more diverse than leaf epiphytic community. There are several reasons why this might be true, and we cannot distinguish them. First, the soil environment is actually heterogeneous at the micrometer scale; it is made up of different components (e.g. sand, silt, clay, organic matter) with different chemical properties which create very different microhabitat along the root [47]. Moreover, the rhizosphere is quite dynamic and complex because of the large and diverse amount of secreted plant exudates [48]. Third, the soil harbors a very diverse bacterial community, which may be a source of endophytic colonizers [49]. In addition, we found differences in the beta-diversity of the leaf and root associated communities, which reveal differences in the composition of microbes associated with these plant organs. Similar differences in the composition of root and shoot associated bacterial communities have been found on potato [15]. These differences in composition might be due to the fact that root and leaf tissues carry different total bacterial population sizes. The bacterial abundance in the phyllosphere is estimated to be 107 cells/cm2 [50] or roughly 106 cells/g [42]. By contrast, bacterial abundance in the rhizosphere may reach up to 108 cells/g dry weight root tissue [51]. Diversity has been shown to be positively correlated with total community size [44].
Communities associated with the outside of the roots were colonized by a greater number of species than in the endophytic compartment, confirming results from a study in poplar [52] and Arabidopsis [21]. Interestingly, the reverse pattern was found in the leaves: communities associated with the outside had lower richness than the endophytic compartment. We expected higher richness in the leaf epiphytic communities based on a previous study in our lab that relied on culturing microbes on A. thaliana [44]. In addition, bacteria are generally thought to first colonize the leaf surface and then colonize the internal space of the leaves [53]. However, there is also evidence that bacteria from the soil first colonize the roots and then migrate to the above-ground part of the plant: for example, GFP-tagged beneficial bacteria such as Rhizobia inoculated in the soil were found in leaves [54]; similarly, pathogenic bacteria such as Dickeya were found in stems [55]. We speculate that over time, bacteria from the root endophytic compartment migrate or are transported to the leaf endophytic compartment, explaining the higher richness in that compartment compared to the leaf epiphytic community. We found several potential movers in the core community: Burkholderiales, which were quite abundant in both root habitats as well as the leaf endophytic community but significantly less abundant in the leaf epiphytic community, as well as Actinomycetales and Actinoplanes, which follow a similar pattern. Indeed, both root and leaf endophytic compartments were colonized by a similar number of OTUs, suggesting that the two compartments may form a continuum.
Supporting Information
Rank abundance of the 50 most heavily sequenced OTUs. Roots (left) and leaves (right). OTU numbers were replaced with GENUS, FAMILY or ORDER name depending on the level at which this OTU could be assigned. Arabidopsis thaliana were collected at 4 sites (purple, Route Marker; blue, North Liberty; green, Michigan Extension; red, Lake Michigan College). DNA was extracted for endophytic fraction (circle) and epiphytic fraction (triangle).
(TIF)
Alpha diversity of the bacterial communities in the leaves and roots of Arabidopsis thaliana . (A) Richness; (B) Diversity; (C) Evenness. Bars represent one standard error of the mean. The letters indicate results from paired t-test (P<0.05, P values adjusted using fdr).
(TIF)
Acknowledgments
We would like to thank Joel Kniskern and Luke Barrett for help with sampling and Lukas Elmiger for help with R.
Funding Statement
This work was supported by two grants to JB: National Science Foundation, NSF, MCB 0603515 entitled Microbial Observatories: Forces shaping microbial communities in the phyllosphere of Arabidopsis thaliana and National Institute of Health, NIH, GM057994 entitled Evolutionary genetics of R gene loci in Arabidopsis. NB acknowledges funding from the Swiss National Science Foundation through the postdoc mobility and Marie Heim-Vögtlin fellowships. MWH acknowledges funding from the National Science Foundation through a predoctoral fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Yang CH, Crowley DE, Borneman J, Keen NT (2001) Microbial phyllosphere populations are more complex than previously realized. Proc Natl Acad Sci U S A 98: 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marschner P, Yang CH, Lieberei R, Crowley DE (2001) Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil Biol Biochem 33: 1437–1445. [Google Scholar]
- 3. Nunan N, Daniell TJ, Singh BK, Papert A, McNicol JW, et al. (2005) Links between plant and rhizoplane bacterial communities in grassland soils, characterized using molecular techniques. Appl Environ Microbiol 71: 6784–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunter PJ, Hand P, Pink D, Whipps JM, Bending GD (2010) Both leaf properties and microbe-microbe interactions influence within-species variation in bacterial population diversity and structure in the lettuce (Lactuca species) phyllosphere. Appl Environ Microbiol 76: 8117–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rasche F, Trondl R, Naglreiter C, Reichenauer T, Sessitsch A (2006) Chilling and cultivar type affect the diversity of bacterial endophytes colonizing sweet pepper (Capsicum anuum L.). Can J Microbiol 52: 1036–1045. [DOI] [PubMed] [Google Scholar]
- 6. van Overbeek L, van Elsas JD (2008) Effects of plant genotype and growth stage on the structure of bacterial communities associated with potato (Solanum tuberosum L.). FEMS Microbiol Ecol 64: 283–296. [DOI] [PubMed] [Google Scholar]
- 7. Redford AJ, Fierer N (2009) Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb Ecol 58: 189–198. [DOI] [PubMed] [Google Scholar]
- 8. Ercolani GL (1991) Distribution of epiphytic bacteria on olive leaves and the influence of leaf age and sampling time. Microb Ecol 21: 35–48. [DOI] [PubMed] [Google Scholar]
- 9. Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68: 1–13. [DOI] [PubMed] [Google Scholar]
- 10.Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA (2010) Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J: 719–728. [DOI] [PubMed]
- 11. Lindow SE, Brandl MT (2003) Microbiology of the phyllosphere. Appl Environ Microbiol 69: 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badri DV, Weir TL, van der Lelie D, Vivanco JM (2009) Rhizosphere chemical dialogues: plant-microbe interactions. Curr Opin Biotechnol 20: 642–650. [DOI] [PubMed] [Google Scholar]
- 13. Hutchinson GE (1961) The paradox of the plankton. American Naturalist 95: 137–145. [Google Scholar]
- 14. Chesson PL, Warner RR (1981) Environmental variability promotes coexistence in lottery competitive-systems. American Naturalist 117: 923–943. [Google Scholar]
- 15. Berg G, Krechel A, Ditz M, Sikora RA, Ulrich A, et al. (2005) Endophytic and ectophytic potato-associated bacterial communities differ in structure and antagonistic function against plant pathogenic fungi. FEMS Microbiol Ecol 51: 215–229. [DOI] [PubMed] [Google Scholar]
- 16.Timms-Wilson TM, Smalla K, Goodall TI, Houlden A, Gallego V, et al. (2006) Microbial diversity in the phyllosphere and rhizosphere of field grown crop plants: Microbial specialisation at the plant surface. In: Bailey MJ, Lilley AK, Timms-Wilson T, M, Spencer-Phililips PTN, editors. Microbial ecology of aerial plant surfaces: CAB International. 21–36.
- 17. Innerebner G, Knief C, Vorholt JA (2011) Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl Environ Microbiol 77: 3202–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Micallef SA, Shiaris MP, Colon-Carmona A (2009) Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot 60: 1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reisberg EE, Hildebrandt U, Riederer M, Hentschel U (2012) Phyllosphere bacterial communities of trichome-bearing and trichomeless Arabidopsis thaliana leaves. Antonie Van Leeuwenhoek International Journal of General and Molecular Microbiology 101: 551–560. [DOI] [PubMed] [Google Scholar]
- 20. Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, et al. (2009) Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci U S A 106: 16428–16433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, et al. (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488: 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, et al. (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488: 91–95. [DOI] [PubMed] [Google Scholar]
- 23. Qvit-Raz N, Jurkevitch E, Belkin S (2008) Drop-size soda lakes: Transient microbial habitats on a salt-secreting desert tree. Genetics 178: 1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chelius MK, Triplett EW (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microbial Ecology. 41: 252–263. [DOI] [PubMed] [Google Scholar]
- 25. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oksanen J, Blanchet FG, Kindt R, Legendre P, O'Hara RB, et al. (2011) vegan: Community Ecology Package. R package version 1.17–10.
- 27. Legendre P (2005) Species associations: The Kendall coefficient of concordance revisited. J Agric Biol Environ Stat 10: 226–245. [Google Scholar]
- 28. Legendre P, Gallagher ED (2001) Ecologically meaningful transformations for ordination of species data. Oecologia 129: 271–280. [DOI] [PubMed] [Google Scholar]
- 29. Jost L (2007) Partitioning diversity into independent alpha and beta components. Ecology 88: 2427–2439. [DOI] [PubMed] [Google Scholar]
- 30.Sheldon AL (1969) Equitability Indices - Dependence on Species Count. Ecology 50: 466-&.
- 31. Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecology 26: 32–46. [Google Scholar]
- 32. Berry KJ, Johnston JE, Mielke PW Jr (2003) Permutation methods for the analysis of matched-pairs experimental designs. Psychol Rep 92: 1141–1150. [DOI] [PubMed] [Google Scholar]
- 33. Clarke KR (1993) Nonparametric Multivariate Analyses of Changes in Community Structure. Australian Journal of Ecology 18: 117–143. [Google Scholar]
- 34. Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Applied and Environmental Microbiology 71: 1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whittaker RH (1972) Evolution and measurement of species diversity. Taxon 21: 213–251. [Google Scholar]
- 36. Manter DK, Delgado JA, Holm DG, Stong RA (2010) Pyrosequencing reveals a highly diverse and cultivar-specific bacterial endophyte community in potato roots. Microb Ecol 60: 157–166. [DOI] [PubMed] [Google Scholar]
- 37. Lopez-Velasco G, Welbaum GE, Boyer RR, Mane SP, Ponder MA (2011) Changes in spinach phylloepiphytic bacteria communities following minimal processing and refrigerated storage described using pyrosequencing of 16S rRNA amplicons. J Appl Microbiol 110: 1203–1214. [DOI] [PubMed] [Google Scholar]
- 38. Becker R, Behrendt U, Hommel B, Kropf S, Ulrich A (2008) Effects of transgenic fructan-producing potatoes on the community structure of rhizosphere and phyllosphere bacteria. FEMS Microbiol Ecol 66: 411–425. [DOI] [PubMed] [Google Scholar]
- 39. Sun L, Qiu FB, Zhang XX, Dai X, Dong XZ, et al. (2008) Endophytic bacterial diversity in rice (Oryza sativa L.) roots estimated by 16S rDNA sequence analysis. Microb Ecol 55: 415–424. [DOI] [PubMed] [Google Scholar]
- 40. Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N (2010) The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12: 2885–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uroz S, Buee M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep 2: 281–288. [DOI] [PubMed] [Google Scholar]
- 42. Rastogi G, Sbodio A, Tech JJ, Suslow TV, Coaker GL, et al. (2012) Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J 6: 1812–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takeuchi M, Hamana K, Hiraishi A (2001) Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 51: 1405–1417. [DOI] [PubMed] [Google Scholar]
- 44. Kniskern JM, Traw MB, Bergelson J (2007) Salicylic acid and jasmonic acid signaling defense pathways reduce natural bacterial diversity on Arabidopsis thaliana . Mol Plant-Microbe Interact 20: 1512–1522. [DOI] [PubMed] [Google Scholar]
- 45. Lee ZMP, Bussema C, Schmidt TM (2009) rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37: D489–D493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crosby LD, Criddle CS (2003) Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. Biotechniques 34: 790–802. [DOI] [PubMed] [Google Scholar]
- 47. Garbeva P, van Veen JA, van Elsas JD (2004) Microbial diversity in soil: selection microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol 42: 243–270. [DOI] [PubMed] [Google Scholar]
- 48. Smalla K, Weinert N, Meincke R, Gottwald C, Heuer H, et al. (2009) Rhizosphere Communities of Genetically Modified Zeaxanthin-Accumulating Potato Plants and Their Parent Cultivar Differ Less than Those of Different Potato Cultivars. Applied and Environmental Microbiology 75: 3859–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosenblueth M, Martinez-Romero E (2006) Bacterial endophytes and their interactions with hosts. Molecular Plant-Microbe Interactions 19: 827–837. [DOI] [PubMed] [Google Scholar]
- 50. Lindow SE, Leveau JH (2002) Phyllosphere microbiology. Current Opinion in Biotechnology 13: 238–243. [DOI] [PubMed] [Google Scholar]
- 51. Hardoim PR, van Overbeek LS, van Elsas JD (2008) Properties of bacterial endophytes and their proposed role in plant growth. Trends in Microbiology 16: 463–471. [DOI] [PubMed] [Google Scholar]
- 52. Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, et al. (2011) Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol 77: 5934–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Beattie GA, Lindow SE (1999) Bacterial colonization of leaves: a spectrum of strategies. Phytopathology 89: 353–359. [DOI] [PubMed] [Google Scholar]
- 54. Chi F, Shen SH, Cheng HP, Jing YX, Yanni YG, et al. (2005) Ascending migration of endophytic rhizobia, from roots to leaves, inside rice plants and assessment of benefits to rice growth physiology. Appl Environ Microbiol 71: 7271–7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Czajkowski R, de Boer WJ, Velvis H, van der Wolf JM (2010) Systemic colonization of potato plants by a soilborne, green fluorescent protein-tagged strain of Dickeya sp. biovar 3. Phytopathology 100: 134–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rank abundance of the 50 most heavily sequenced OTUs. Roots (left) and leaves (right). OTU numbers were replaced with GENUS, FAMILY or ORDER name depending on the level at which this OTU could be assigned. Arabidopsis thaliana were collected at 4 sites (purple, Route Marker; blue, North Liberty; green, Michigan Extension; red, Lake Michigan College). DNA was extracted for endophytic fraction (circle) and epiphytic fraction (triangle).
(TIF)
Alpha diversity of the bacterial communities in the leaves and roots of Arabidopsis thaliana . (A) Richness; (B) Diversity; (C) Evenness. Bars represent one standard error of the mean. The letters indicate results from paired t-test (P<0.05, P values adjusted using fdr).
(TIF)